
Synthesis, Biological, Spectroscopic and Computational Investigations of Novel N-Acylhydrazone Derivatives of Pyrrolo[3,4-d]pyridazinone as Dual COX/LOX Inhibitors
 ,
,  , , , ,
, , , ,  , ,
, ,  and
and
Abstract
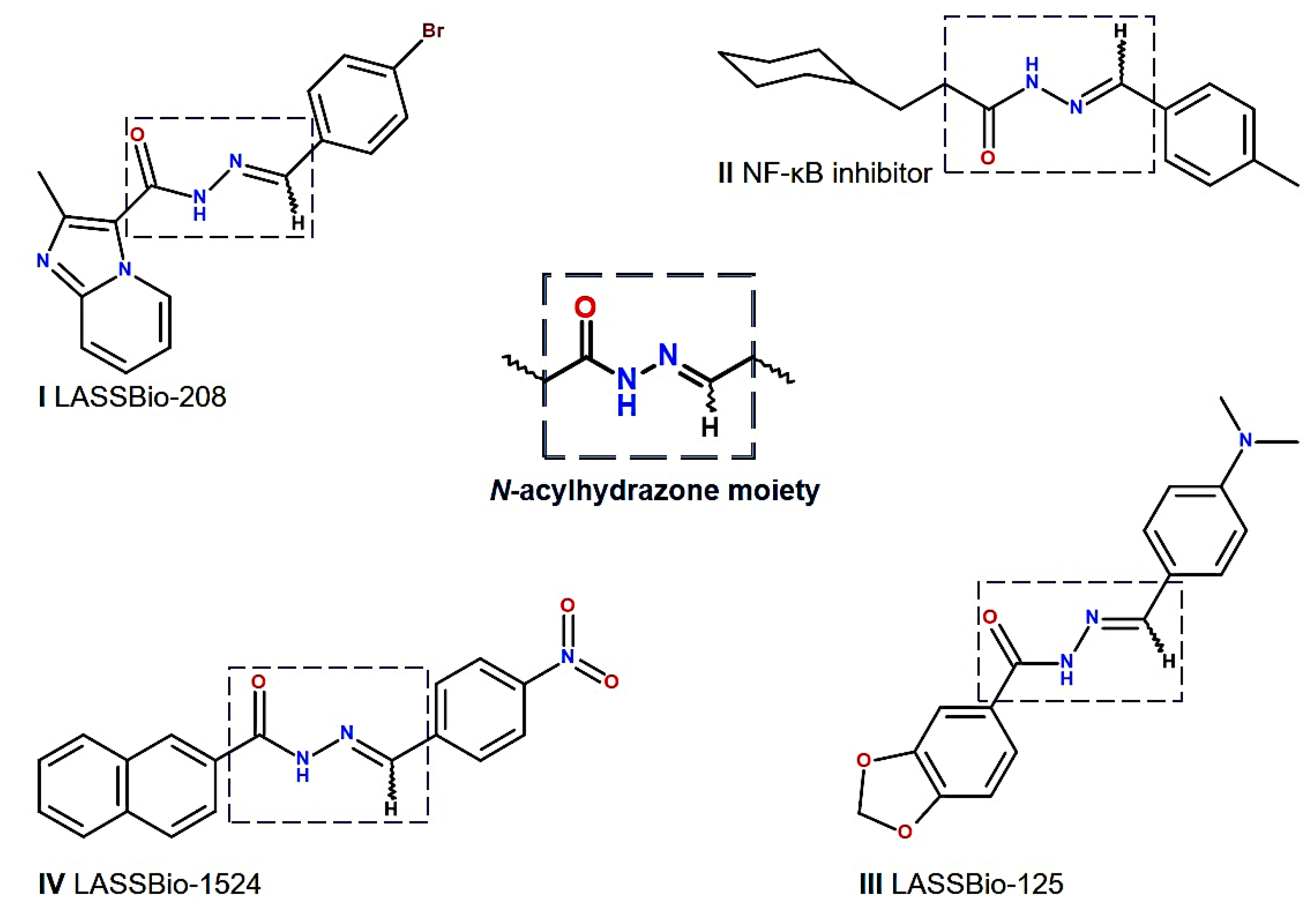
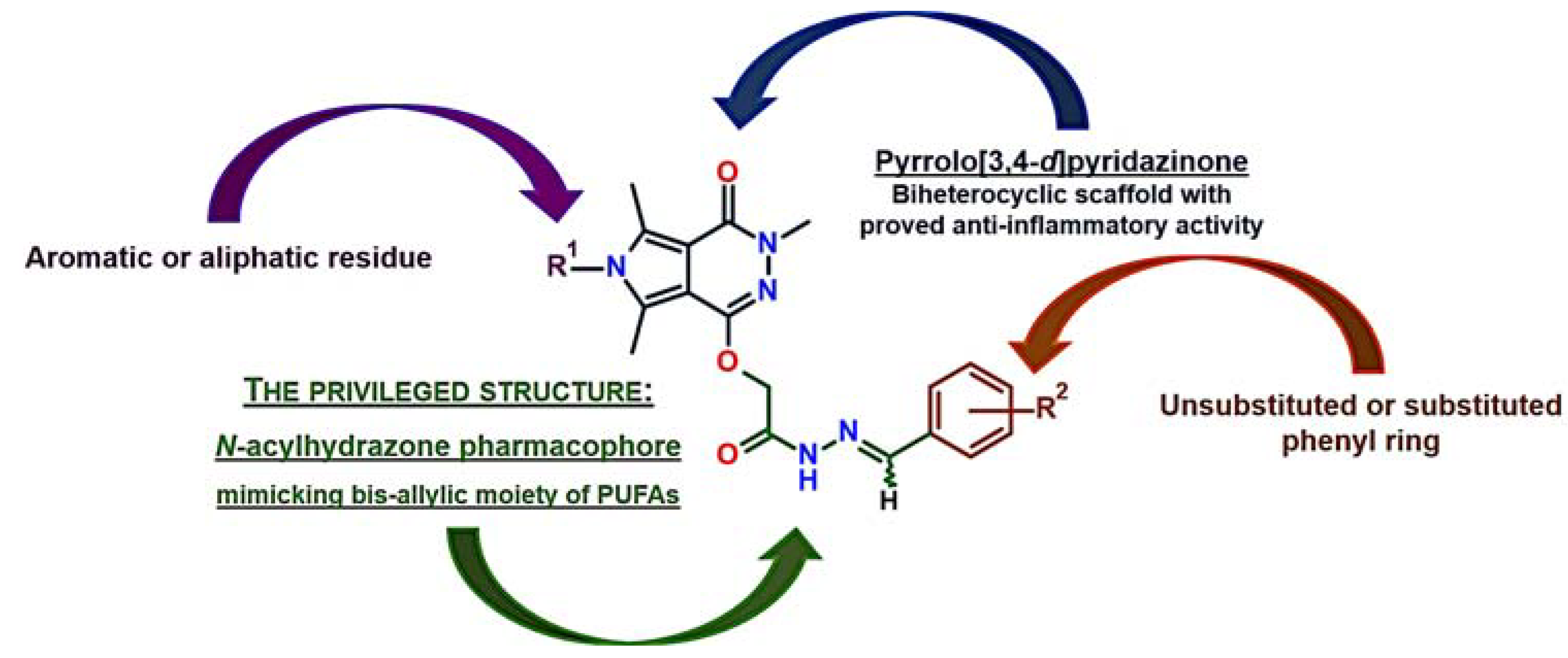
1. Introduction
2. Results
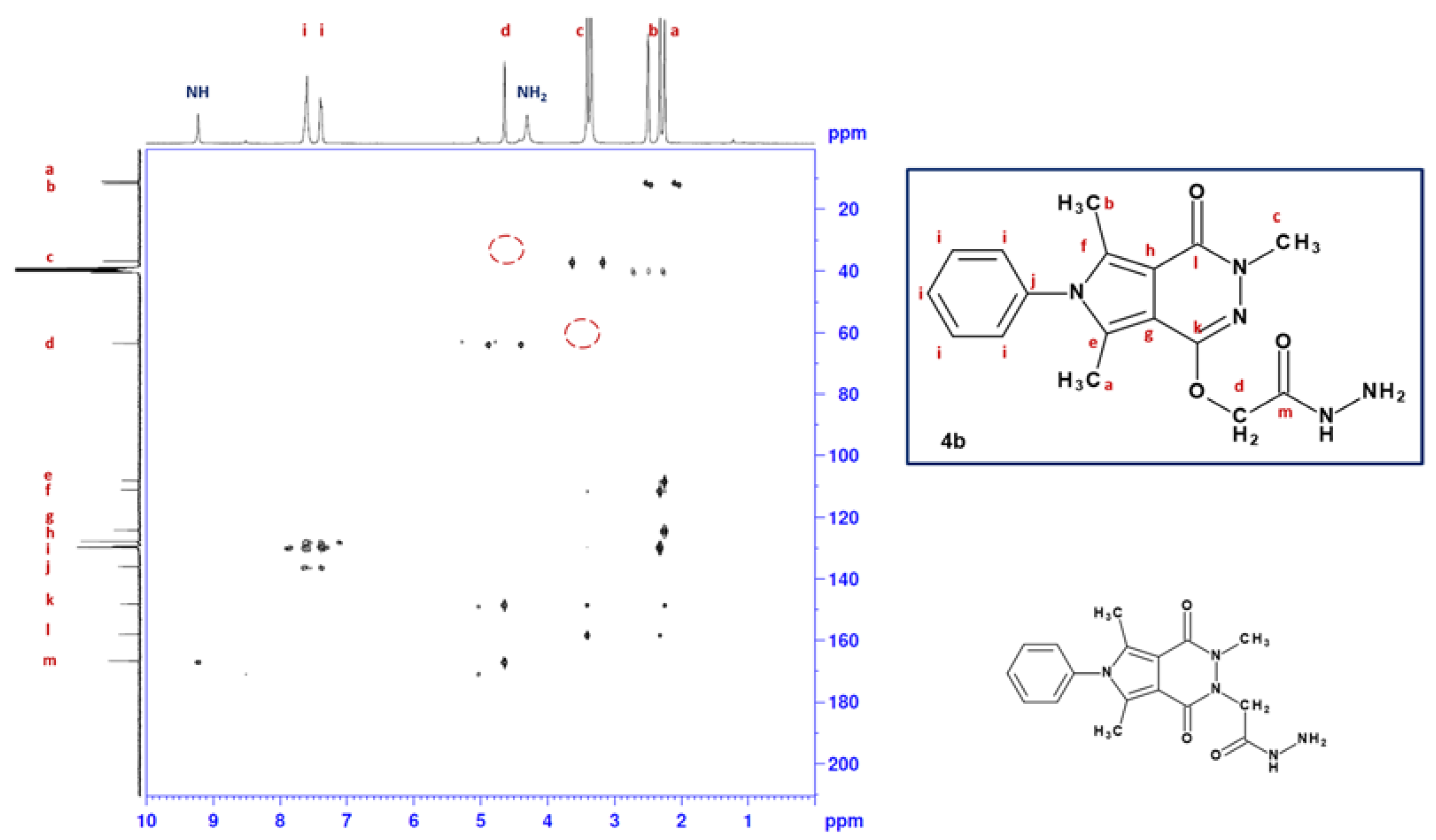
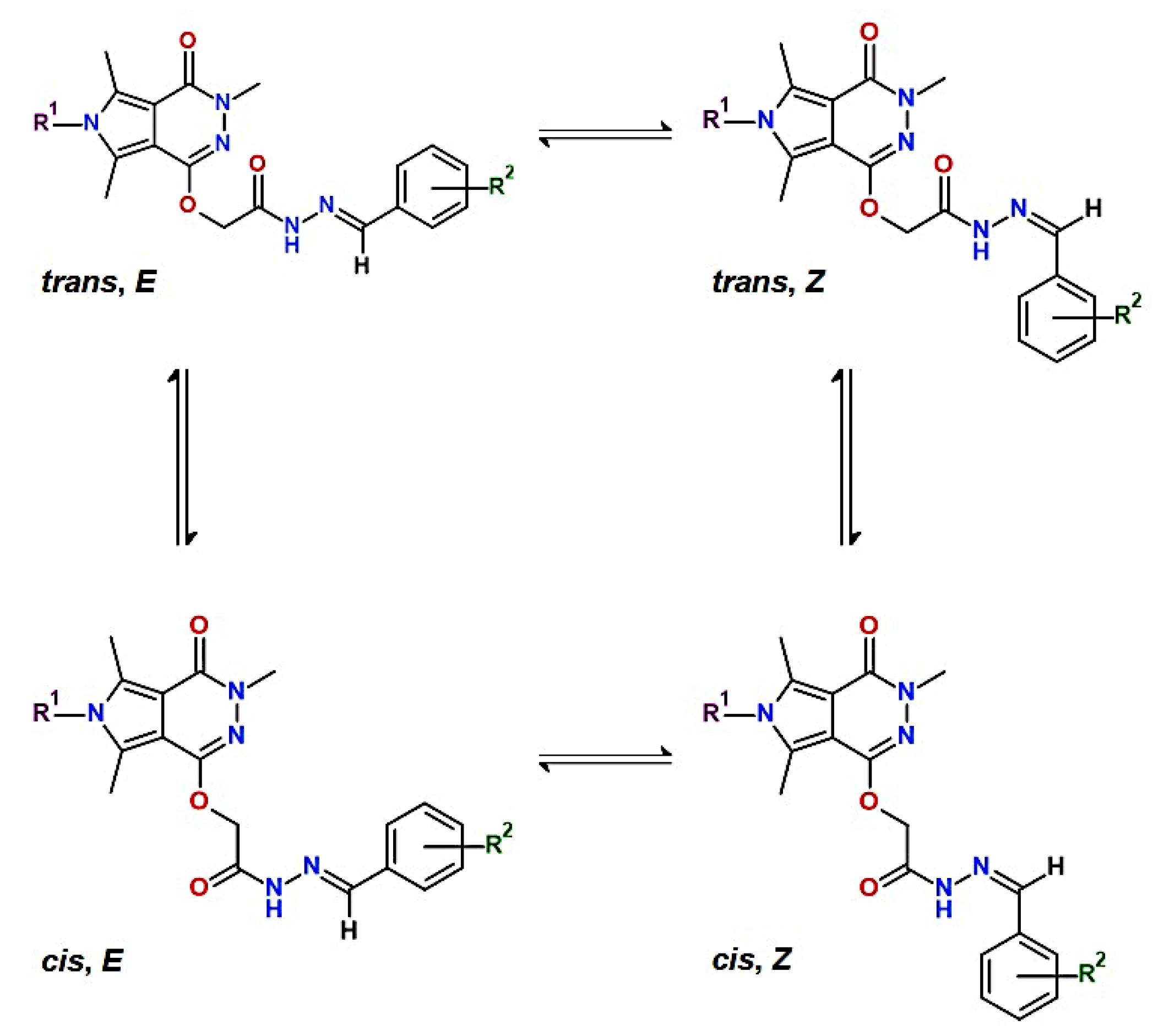
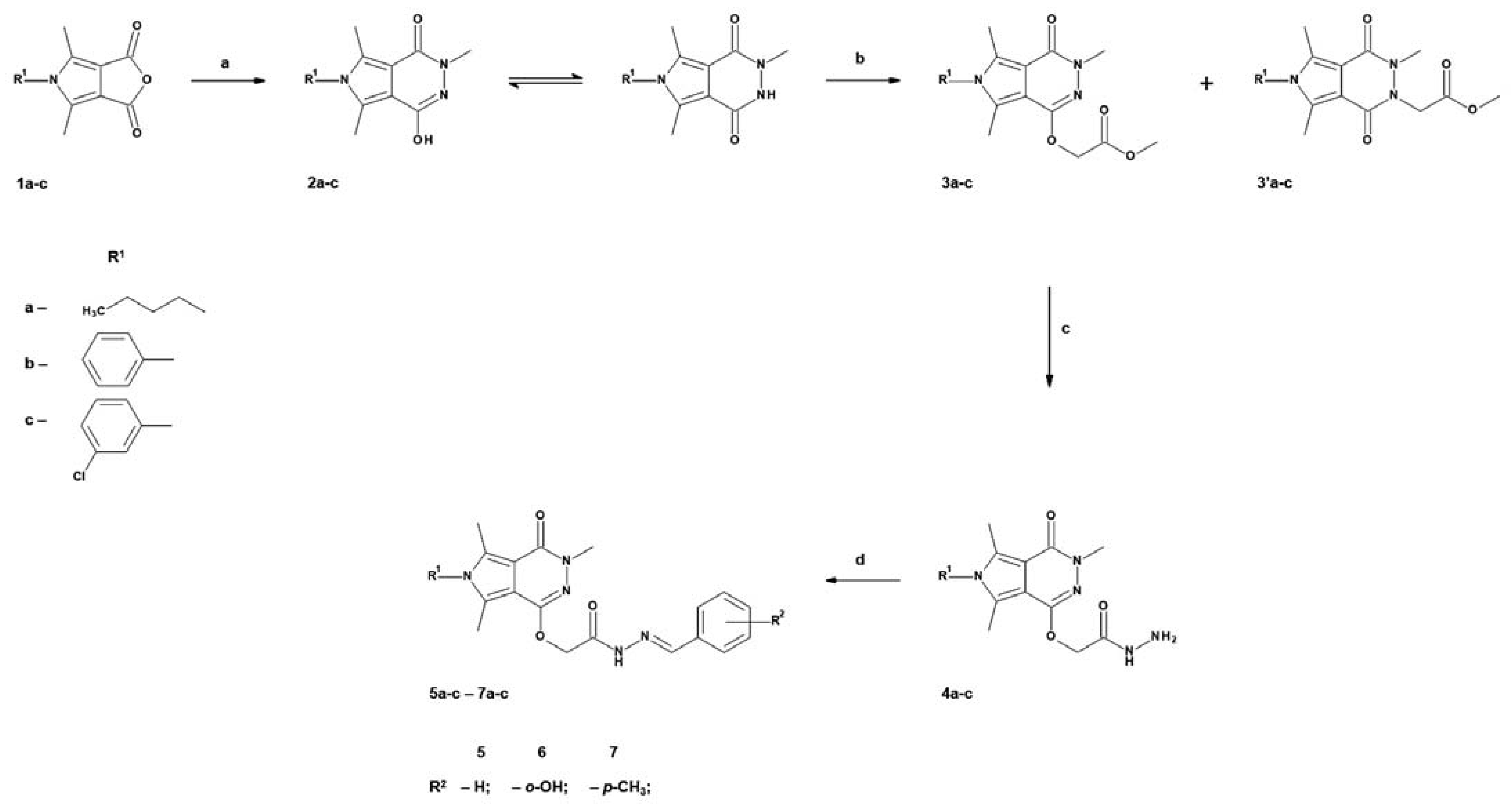
2.1. Chemistry
2.2. Evaluation of Viability
2.3. Cyclooxygenase (COX-1 and COX-2) and 15-Lipoxygenase (15-LOX) Inhibition Studies
2.3.1. In Vitro Inhibition Assay
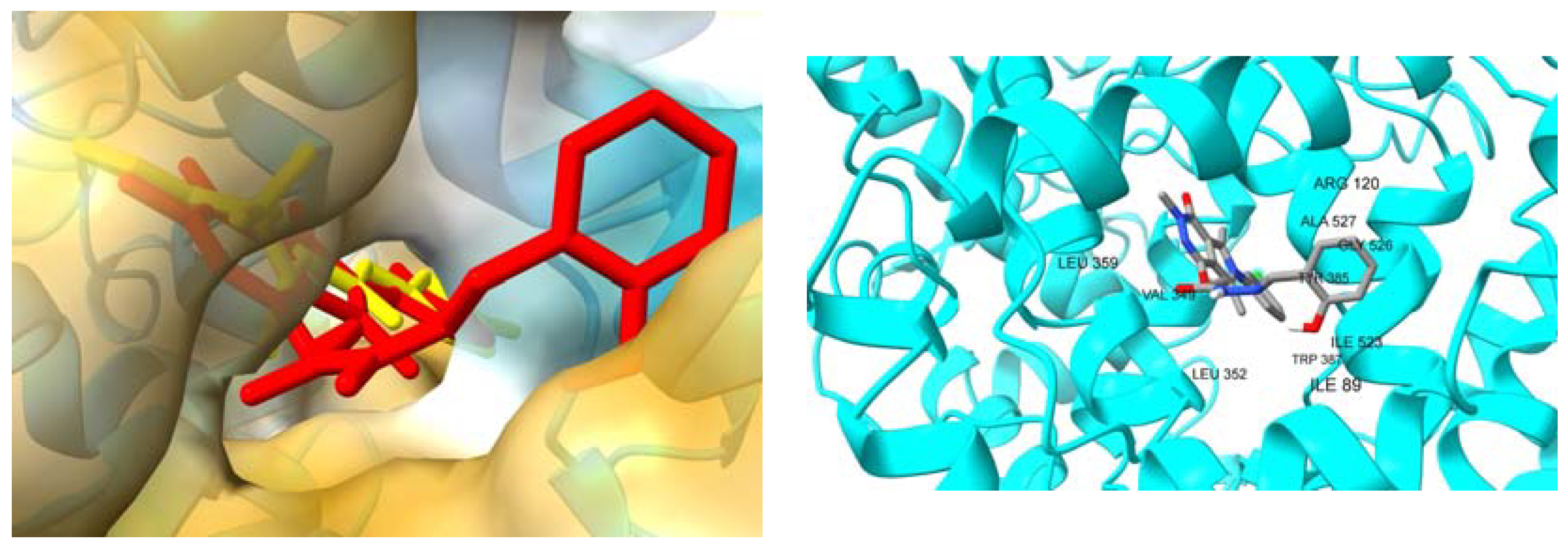
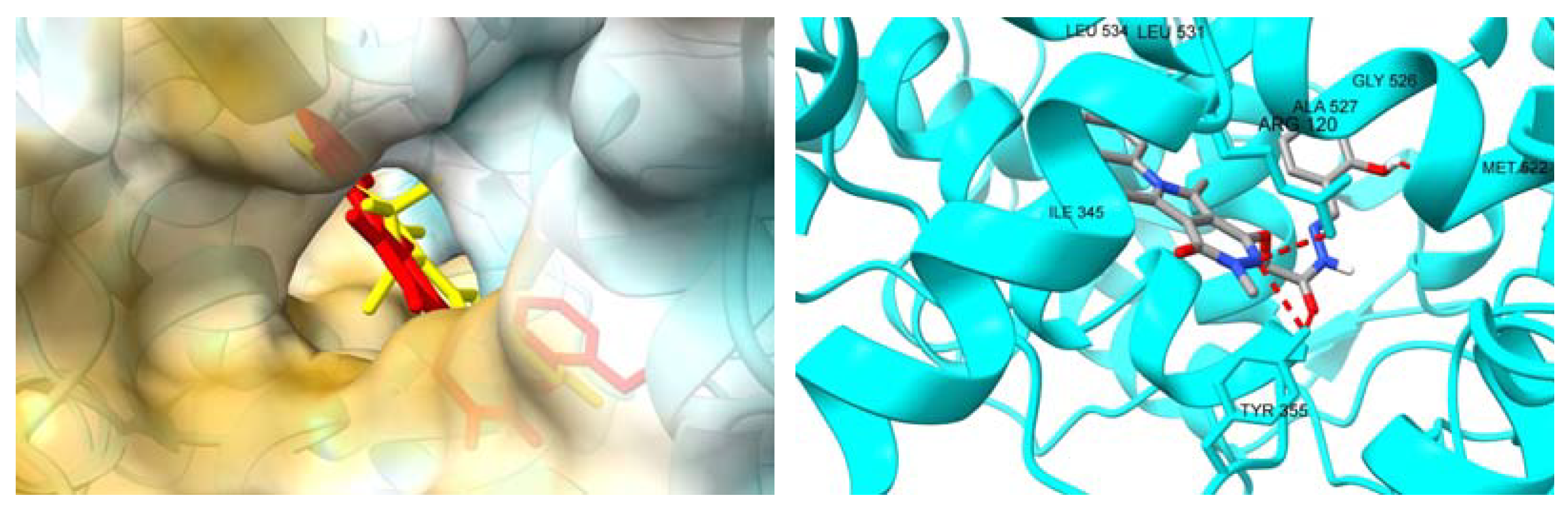
2.3.2. Molecular Docking Studies
2.4. Interactions with Plasma Proteins
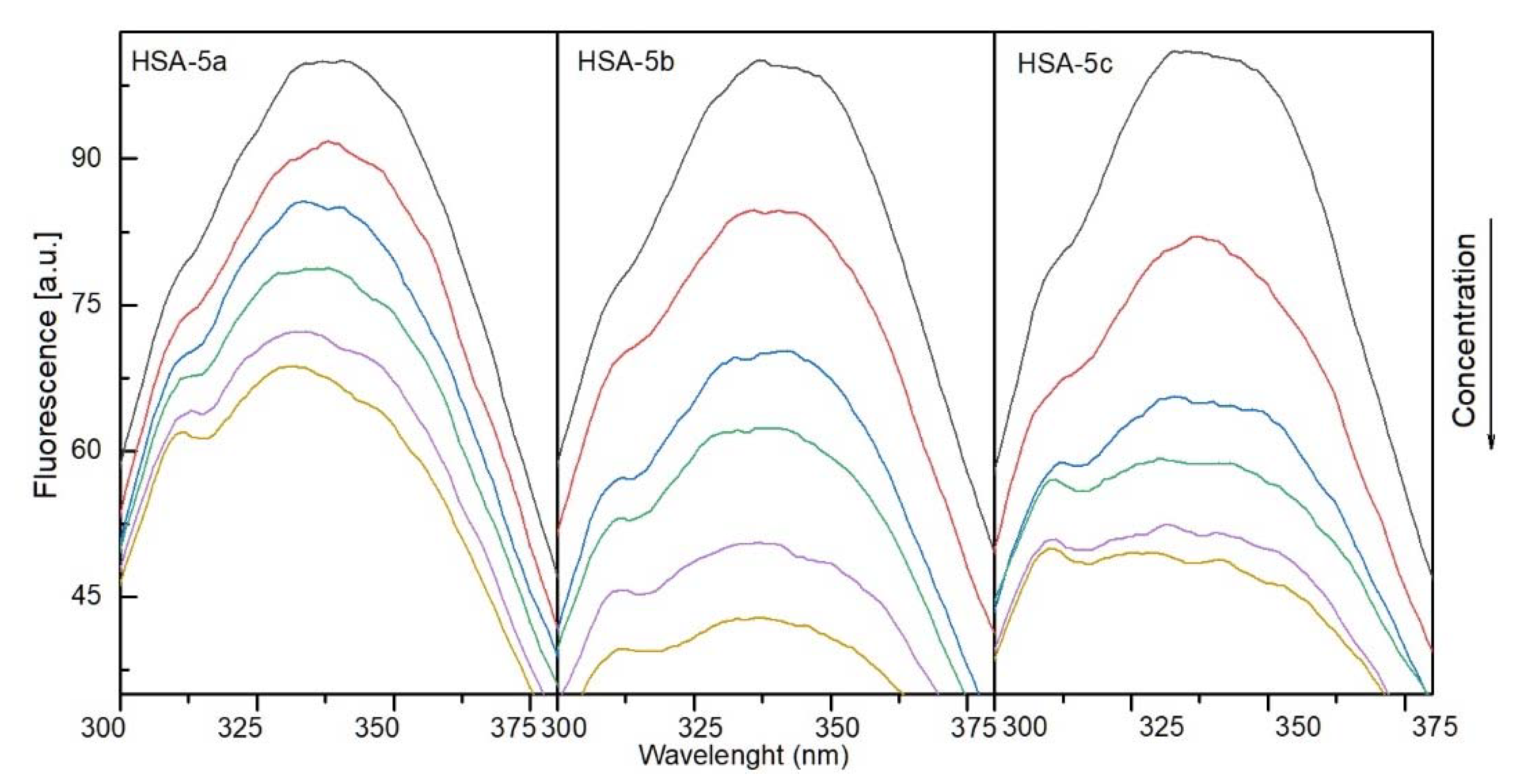
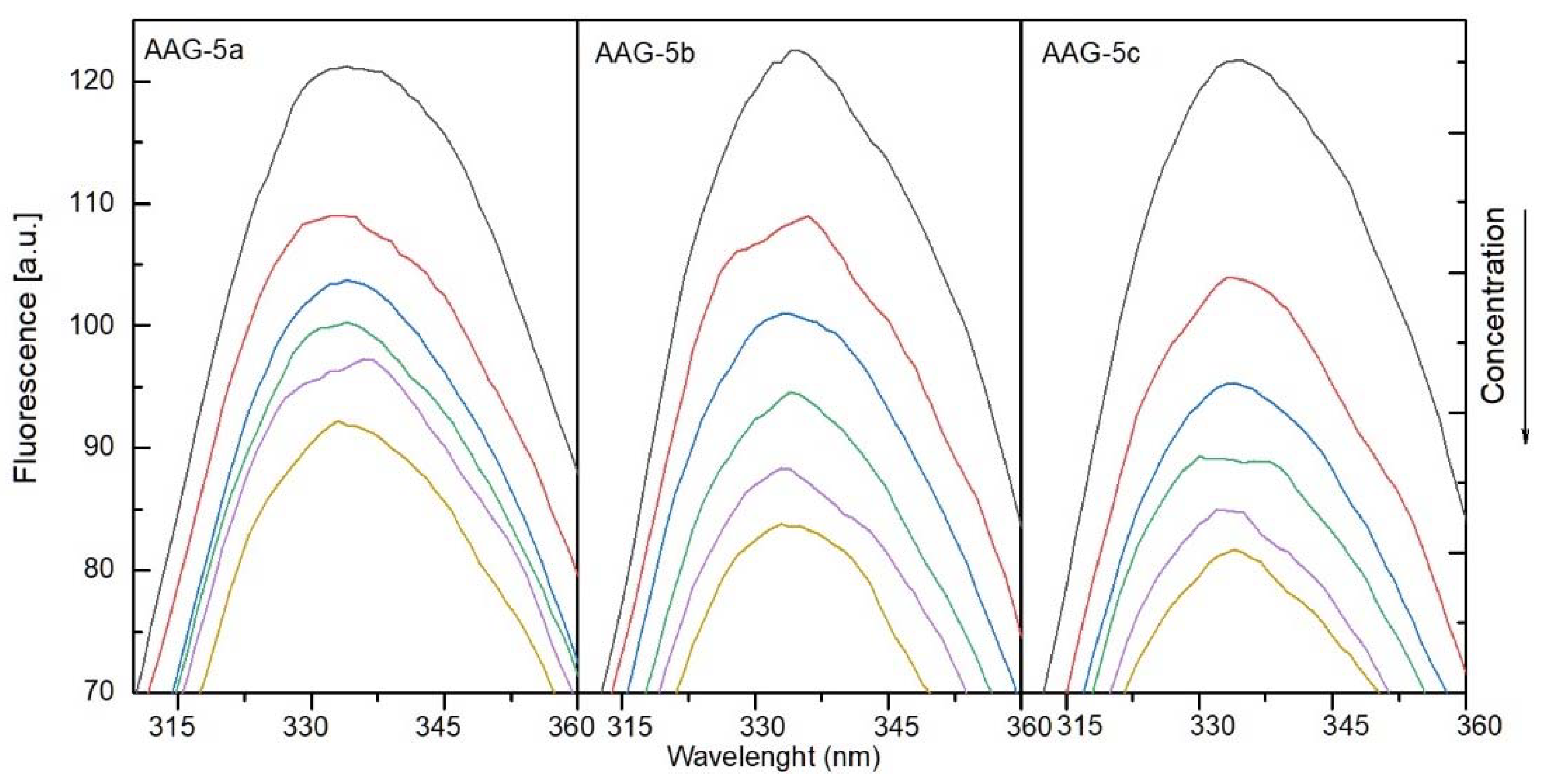
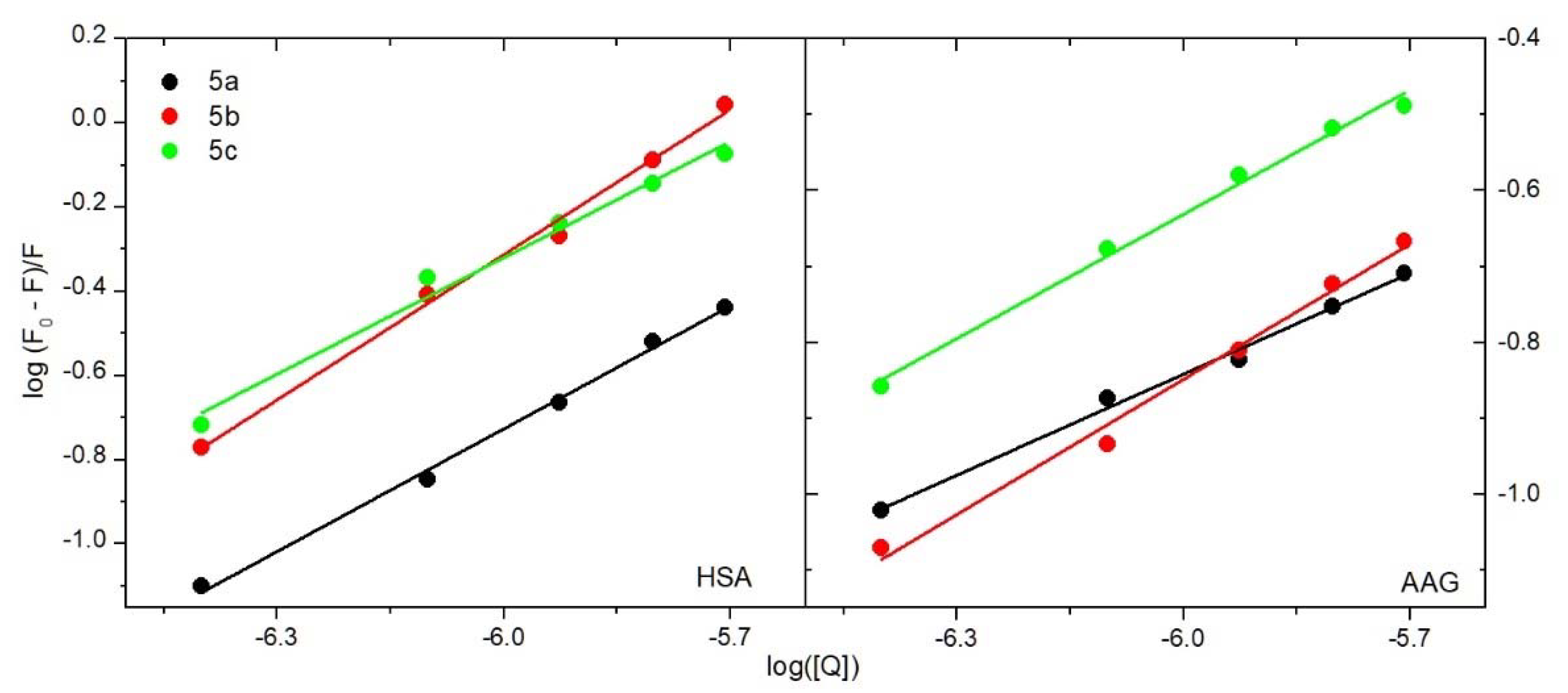
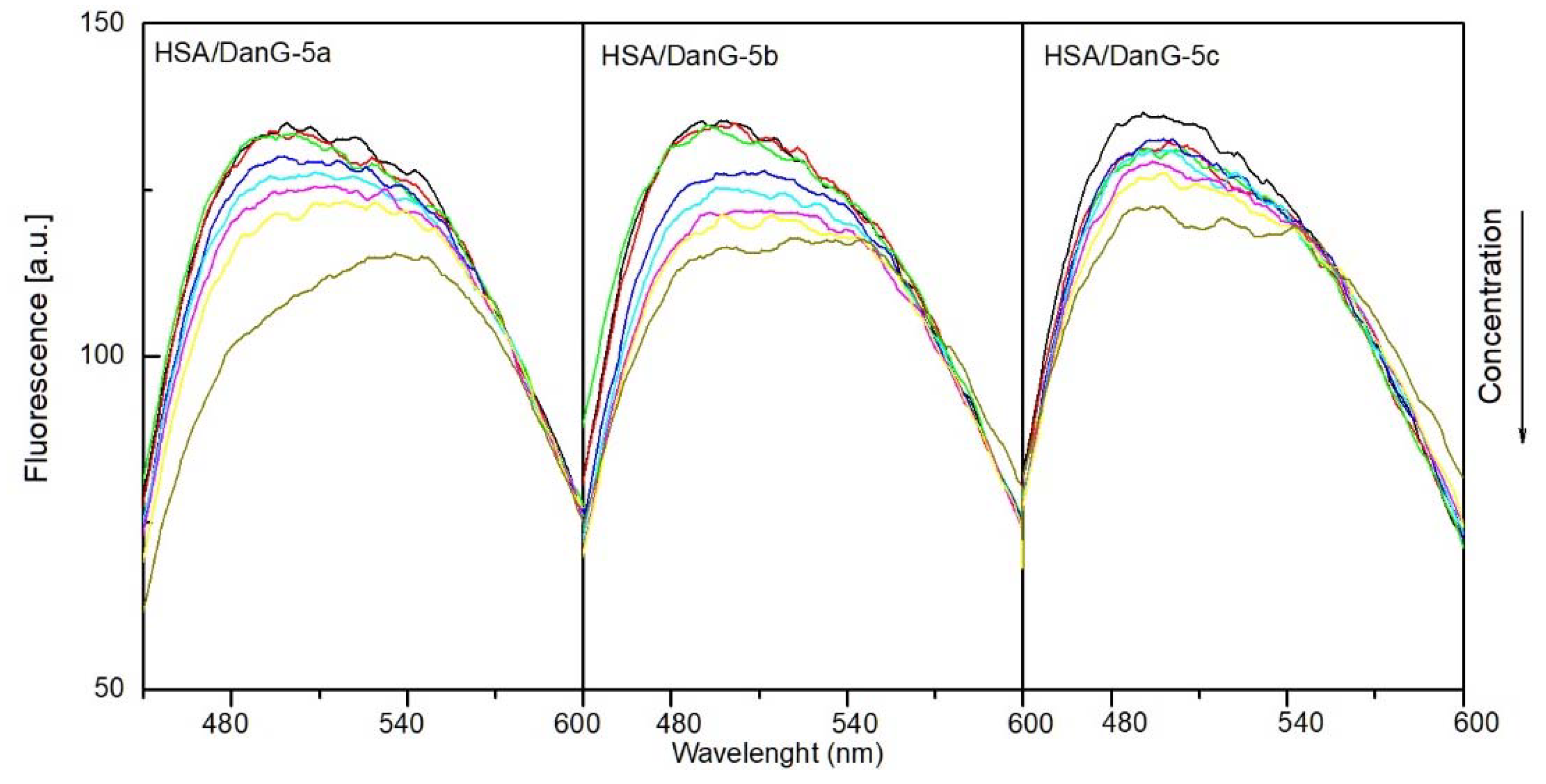
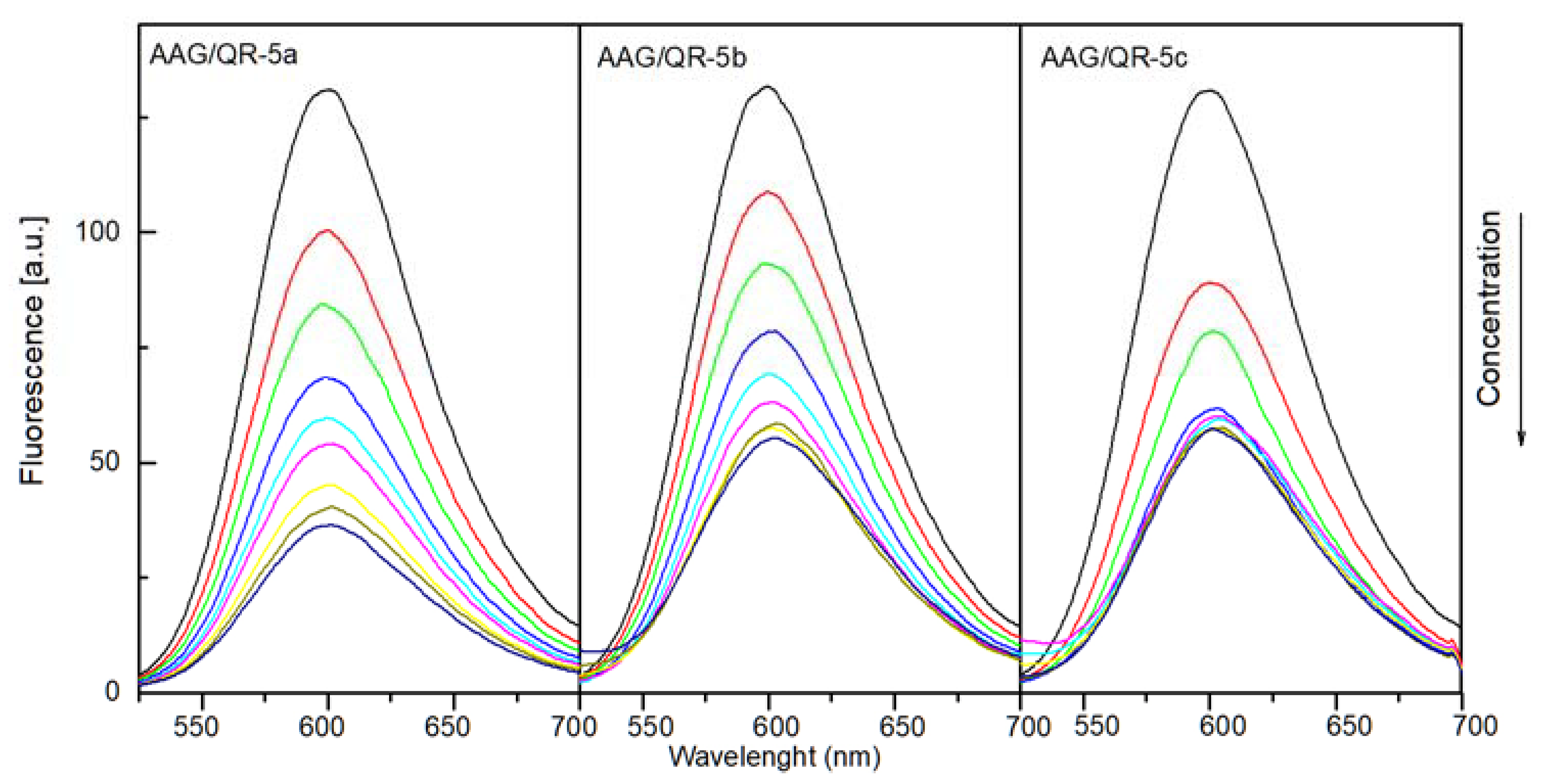
2.4.1. The Fluorescence Spectroscopic Studies
2.4.2. Binding Site Studies with HSA and AAG
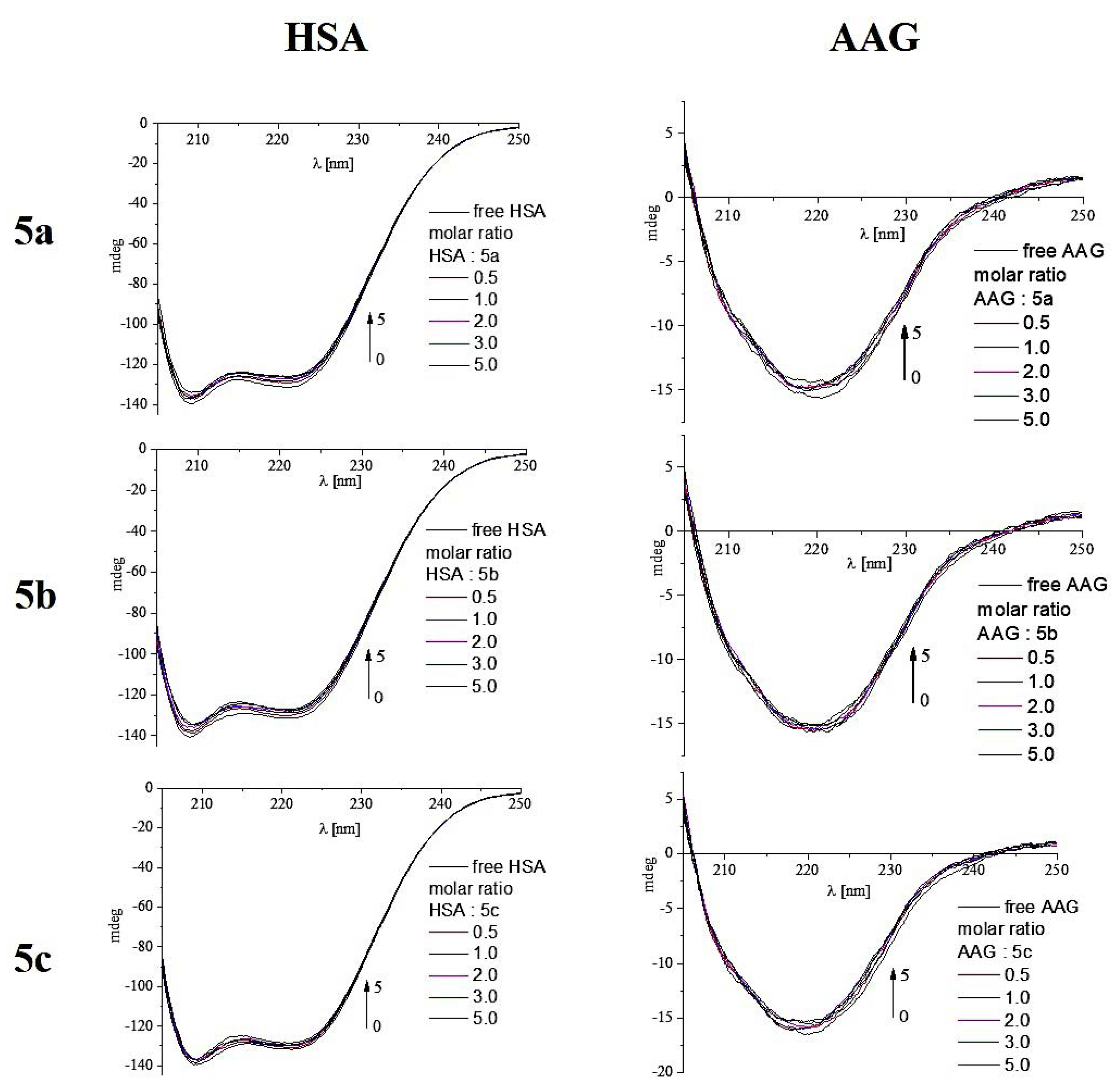
2.4.3. Circular Dichroism Spectroscopy
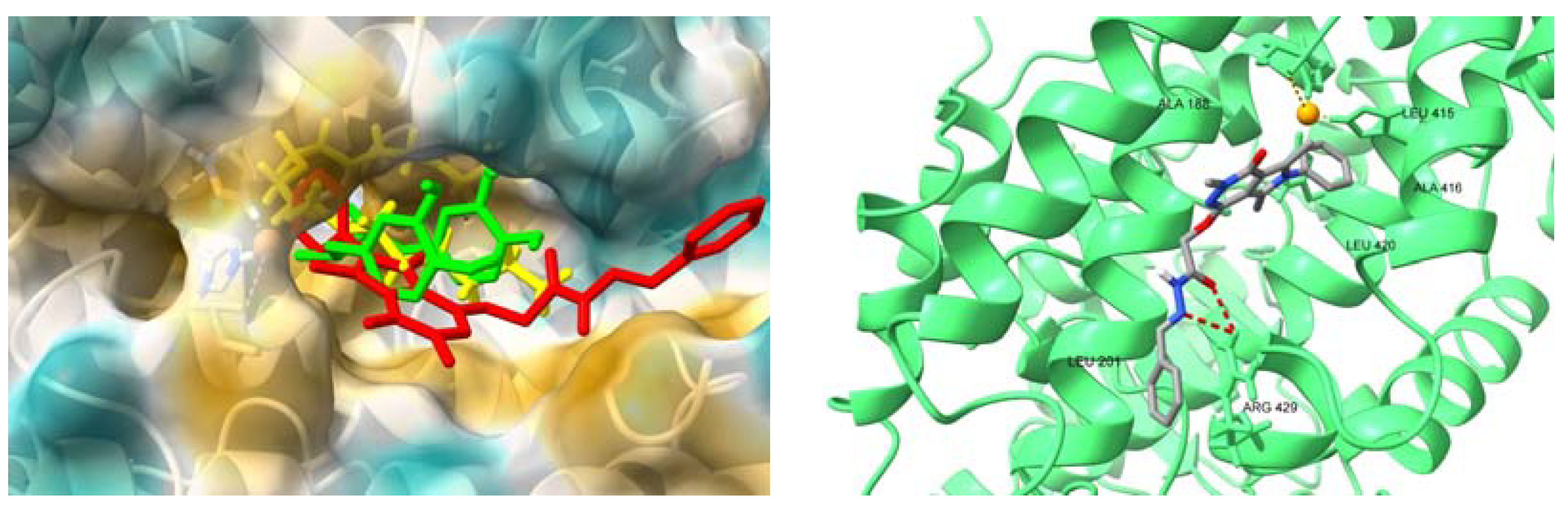
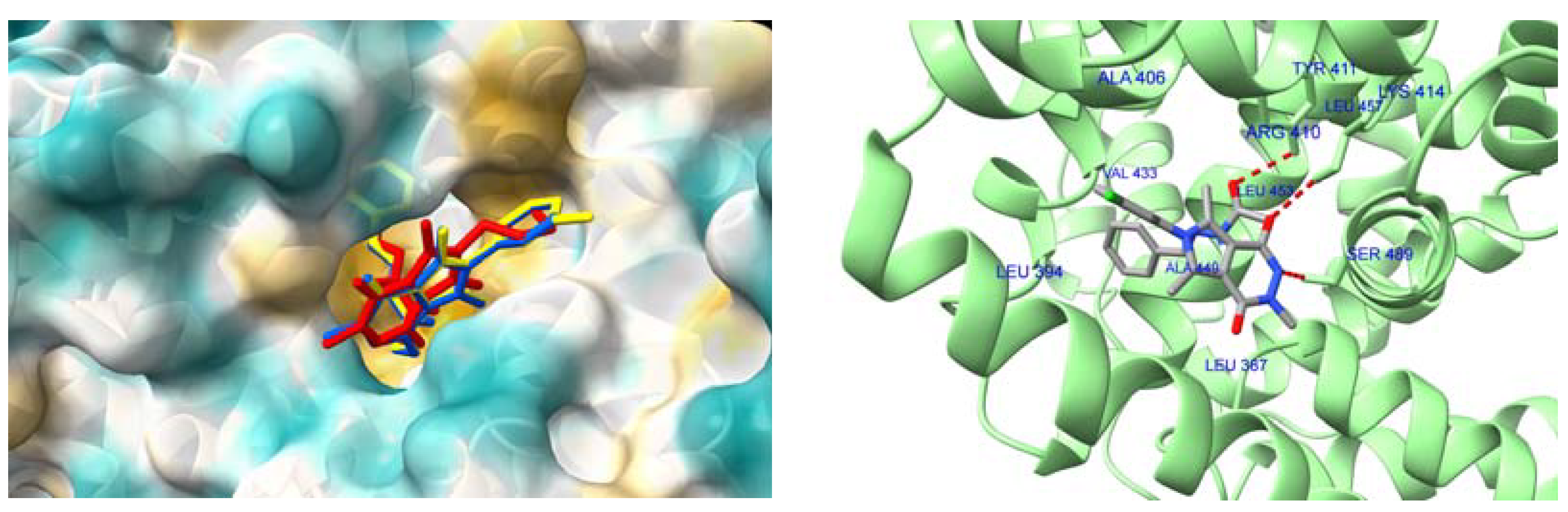
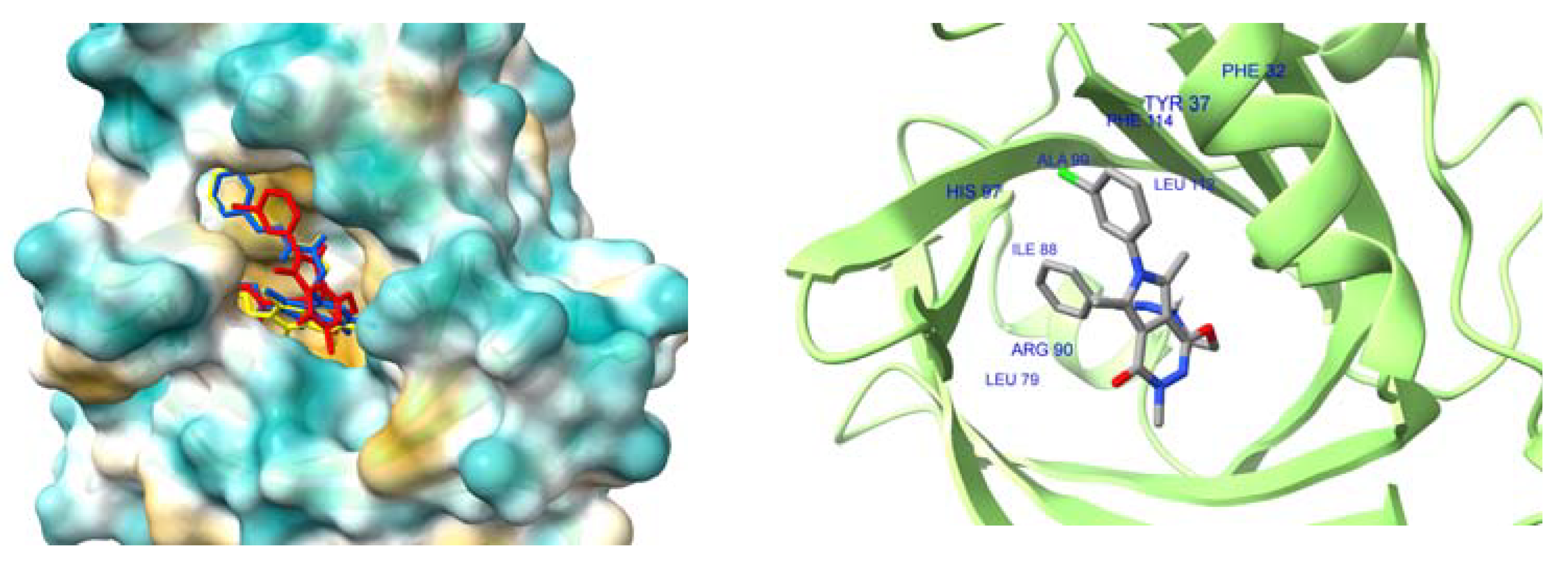
2.4.4. Interactions with HSA and AAG–Molecular Docking Studies
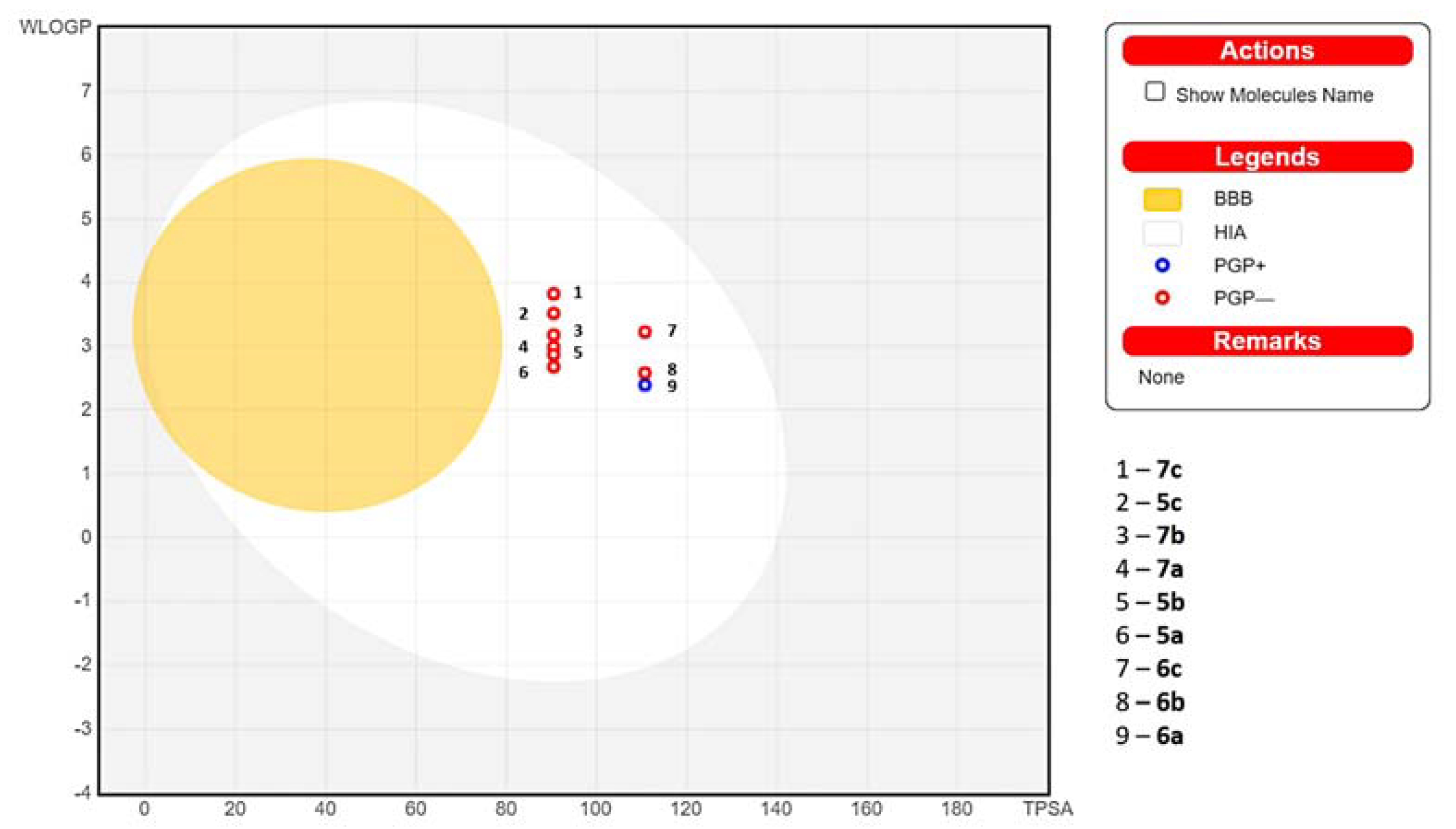
2.5. In Silico Absorption Distribution Metabolism and Excretion (ADME) Pharmacokinetic and Drug-Likeness Predictions
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Instrumentation and Chemicals
4.1.2. Chemical Synthesis
The Synthetic Procedure for 6-(3-chlorophenyl)-3,5,7-trimethyl-2H-pyrrolo[3,4-d]pyridazine-1,4-dione (2c):
- 2c: 6-(3-chlorophenyl)-3,5,7-trimethyl-2-[H]-pyrrolo[3,4-d]pyridazine-1,4-dione
The Synthetic Procedure for methyl 2-[6-(3-chlorophenyl)-3,5,7-trimethyl-4-oxo-Pyrrolo[3,4-d]pyridazin-1-yl]oxyacetate (3c):
- 3c: Methyl 2-[6-(3-chlorophenyl)-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl]oxyacetate
The Synthetic Procedure for 2-[6-(3-chlorophenyl)-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl]oxyacetohydrazide (4c):
- 4c: 2-[6-(3-chlorophenyl)-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl]oxyacetohydrazide
The General Procedure for Preparation of Title N-acylhydrazones (5a-c–7a-c)
- 5a: N-[(Z/E)-benzylideneamino]-2-(6-butyl-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl)oxy-acetamide
- 6a: 2-(6-butyl-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl)oxy-N-[(Z/E)-(2-hydroxyphenyl)methyleneamino]acetamide
- 7a: 2-(6-butyl-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl)oxy-N-[(Z/E)-p-tolylmethyleneamino]acetamide
- 5b: N-[(Z/E)-benzylideneamino]-2-(3,5,7-trimethyl-4-oxo-6-phenyl-pyrrolo[3,4-d]pyridazin-1-yl)oxy-acetamide
- 6b: N-[(Z/E)-(2-hydroxyphenyl)methyleneamino]-2-(3,5,7-trimethyl-4-oxo-6-phenyl-pyrrolo[3,4-d]pyridazin-1-yl)oxy-acetamide
- 7b: N-[(Z/E)-p-tolylmethyleneamino]-2-(3,5,7-trimethyl-4-oxo-6-phenyl-pyrrolo[3,4-d]pyridazin-1-yl)oxy-acetamide
- 5c: N-[(Z/E)-benzylideneamino]-2-[6-(3-chlorophenyl)-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl]oxyacetamide
- 6c: 2-[6-(3-chlorophenyl)-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl]oxy-N-[(Z/E)-(2-hydroxyphenyl)methyleneamino]acetamide
- 7c: 2-[6-(3-chlorophenyl)-3,5,7-trimethyl-4-oxo-pyrrolo[3,4-d]pyridazin-1-yl]oxy-N-[(Z/E)-p-tolylmethyleneamino]acetamide
4.2. Biological Evaluation
4.2.1. Cell Line and Culture Conditions
4.2.2. Tested Compounds
4.2.3. Cytotoxicity Assay
4.2.4. Evaluation of COX-1, COX-2 and 15-LOX Inhibitory Activity
4.2.5. Statistical Analysis
4.3. Molecular Docking Studies
4.4. Spectroscopic Studies
4.4.1. Fluorescence Spectroscopy
4.4.2. Circular Dichroism Spectroscopy
4.5. In Silico Pharmacokinetic, Physicochemical and Drug-Likeness Predictions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nathan, C. Points of Control in Inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Savill, J. Resolution of Inflammation: The Beginning Programs the End. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef]
- Leuti, A.; Fazio, D.; Fava, M.; Piccoli, A.; Oddi, S.; Maccarrone, M. Bioactive Lipids, Inflammation and Chronic Diseases. Adv. Drug Deliv. Rev. 2020, 159, 133–169. [Google Scholar] [CrossRef]
- Marnett, L.J. Cyclooxygenase Mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L.; Marnett, L.J.; Hancock, A.B. Perspective Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2006, 50, 1425–1441. [Google Scholar] [CrossRef]
- Chandrasekharan, N.; Simmons, D.L. The Cyclooxygenases. Genome Biol. 2004, 5, 241. [Google Scholar] [CrossRef]
- Omar, Y.M.; Abdel-Moty, S.G.; Abdu-Allah, H.H.M. Further Insight into the Dual COX-2 and 15-LOX Anti-Inflammatory Activity of 1,3,4-Thiadiazole-Thiazolidinone Hybrids: The Contribution of the Substituents at 5th Positions Is Size Dependent. Bioorg. Chem. 2020, 97, 103657. [Google Scholar] [CrossRef]
- Omar, Y.M.; Abdu-Allah, H.H.M.; Abdel-Moty, S.G. Synthesis, Biological Evaluation and Docking Study of 1,3,4-Thiadiazole-Thiazolidinone Hybrids as Anti-Inflammatory Agents with Dual Inhibition of COX-2 and 15-LOX. Bioorg. Chem. 2018, 80, 461–471. [Google Scholar] [CrossRef]
- Jo-Watanabe, A.; Okuno, T.; Yokomizo, T. Molecular Sciences The Role of Leukotrienes as Potential Therapeutic Targets in Allergic Disorders. Int. J. Mol. Sci. 2019, 20, 3580. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Botting, R.M. Mechanism of Action of Nonsteroidal Anti-Inflammatory Drugs. Am. J. Med. 1998, 104, 25–85. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.N. The Mechanisms of Action of NSAIDs in Analgesia. Drugs 1996, 52, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Soll, A.H.; McCarthy, D. NSAID-Related Gastrointestinal Complications. Clin. Cornerstone 1999, 1, 42–56. [Google Scholar] [CrossRef]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse Effects of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs, Aspirin and Coxibs) on Upper Gastrointestinal Tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef]
- Laine, L. Gastrointestinal Effects of NSAIDs and Coxibs. J. Pain Symptom Manag. 2003, 25, 32–40. [Google Scholar] [CrossRef]
- Cannon, C.P.; Cannon, P.J. Physiology. COX-2 Inhibitors and Cardiovascular Risk. Science 2012, 336, 1386–1387. [Google Scholar] [CrossRef]
- Dogné, J.-M.; Supuran, C.T.; Pratico, D. Adverse Cardiovascular Effects of the Coxibs. J. Med. Chem. 2005, 48, 2251–2257. [Google Scholar] [CrossRef]
- Gedawy, E.M.; Kassab, A.E.; El Kerdawy, A.M. Design, Synthesis and Biological Evaluation of Novel Pyrazole Sulfonamide Derivatives as Dual COX-2/5-LOX Inhibitors. Eur. J. Med. Chem. 2020, 189, 112066. [Google Scholar] [CrossRef]
- Jacob, P.J.; Manju, S.L. Identification and Development of Thiazole Leads as COX-2/5-LOX Inhibitors through in-Vitro and in-Vivo Biological Evaluation for Anti-Inflammatory Activity. Bioorg. Chem. 2020, 100, 103882. [Google Scholar] [CrossRef]
- Chen, W.; Xu, Q.; Ma, X.; Mo, J.; Lin, G.; He, G.; Chu, Z.; Li, J. Synthesis and Biological Evaluation of N-(Benzene Sulfonyl)acetamide Derivatives as Anti-Inflammatory and Analgesic Agents with COX-2/5-LOX/ TRPV1 Multifunctional Inhibitory Activity. Bioorg. Med. Chem. Lett 2023, 80, 129101. [Google Scholar] [CrossRef] [PubMed]
- Braeckman, R.A.; Granneman, G.R.; Locke, C.S.; Machinist, J.M.; Cavanaugh, J.H.; Awni, W.M. The Pharmacokinetics of Zileuton in Healthy Young and Elderly Volunteers. Clin. Pharmacokinet. 1995, 29, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, T.F.; Júnior, W.B.; Alexandre-Moreira, M.S.; Costa, F.N.; Da, C.E.; Monteiro, S.; Ferreira, F.F.; Cely, R.; Barroso, R.; Noël, F.; et al. Molecules Novel Orally Active Analgesic and Anti-Inflammatory Cyclohexyl-N-Acylhydrazone Derivatives. Molecules 2015, 20, 3067–3088. [Google Scholar] [CrossRef] [PubMed]
- Rollas, S.; Küçükgüzel, S. Biological Activities of Hydrazone Derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed]
- Chelucci, R.C.; Dutra, L.A.; Pires, M.E.L.; de Melo, T.R.F.; Bosquesi, P.L.; Chung, M.C.; dos Santos, J.L. Molecules Antiplatelet and Antithrombotic Activities of Non-Steroidal Anti-Inflammatory Drugs Containing an N-Acyl Hydrazone Subunit. Molecules 2014, 19, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Horchani, M.; Della Sala, G.; Caso, A.; Esposito, G.; Laurenzana, I.; Giancola, C.; Costantino, V.; Ben Jannet, H.; Romdhane, A. Molecular Docking and Biophysical Studies for Antiproliferative Assessment of Synthetic Pyrazolo-Pyrimidinones Tethered with Hydrazide-Hydrazones. Int. J. Mol. Sci 2021, 22, 2742. [Google Scholar] [CrossRef]
- Beteck, R.M.; Seldon, R.; Jordaan, A.; Warner, D.F.; Hoppe, H.C.; Laming, D.; Legoabe, L.J.; Khanye, S.D. MedChemComm Quinolone-Isoniazid Hybrids: Synthesis and Preliminary in Vitro Cytotoxicity and Anti-Tuberculosis Evaluation †. Cite this Med. Chem. Commun 2019, 10, 326. [Google Scholar] [CrossRef]
- Kaushik-Basu, N.; Durmaz, I.; Manvar, D.; Basu, A.; Atalay, R.; Güniz Küçükgüzel, S. Synthesis of Novel Diflunisal Hydrazideehydrazones as Anti-Hepatitis C Virus Agents and Hepatocellular Carcinoma Inhibitors*. Eur. J. Med. Chem. 2015, 108, 301–308. [Google Scholar] [CrossRef]
- De Miranda, A.S.; Bispo, W.; Da Silva, Y.K.C.; Alexandre-Moreira, M.S.; De Paula Castro, R.; Sabino, J.R.; Lião, L.M.; Lima, L.M.; Barreiro, E.J. Design, Synthesis, Antinociceptive and Anti-Inflammatory Activities of Novel Piroxicam Analogues. Molecules 2012, 17, 14126–14145. [Google Scholar] [CrossRef]
- Hernández, P.; Cabrera, M.; Lavaggi, L.; Celano, L.; Tiscornia, I.; Da Costa, T.R.; Thomson, L.; Bollati-Fogolín, M.; Luisa, A.; Miranda, P.; et al. Discovery of New Orally Effective Analgesic and Anti-Inflammatory Hybrid Furoxanyl N-Acylhydrazone Derivatives. Bioorg. Med. Chem. 2012, 20, 2158–2171. [Google Scholar] [CrossRef]
- Cordeiro, N.; Freitas, R.H.C.N.; Fraga, C.A.M.; Fernandes, P.D. Therapeutic Effects of Anti-Inflammatory N-Acylhydrazones in the Resolution of Experimental Colitis. J. Pharmacol. Exp. Ther. 2020, 374, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Tributino, J.L.M.; Duarte, C.D.; Corrêa, R.S.; Doriguetto, A.C.; Ellena, J.; Romeiro, N.C.; Castro, N.G.; Luisa, A.; Miranda, P.; Barreiro, J.; et al. Novel 6-Methanesulfonamide-3,4-Methylenedioxyphenyl-N-Acylhydrazones: Orally Effective Anti-Inflammatory Drug Candidates. Bioorg. Med. Chem. 2008, 17, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, N.D.M.; Freitas, R.H.; Fraga, C.A.M.; Fernandes, P.D. New 2-Amino-Pyridinyl-N-Acylhydrazones: Synthesis and Identification of Their Mechanism of Anti-Inflammatory Action. Biomed. Pharmacother. 2019, 123, 109739. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, N.M.; Freitas, R.H.C.N.; Fraga, C.A.M.; Fernandes, P.D. Discovery of Novel Orally Active Tetrahydro-Naphthyl-N-Acylhydrazones with In Vivo Anti-TNF-α Effect and Remarkable Anti-Inflammatory Properties. PLoS ONE 2016, 11, e0156271. [Google Scholar] [CrossRef]
- Da Silva, Y.K.C.; Reyes, C.T.M.; Rivera, G.; Alves, M.A.; Barreiro, E.J.; Moreira, M.S.A.; Lima, L.M. 3-Aminothiophene-2-Acylhydrazones: Non-Toxic, Analgesic and Anti-Inflammatory Lead-Candidates. Molecules 2014, 19, 8456–8471. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Redzicka, A.; Wiatrak, B.; Krzyżak, E.; Marciniak, A.; Gębczak, K.; Gębarowski, T.; Świątek, P. Design, Synthesis, Biological Evaluation and in Silico Studies of Novel pyrrolo[3,4-D]pyridazinone Derivatives with Promising Anti-Inflammatory and Antioxidant Activity. Bioorg. Chem. 2020, 102, 104035. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Krzyżak, E.; Zborowska, A.; Zając, P.; Potyrak, K.; Peregrym, K.; Wiatrak, B.; Marciniak, A.; Świątek, P. Design, Synthesis and Comprehensive Investigations of Pyrrolo[3,4-D]pyridazinone-Based 1,3,4-Oxadiazole as New Class of Selective COX-2 Inhibitors. Int. J. Mol. Sci. 2020, 21, 9623. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Krzyżak, E.; Wiatrak, B.; Jawień, P.; Marciniak, A.; Kotynia, A.; Świątek, P. New n-substituted-1,2,4-triazole Derivatives of pyrrolo[3,4-d]pyridazinone with Significant Anti-inflammatory Activity—Design, Synthesis and Complementary in Vitro, Computational and Spectroscopic Studies. Int. J. Mol. Sci. 2021, 22, 11235. [Google Scholar] [CrossRef]
- Peregrym, K.; Szczukowski, Ł.; Wiatrak, B.; Potyrak, K.; Czyżnikowska, Ż.; Świątek, P. In Vitro and In Silico Evaluation of New 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-D]pyridazinone as Promising Cyclooxygenase Inhibitors. Int. J. Mol. Sci. 2021, 22, 9130. [Google Scholar] [CrossRef]
- Szandruk-Bender, M.; Wiatrak, B.; Szczukowski, Ł.; Świątek, P.; Rutkowska, M.; Dzimira, S.; Merwid-Ląd, A.; Danielewski, M.; Szeląg, A. Oxadiazole Derivatives of Pyrrolo[3,4-D]pyridazinone Exert Antinociceptive Activity in the Tail-Flick and Formalin Test in Rodents and Reveal Reduced Gastrotoxicity. Int. J. Mol. Sci. 2020, 21, 9685. [Google Scholar] [CrossRef]
- Szandruk-Bender, M.; Merwid-Ląd, A.; Wiatrak, B.; Danielewski, M.; Dzimira, S.; Szkudlarek, D.; Szczukowski, Ł.; Świątek, P.; Szeląg, A. Novel 1,3,4-Oxadiazole Derivatives of Pyrrolo [3,4-d]Pyridazinone Exert Anti-Inflammatory Activity without Acute Gastrotoxicity in the Carrageenan-Induced Rat Paw Edema Test. J. Inflamm. Res. 2021, 14, 5739–5756. [Google Scholar] [CrossRef] [PubMed]
- Redzicka, A.; Szczukowski, Ł.; Kochel, A.; Wiatrak, B.; Gębczak, K.; Czyżnikowska, Ż. COX-1/COX-2 Inhibition Activities and Molecular Docking Study of Newly Designed and Synthesized pyrrolo[3,4-C]pyrrole Mannich Bases. Bioorg. Med. Chem. 2019, 27, 3918–3928. [Google Scholar] [CrossRef]
- Malinka, W. Synthesis of Some pyrrolo[3,4-D]pyridazinones and Their Preliminary Anticancer, Antimycobacterial and CNS Screening. Pharmazie 2001, 56, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Malinka, W.; Redzicka, A.; Jastrzbska Wiȩsek, M.; Filipek, B.; Dybała, M.; Karczmarzyk, Z.; Urbańczyk-Lipkowska, Z.; Kalicki, P. Derivatives of pyrrolo[3,4-D]pyridazinone, a New Class of Analgesic Agents. Eur. J. Med. Chem. 2011, 46, 4992–4999. [Google Scholar] [CrossRef] [PubMed]
- Wyrzykiewicz, E.; Prukała, D. New Isomeric N -Substituted Hydrazones of 2-, 3-and 4-Pyridinecarboxaldehydes. J. Heterocycl. Chem. 1998, 35, 381–387. [Google Scholar] [CrossRef]
- Palla, G.; Predieri, G.; Domiano, P.; Vignali, C.; Turner, W. Conformational Behaviour and E/Z Isomerization of N-Acyl and N-Aroylhydrazones. Tetrahedron 1986, 42, 3649–3654. [Google Scholar] [CrossRef]
- Galić, N.; Perić, B.; Kojić-Prodić, B.; Cimerman, Z. Structural and Spectroscopic Characteristics of Aroylhydrazones Derived from Nicotinic Acid Hydrazide. J. Mol. Struct. 2001, 559, 187–194. [Google Scholar] [CrossRef]
- Vicini, P.; Incerti, M.; Doytchinova, I.A.; La Colla, P.; Busonera, B.; Loddo, R. Synthesis and Antiproliferative Activity of Benzo[d]isothiazole Hydrazones. Eur. J. Med. Chem. 2006, 41, 624–632. [Google Scholar] [CrossRef]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams Bind in a Novel Mode to the Cyclooxygenase Active Site via a Two-Water-Mediated H-Bonding Network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef]
- Kobe, M.J.; Neau, D.B.; Mitchell, C.E.; Bartlett, S.G.; Newcomer, M.E. The Structure of Human 15-Lipoxygenase-2 with a Substrate Mimic. J. Biol. Chem. 2014, 289, 8562. [Google Scholar] [CrossRef]
- Tsai, W.C.; Gilbert, N.C.; Ohler, A.; Armstrong, M.; Perry, S.; Kalyanaraman, C.; Yasgar, A.; Rai, G.; Simeonov, A.; Jadhav, A.; et al. Kinetic and Structural Investigations of Novel Inhibitors of Human Epithelial 15-Lipoxygenase-2. Bioorg. Med. Chem. 2021, 46, 116349. [Google Scholar] [CrossRef] [PubMed]
- Orafaie, A.; Mousavian, M.; Orafai, H.; Sadeghian, H. An Overview of Lipoxygenase Inhibitors with Approach of in Vivo Studies. Prostaglandins Other Lipid Mediat. 2020, 148, 106411. [Google Scholar] [CrossRef]
- Ng, C.H.; Rullah, K.; Aluwi, M.F.F.M.; Abas, F.; Lam, K.W.; Ismail, I.S.; Narayanaswamy, R.; Jamaludin, F.; Shaari, K. Synthesis and Docking Studies of 2,4,6-Trihydroxy-3-Geranylacetophenone Analogs as Potential Lipoxygenase Inhibitor. Molecules 2014, 19, 11645. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.; Shirazi, M.S.; Taherkhani, R.; Saeedi, M.; Alipour, E.; Moghadam, F.H.; Moradi, A.; Nadri, H.; Emami, S.; Firoozpour, L.; et al. Synthesis, Biological Evaluation and Docking Study of 3-Aroyl-1-(4- Sulfamoylphenyl)thiourea Derivatives as 15-Lipoxygenase Inhibitors. Eur. J. Med. Chem. 2014, 82, 308–313. [Google Scholar] [CrossRef] [PubMed]
- ElBordiny, H.S.; El-Miligy, M.M.; Kassab, S.E.; Daabees, H.; Abdelhamid Mohamed El-Hawash, S.; Mohamed Ali, W.A. Design, Synthesis, Biological Evaluation and Docking Studies of New 3-(4,5-Dihydro-1H-Pyrazol/isoxazol-5-Yl)-2-Phenyl-1H-Indole Derivatives as Potent Antioxidants and 15-Lipoxygenase Inhibitors. Eur. J. Med. Chem. 2018, 145, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Awad, S.M.; Said, A.M.; Mahgoub, S.; Taha, H.; Ahmed, N.M. Design, Synthesis, Molecular Modelling and Biological Evaluation of Novel 3-(2-Naphthyl)-1-Phenyl-1H-Pyrazole Derivatives as Potent Antioxidants and 15-Lipoxygenase Inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 847. [Google Scholar] [CrossRef]
- Li, Y.; He, W.; Liu, J.; Sheng, F.; Hu, Z.; Chen, X. Binding of the Bioactive Component Jatrorrhizine to Human Serum Albumin. Biochim. Biophys. Acta Gen. Subj. 2005, 1722, 15–21. [Google Scholar] [CrossRef]
- Meloun, B.; Morávek, L.; Kostka, V. Complete Amino Acid Sequence of Human Serum Albumin. FEBS Lett. 1975, 58, 134–138. [Google Scholar] [CrossRef]
- Lawn, R.M.; Adelman, J.; Bock, S.C.; Franke, A.E.; Houck, C.M.; Najarian, R.; Seeburg, P.H.; Wion, K.L. The Sequence of Human Serum Albumin cDNA and Its Expression in E. Coli. Nucleic Acids Res. 1981, 9, 6103–6114. [Google Scholar] [CrossRef]
- Chen, G.Z.; Huang, X.Z.; Zheng, Z.Z.; Xu, J.G.; Wang, Z.B. The Methods of Fluorescence Analysis, 3rd ed.; Science Press: Beijing, China, 1990. [Google Scholar]
- Lakowicz, J.R.; Weber, G. Quenching of Fluorescence by Oxygen. Probe for Structural Fluctuations in Macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, MA, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Wani, T.A.; Bakheit, A.H.; Zargar, S.; Bhat, M.A.; Al-Majed, A.A. Molecular Docking and Experimental Investigation of New Indole Derivative Cyclooxygenase Inhibitor to Probe Its Binding Mechanism with Bovine Serum Albumin. Bioorg. Chem. 2019, 89, 103010. [Google Scholar] [CrossRef] [PubMed]
- Ware, W.R. Oxygen Quenching of Fluorescence in Solution: An Experimental Study of the Diffusion Process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, Y.; Li, Y.; Zhang, X. Interactions between Human Serum Albumin and Sulfadimethoxine Determined Using Spectroscopy and Molecular Docking. Molecules 2022, 27, 1526. [Google Scholar] [CrossRef] [PubMed]
- El Gammal, R.N.; Elmansi, H.; El-Emam, A.A.; Belal, F.; Hammouda, M.E.A. Exploring the Molecular Interaction of Mebendazole with Bovine Serum Albumin Using Multi-Spectroscopic Approaches and Molecular Docking. Sci. Rep. 2022, 12, 11582. [Google Scholar] [CrossRef]
- Liu, J.; He, Y.; Liu, D.; He, Y.; Tang, Z.; Lou, H.; Huo, Y.; Cao, X. Characterizing the Binding Interaction of Astilbin with Bovine Serum Albumin: A Spectroscopic Study in Combination with Molecular Docking Technology. RSC Adv. 2018, 8, 7280–7286. [Google Scholar] [CrossRef]
- Ateba, B.A.; Lissouck, D.; Mbazé, L.M.; Tjegbe, M.J.M.; Azébazé, A.G.B.; Kenfack, C.A. Binding of Mammea A/AA (MA) to Plasma Proteins: UV-Visible Spectroscopy and Molecular Dynamics Simulations Study. ACS Omega 2019, 4, 4592–4603. [Google Scholar] [CrossRef]
- Klotz, I.M. Physiochemical Aspects of Drug-Protein Interactions: A General Perspective. Ann. N. Y. Acad. Sci. 1973, 226, 18–35. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of Protein Association Reactions: Forces Contributing to Stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Lee, P.; Wu, X. Review: Modifications of Human Serum Albumin and Their Binding Effect. Curr. Pharm. Des. 2015, 21, 1862–1865. [Google Scholar] [CrossRef]
- Owczarzy, A.; Zięba, A.; Pożycka, J.; Kulig, K.; Rogóż, W.; Szkudlarek, A.; Maciążek-jurczyk, M. Spectroscopic Studies of Quinobenzothiazine Derivative in Terms of the in Vitro Interaction with Selected Human Plasma Proteins. Part 1. Molecules 2021, 26, 4776. [Google Scholar] [CrossRef]
- Ryan, A.J.; Ghuman, J.; Zunszain, P.A.; Chung, C.-W.; Curry, S. Structural Basis of Binding of Fluorescent, Site-Specific Dansylated Amino Acids to Human Serum Albumin. J. Struct. Biol. 2011, 174, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Israili, Z.H.; Dayton, P.G. Human Alpha-1-Glycoprotein and Its Interactions with Drugs. Drug Metab. Rev. 2001, 33, 161–235. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Maruyama, T.; Otagiri, M. Evaluation of Quinaldine Red as a Fluorescent Probe for Studies of Drug-Alpha 1-Acid Glycoprotein Interaction. Biol. Pharm. Bull. 1993, 16, 926–929. [Google Scholar] [CrossRef]
- Yao, H.; Wynendaele, E.; Xu, X.; Kosgei, A.; De Spiegeleer, B. Circular dichroism in Functional quality evaluation of medicines. J. Pharm. Biomed. Anal. 2018, 147, 50–64. [Google Scholar] [CrossRef]
- Maciążek-Jurczyk, M.; Morak-Młodawska, B.; Jeleń, M.; Kopeć, W.; Szkudlarek, A.; Owczarzy, A.; Kulig, K.; Rogóż, W.; Pożycka, J. The Influence of Oxidative Stress on Serum Albumin Structure as a Carrier of Selected Diazaphenothiazine with Potential Anticancer Activity. Pharmaceuticals 2021, 14, 285. [Google Scholar] [CrossRef]
- Schönfeld, D.L.; Ravelli, R.B.G.; Mueller, U.; Skerra, A. The 1.8-Å Crystal Structure of α1-Acid Glycoprotein (Orosomucoid) Solved by UV RIP Reveals the Broad Drug-Binding Activity of This Human Plasma Lipocalin. J. Mol. Biol. 2008, 384, 393–405. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominny, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian Revision 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 [µM] | Cell Morphology in Culture |
|---|---|---|
| 5a | 85.6 ± 2.5 | Cells with an elongated shape characteristic of fibroblasts, the granularities were observed in 3–5 fields of view from 10 analyzed fields. |
| 6a | 88.7 ± 4.1 | Cells with an elongated shape characteristic of fibroblasts, the granularities were observed in 3–5 fields of view from 10 analyzed fields. |
| 7a | 116.4 ± 3.6 | Cells with an elongated shape characteristic of fibroblasts, the granularities were observed in 3–5 fields of view from 10 analyzed fields. |
| 5b | 486.5 ± 6.5 | Normal morphology of fibroblasts-elongated cells, single granular cells in 1 of 10 assessed fields of view. |
| 6b | 80.7 ± 1.9 | Cells with an elongated shape characteristic of fibroblasts, the granularities were observed in 3–5 fields of view from 10 analyzed fields. |
| 7b | 180.7 ± 8.7 | Normal morphology of fibroblasts-elongated cells, single granular cells in 1 of 10 assessed fields of view. |
| 5c | 95.2 ± 1.5 | Cells with an elongated shape characteristic of fibroblasts, the granularities were observed in 3–5 fields of view from 10 analyzed fields. |
| 6c | 210.6 ± 9.6 | Normal morphology of fibroblasts–elongated cells, single granular cells in 1 of 10 assessed fields of view. |
| 7c | 81.6 ± 5.1 | Cells with an elongated shape characteristic of fibroblasts, the granularities were observed in 3–5 fields of view from 10 analyzed fields. |
| Compound | IC50 [µM] | Selectivity Index (SI) b | ||
|---|---|---|---|---|
| 15-LOX | COX-1 | COX-2 | ||
| 5a | 12.9 ± 0.06 * | 81.9 ± 2.1 # ^ & | 17.2 ± 1.4 # ^ | 0.21 |
| 6a | 13.6 ± 0.03 * | 89.2 ± 4.1 # ^ & | 16.4 ± 1.1 # ^ | 0.18 |
| 7a | 15.3 ± 2.1 | 93.8 ± 4.2 ^ & | 16.1 ± 1.8 # ^ | 0.17 |
| 5b | 13.5 ± 0.02 | 90.1 ± 1.8 # ^ & | 22.7 ± 1.2 # ^ & | 0.25 |
| 6b | 13.7 ± 0.03 * | 97.7 ± 6.8 ^ & | 27.7 ± 2.5 # ^ & | 0.28 |
| 7b | 13.7 ± 0.07 * | 91.5 ± 5.1 ^ & | 24.5 ± 2.1 # ^ & | 0.27 |
| 5c | 12.7 ± 0.04 * | 98.9 ± 1.6 ^ & | 23.6 ± 2.5 # ^ & | 0.23 |
| 6c | 14.7 ± 0.02 * | 76.8 ± 6.0 # ^ & | 9.8 ± 3.1 # & | 0.13 |
| 7c | 13.3 ± 0.02 | N/A a | 22.7 ± 2.5 # ^ & | - |
| Meloxicam | - | 101.4 ± 1.3 | 60.1 ± 2.4 | 0.59 |
| Celecoxib | - | 53.2 ± 1.9 | 0.28 ± 2.5 | 0.005 |
| Diclofenac | - | 3.5 ± 2.2 | 15.4 ± 1.4 | 4.4 |
| Zileuton | 13.41 ± 0.04 | - | - | 0.21 |
| Compound | 15-LOX | COX-1 | COX-2 |
|---|---|---|---|
| 5a | −8.0 | −7.4 | −8.7 |
| 6a | −8.6 | −7.6 | −9.1 |
| 7a | −8.4 | −5.9 | −8.0 |
| 5b | −10.3 | −7.6 | −9.3 |
| 6b | −8.7 | −8.8 | −10.9 |
| 7b | −10.2 | −5.7 | −9.1 |
| 5c | −9.9 | −8.5 | −9.6 |
| 6c | −8.4 | −9.4 | −10.4 |
| 7c | −9.8 | −6.2 | −9.5 |
| Meloxicam | - | −8.8 | −8.8 |
| Celecoxib | - | −8.2 | −9.5 |
| Diclofenac | - | −7.3 | −7.7 |
| Nordihydroguaiaretic Acid (ndga) | −7.1 | - | - |
| C8B | −5.6 | - | - |
| Baicalein | −8.3 | - | - |
| Quenching | Binding | Thermodynamic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T [K] | Ksv × 105 [dm3·mol−1] | kq × 1013 [dm3·mol−1·s−1] | logKb | Kb × 105 [dm3·mol−1] | n | ΔG° [kJ·mol−1] | ΔH° [kJ·mol−1] | ΔS° [J·mol−1·K−1] | |
| 5a | 297 303 308 | 1.87 ± 0.04 1.71 ± 0.03 1.41 ± 0.03 | 1.87 1.71 1.41 | 5.10 ± 0.20 4.95 ± 0.10 4.68 ± 0.09 | 1.26 0.89 0.48 | 0.97 ± 0.04 0.95 ± 0.02 0.92 ± 0.02 | −29.20 | −67.55 | −129.11 |
| 5b | 297 303 308 | 5.55 ± 0.33 5.26 ± 0.28 4.75 ± 0.23 | 5.55 5.26 4.75 | 6.58 ± 0.31 6.31 ± 0.56 5.84 ± 0.30 | 38.02 20.42 6.92 | 1.15 ± 0.06 1.10 ± 0.10 1.02 ± 0.05 | −37.66 | −116.19 | −264.38 |
| 5c | 297 303 308 | 4.33± 0.25 3.86 ± 0.13 2.87 ± 0.12 | 4.33 3.86 2.87 | 5.21 ± 0.38 5.05 ± 0.14 4.58 ± 0.23 | 1.62 1.12 0.38 | 0.92 ± 0.06 0.90 ± 0.02 0.85 ± 0.04 | −29.97 | −98.113 | −229.47 |
| Quenching | Binding | Thermodynamic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T [K] | Ksv·104 [dm3·mol−1] | kq·1012 [dm3·mol−1·s−1] | logKb | Kb·101 [dm3·mol−1] | n | ΔG° [kJ·mol−1] | ΔH° [kJ·mol−1] | ΔS° [J·mol−1·K−1] | |
| 5a | 297 303 308 | 9.04 ± 0.16 3.15 ± 0.41 1.11 ± 0.16 | 9.04 3.15 1.11 | 1.82 ± 0.12 1.73 ± 0.27 1.62 ± 0.26 | 6.61 5.37 4.17 | 0.44 ± 0.02 0.51 ± 0.05 0.57 ± 0.04 | −10.38 | −31.59 | −71.42 |
| 5b | 297 303 308 | 10.41 ± 1.08 11.41 ± 1.42 8.39 ± 1.24 | 10.41 11.41 8.39 | 2.71 ± 0.21 2.53 ± 0.15 2.35 ± 0.12 | 51.28 33.88 22.39 | 0.59 ± 0.04 0.55 ± 0.03 0.54 ± 0.03 | −15.49 | −57.20 | −140.43 |
| 5c | 297 303 308 | 15.87± 2.41 14.60 ± 2.05 9.99 ± 1.60 | 15.87 14.60 9.99 | 2.64 ± 0.16 2.47 ± 0.17 2.31 ± 0.20 | 43.65 29.51 20.42 | 0.55 ± 0.03 0.53 ± 0.03 0.52 ± 0.03 | −15.04 | −53.91 | −130.86 |
| HSA:Marker:Analyzed Compound Molar Ratio | DanG | DanF | ||||
|---|---|---|---|---|---|---|
| 5a | 5b | 5c | 5a | 5b | 5c | |
| 1:1:0 | - | - | - | - | - | - |
| 1:1:0.5 | 0.7% | 0% | 2.9% | 2.3% | 5.2% | 6.2% |
| 1:1:1 | 0.7% | 0.7% | 2.9% | 2.7% | 9.1% | 9.5% |
| 1:1:2 | 3.7% | 5.6% | 4.3% | 5.9% | 15.7% | 13.8% |
| 1:1:3 | 5.2% | 7.7% | 5.0% | 9.0% | 20.4% | 18.1% |
| 1:1:4 | 6.7% | 9.8% | 5.7% | 11.7% | 23.5% | 19.0% |
| 1:1:5 | 8.5% | 11.2% | 6.4% | 14.9% | 27.4% | 21.0% |
| 1:1:10 | 15.7% | 14.0% | 11.4% | 28.8% | 32.2% | 23.3% |
| AAG:Marker:Analyzed Compound Molar Ratio | QR | ||
|---|---|---|---|
| 5a | 5b | 5c | |
| 1:1:0 | - | - | - |
| 1:1:0.5 | 23.7% | 17.2% | 31.7% |
| 1:1:1 | 35.5% | 29.1% | 39.7% |
| 1:1:2 | 48.1% | 40.3% | 52.4% |
| 1:1:3 | 55.0% | 47.0% | 54.0% |
| 1:1:4 | 58.8% | 52.2% | 54.8% |
| 1:1:6 | 65.6% | 55.2% | 55.6% |
| 1:1:8 | 69.5% | 56.7% | 55.6% |
| 1:1:10 | 72.5% | 58.2% | 55.6% |
| HSA: Analyzed Compound Molar Ratio | % α-Helix | % β-Sheet | % β-Turn | % Other |
|---|---|---|---|---|
| 5a | ||||
| 1:0 | 68.1% | 1.8% | 9.2% | 20.8% |
| 1:0.5 | 67.3% | 2.8% | 9.2% | 20.7% |
| 1:1 | 66.9% | 2.8% | 9.3% | 20.9% |
| 1:2 | 66.3% | 2.7% | 9.4% | 21.5% |
| 1:3 | 66.2% | 2.8% | 9.5% | 21.6% |
| 1:5 | 66.1% | 4.3% | 9.3% | 20.3% |
| Δ = (1:0)–(1:5) | −2.0% | |||
| 5b | ||||
| 1:0 | 68.7% | 2.0% | 9.2% | 20.1% |
| 1:0.5 | 67.7% | 2.2% | 9.3% | 20.9% |
| 1:1 | 67.1% | 2.5% | 9.3% | 21.0% |
| 1:2 | 66.9% | 3.2% | 9.3% | 20.6% |
| 1:3 | 66.6% | 3.7% | 9.3% | 20.5% |
| 1:5 | 66.3% | 4.3% | 9.2% | 20.2% |
| Δ = (1:0)–(1:5) | −2.4% | |||
| 5c | ||||
| 1:0 | 68.9% | 2.1% | 9.2% | 19.8% |
| 1:0.5 | 68.4% | 2.9% | 9.1% | 19.6% |
| 1:1 | 68.0% | 2.5% | 9.2% | 20.3% |
| 1:2 | 67.8% | 3.2% | 9.2% | 19.8% |
| 1:3 | 67.8% | 3.3% | 9.1% | 19.7% |
| 1:5 | 67.3% | 3.5% | 9.2% | 20.0% |
| Δ = (1:0)–(1:5) | −1.6% | |||
| AAG: Analyzed Compound Molar Ratio | % α-Helix | % β-Sheet | % β-Turn | % Other |
|---|---|---|---|---|
| 5a | ||||
| 1:0 | 20.7% | 35.7% | 10.8% | 32.8% |
| 1:0.5 | 19.8% | 35.6% | 10.9% | 33.7% |
| 1:1 | 19.8% | 36.4% | 10.8% | 33.0% |
| 1:2 | 19.7% | 36.8% | 10.8% | 32.7% |
| 1:3 | 19.2% | 36.5% | 10.8% | 33.4% |
| 1:5 | 19.0% | 36.4% | 10.9% | 33.7% |
| Δ = (1:0)–(1:5) | −1.7% | +0.7% | ||
| 5b | ||||
| 1:0 | 21.2% | 35.6% | 10.8% | 32.4% |
| 1:0.5 | 20.7% | 36.3% | 10.7% | 32.3% |
| 1:1 | 20.5% | 36.1% | 10.7% | 32.6% |
| 1:2 | 20.4% | 36.5% | 10.7% | 32.5% |
| 1:3 | 20.1% | 37.0% | 10.6% | 32.3% |
| 1:5 | 19.9% | 36.4% | 10.8% | 32.9% |
| Δ = (1:0)–(1:5) | −1.3% | +0.8% | ||
| 5c | ||||
| 1:0 | 22.3% | 35.6% | 10.6% | 31.4% |
| 1:0.5 | 21.5% | 36.2% | 10.6% | 31.7% |
| 1:1 | 21.2% | 36.3% | 10.6% | 31.9% |
| 1:2 | 21.0% | 37.4% | 10.5% | 31.2% |
| 1:3 | 20.9% | 37.0% | 10.6% | 31.6% |
| 1:5 | 20.4% | 36.7% | 10.7% | 32.3% |
| Δ = (1:0)–(1:5) | −1.9% | +1.1% | ||
| Compound | HSA IIIA (DanF) | AAG |
|---|---|---|
| 5a | −8.2 | −8.9 |
| 6a | −8.5 | −8.7 |
| 7a | −8.5 | −8.5 |
| 5b | −8.9 | −9.7 |
| 6b | −9.0 | −9.2 |
| 7b | −9.1 | −9.7 |
| 5c | −8.9 | −9.7 |
| 6c | −9.0 | −9.3 |
| 7c | −9.3 | −9.5 |
| Compound | Physicochemical Properties–Lipinski’s Rule of Five (Ro5) | ||||
|---|---|---|---|---|---|
| #H-Bond Acceptors | #H-Bond Donors | Log Po/w (MLOGP) | MW [g/mol] | #Violations | |
| 5a | 5 | 1 | 2.62 | 409.48 | 0 |
| 6a | 6 | 2 | 2.11 | 425.48 | 0 |
| 7a | 5 | 1 | 2.84 | 423.51 | 0 |
| 5b | 5 | 1 | 3.10 | 429.47 | 0 |
| 6b | 6 | 2 | 2.59 | 445.47 | 0 |
| 7b | 5 | 1 | 3.31 | 443.50 | 0 |
| 5c | 5 | 1 | 3.58 | 463.93 | 0 |
| 6c | 6 | 2 | 3.07 | 479.92 | 0 |
| 7c | 5 | 1 | 3.78 | 477.94 | 0 |
| Compound | Pharmacokinetics | |||
|---|---|---|---|---|
| GI Absorption | BBB Permeability | P-gp Substrate | Water Solubility (ESOL) | |
| 5a | High | No | No | Moderately soluble |
| 6a | High | No | Yes | Soluble |
| 7a | High | No | No | Moderately soluble |
| 5b | High | No | No | Moderately soluble |
| 6b | High | No | No | Moderately soluble |
| 7b | High | No | No | Moderately soluble |
| 5c | High | No | No | Moderately soluble |
| 6c | High | No | No | Moderately soluble |
| 7c | High | No | No | Moderately soluble |
| Compound | Drug-Likeness | |||
|---|---|---|---|---|
| Lipinski | Veber | Bioavailability Score | TPSA [Å2] | |
| 5a | Yes, 0 violation | Yes | 0.55 | 90.51 |
| 6a | Yes, 0 violation | Yes | 0.55 | 110.74 |
| 7a | Yes, 0 violation | Yes | 0.55 | 90.51 |
| 5b | Yes, 0 violation | Yes | 0.55 | 90.51 |
| 6b | Yes, 0 violation | Yes | 0.55 | 110.74 |
| 7b | Yes, 0 violation | Yes | 0.55 | 90.51 |
| 5c | Yes, 0 violation | Yes | 0.55 | 90.51 |
| 6c | Yes, 0 violation | Yes | 0.55 | 110.74 |
| 7c | Yes, 0 violation | Yes | 0.55 | 90.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikus, J.; Świątek, P.; Przybyła, P.; Krzyżak, E.; Marciniak, A.; Kotynia, A.; Redzicka, A.; Wiatrak, B.; Jawień, P.; Gębarowski, T.; et al. Synthesis, Biological, Spectroscopic and Computational Investigations of Novel N-Acylhydrazone Derivatives of Pyrrolo[3,4-d]pyridazinone as Dual COX/LOX Inhibitors. Molecules 2023, 28, 5479. https://doi.org/10.3390/molecules28145479
Mikus J, Świątek P, Przybyła P, Krzyżak E, Marciniak A, Kotynia A, Redzicka A, Wiatrak B, Jawień P, Gębarowski T, et al. Synthesis, Biological, Spectroscopic and Computational Investigations of Novel N-Acylhydrazone Derivatives of Pyrrolo[3,4-d]pyridazinone as Dual COX/LOX Inhibitors. Molecules. 2023; 28(14):5479. https://doi.org/10.3390/molecules28145479
Chicago/Turabian StyleMikus, Jakub, Piotr Świątek, Patrycja Przybyła, Edward Krzyżak, Aleksandra Marciniak, Aleksadra Kotynia, Aleksandra Redzicka, Benita Wiatrak, Paulina Jawień, Tomasz Gębarowski, and et al. 2023. "Synthesis, Biological, Spectroscopic and Computational Investigations of Novel N-Acylhydrazone Derivatives of Pyrrolo[3,4-d]pyridazinone as Dual COX/LOX Inhibitors" Molecules 28, no. 14: 5479. https://doi.org/10.3390/molecules28145479
APA StyleMikus, J., Świątek, P., Przybyła, P., Krzyżak, E., Marciniak, A., Kotynia, A., Redzicka, A., Wiatrak, B., Jawień, P., Gębarowski, T., & Szczukowski, Ł. (2023). Synthesis, Biological, Spectroscopic and Computational Investigations of Novel N-Acylhydrazone Derivatives of Pyrrolo[3,4-d]pyridazinone as Dual COX/LOX Inhibitors. Molecules, 28(14), 5479. https://doi.org/10.3390/molecules28145479