
Conversion of Similar Xenochemicals to Dissimilar Products: Exploiting Competing Reactions in Whole-Cell Catalysis
 ,
,
Abstract
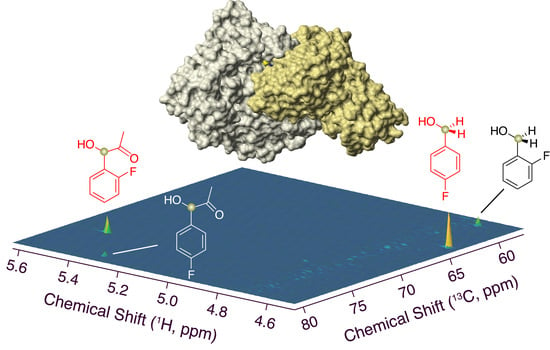
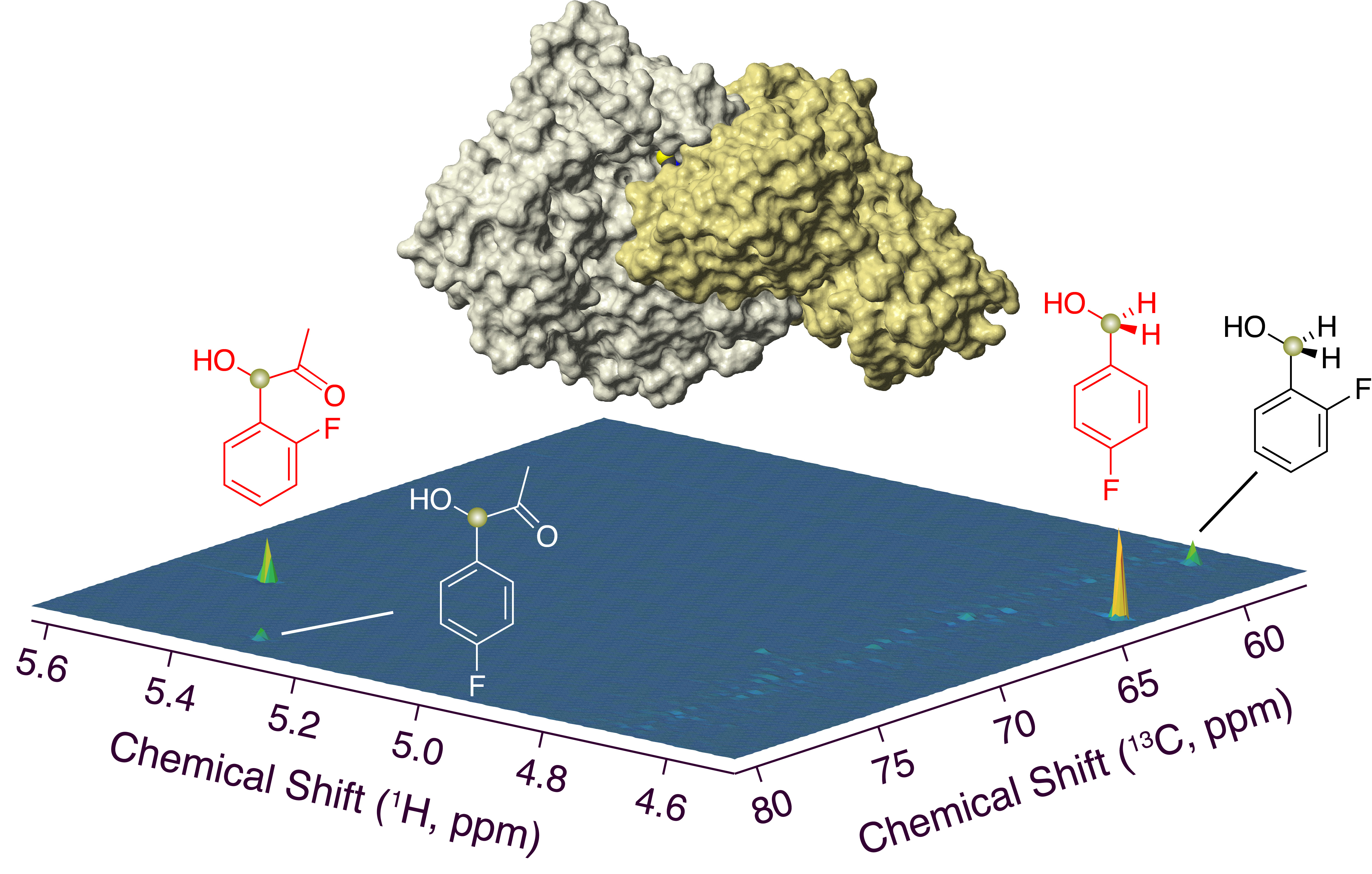{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
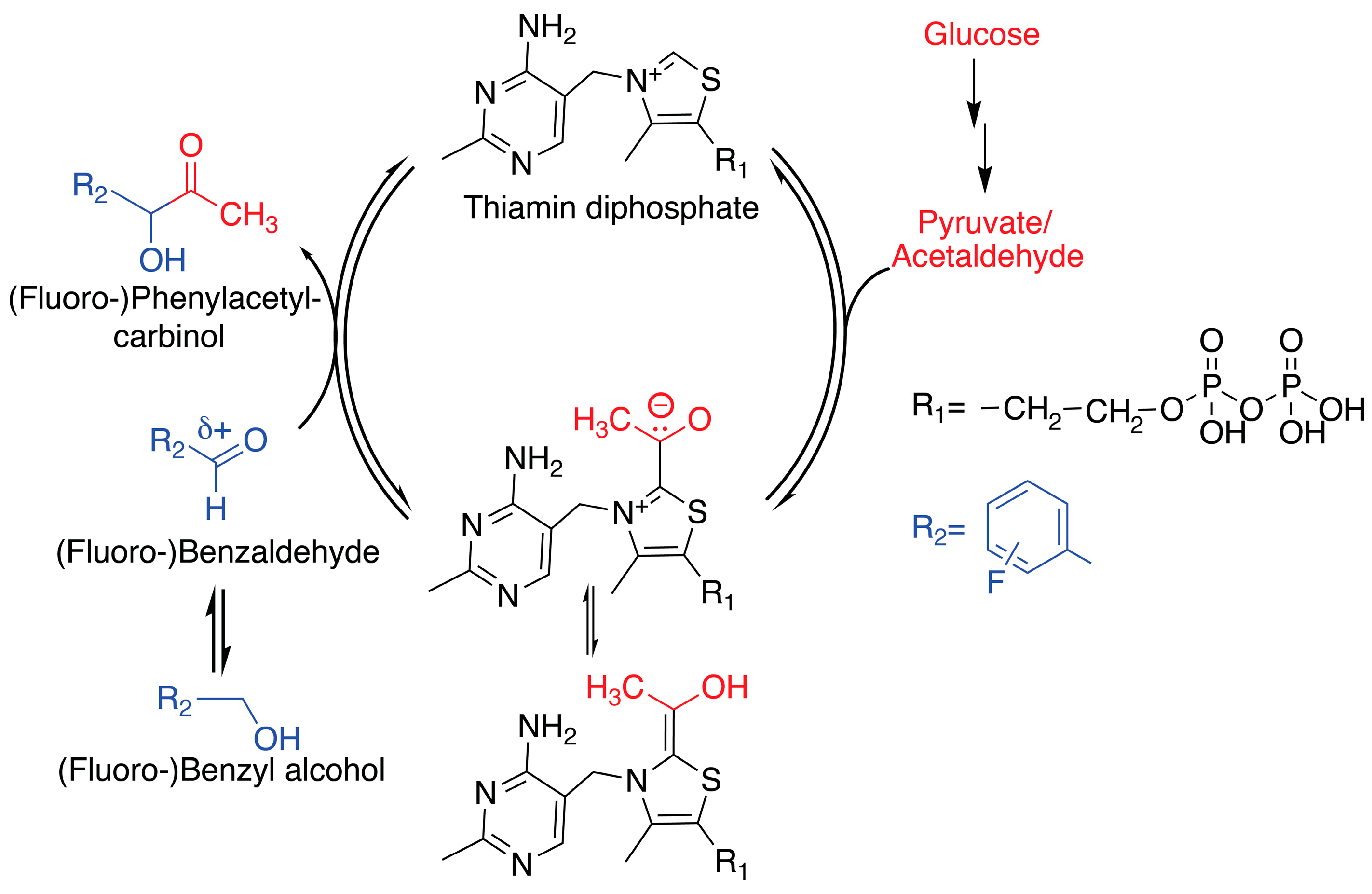
2. Results and Discussion
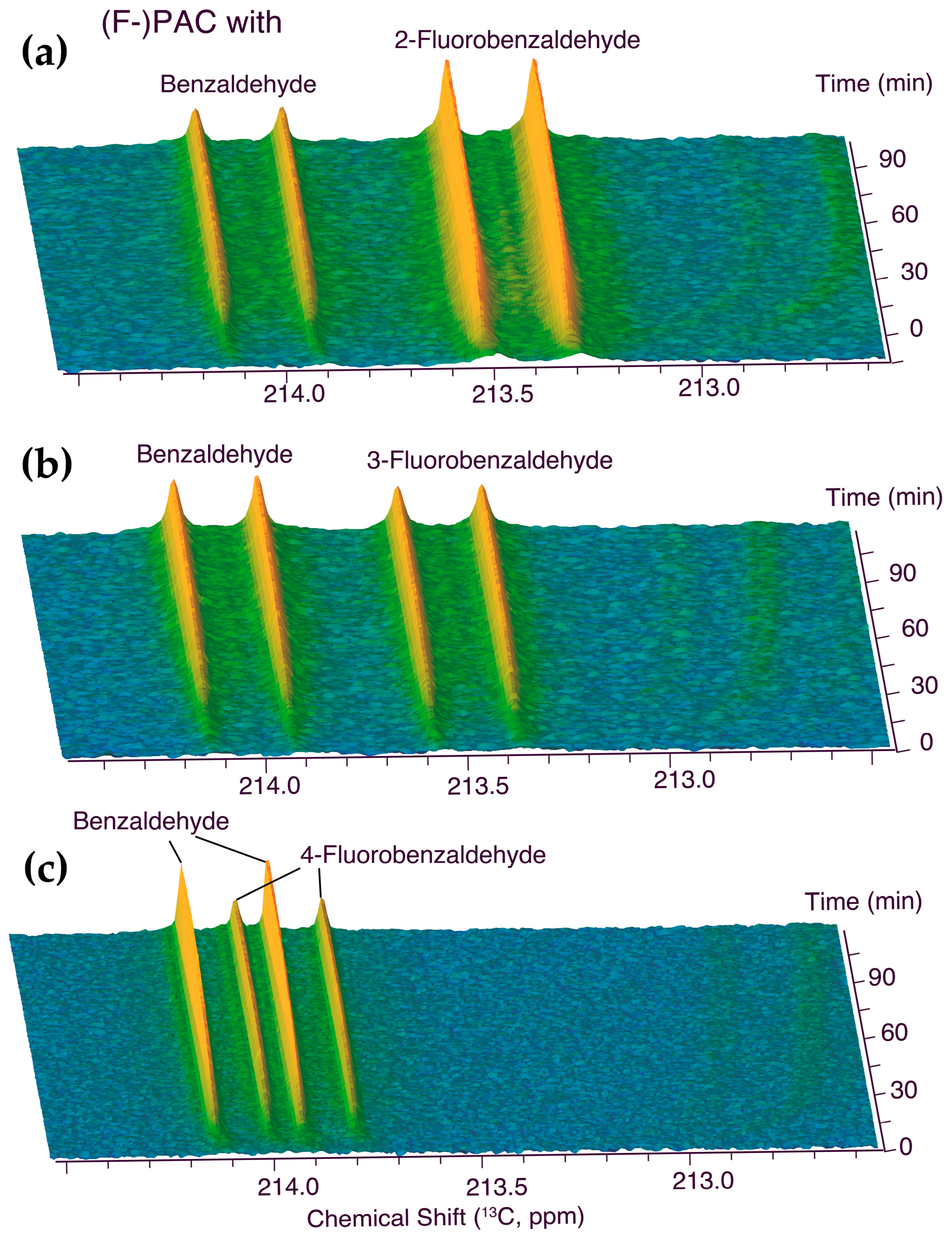
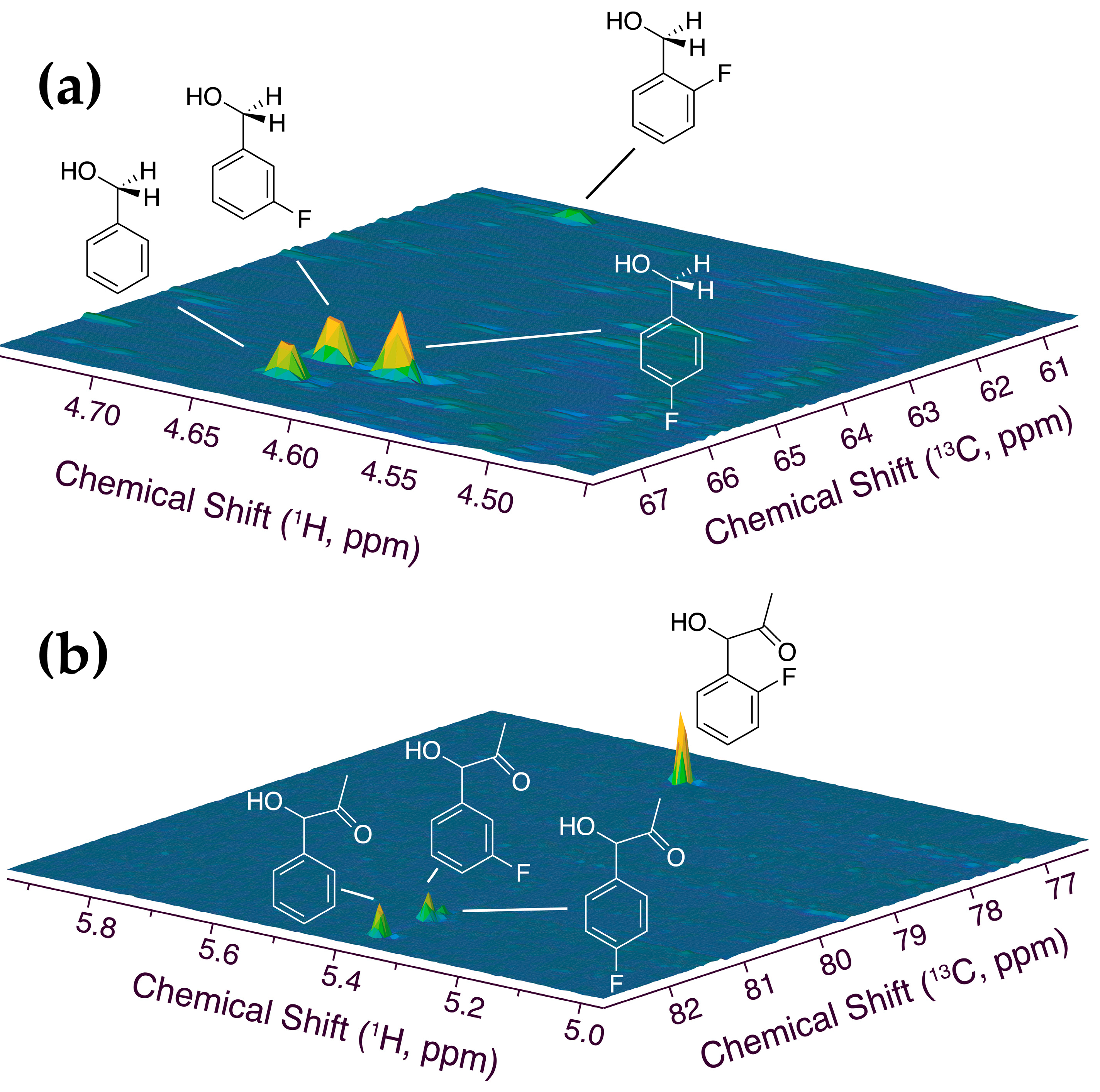
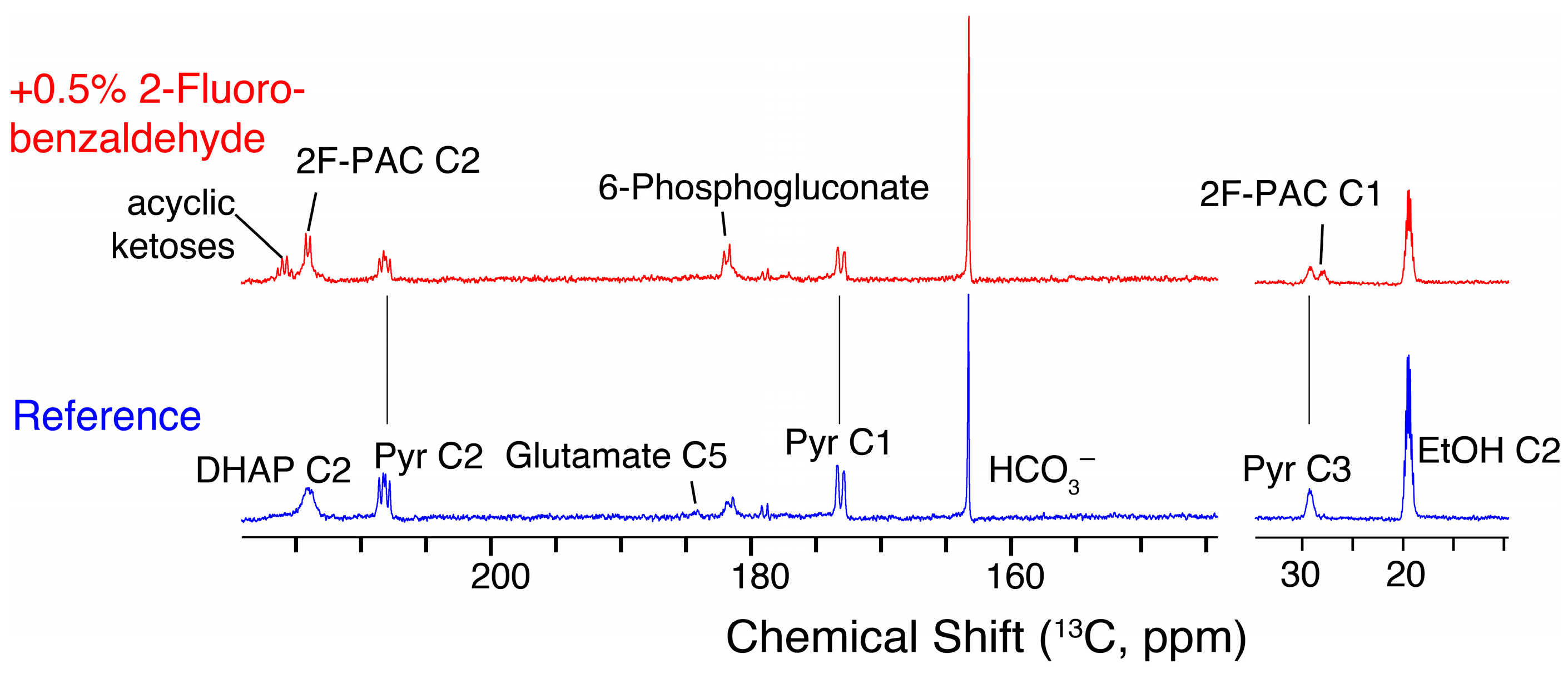
2.1. In-cell NMR Competition Assays of Carboligation with Benzaldehyde and Fluorinated Forms
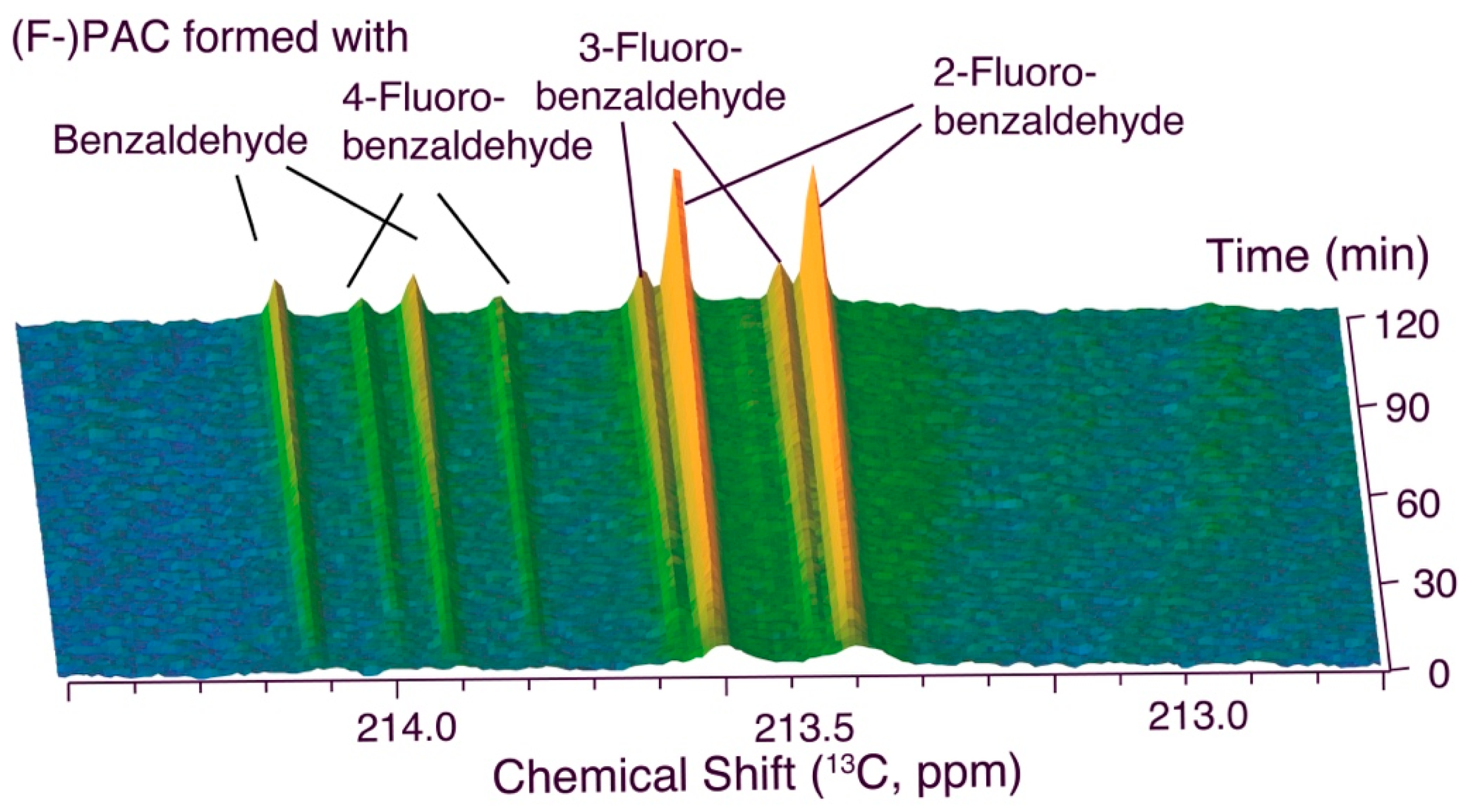
2.2. Competition in a Quinary Mixture Yields Different Results in the Cell and in Enzyme Catalysis
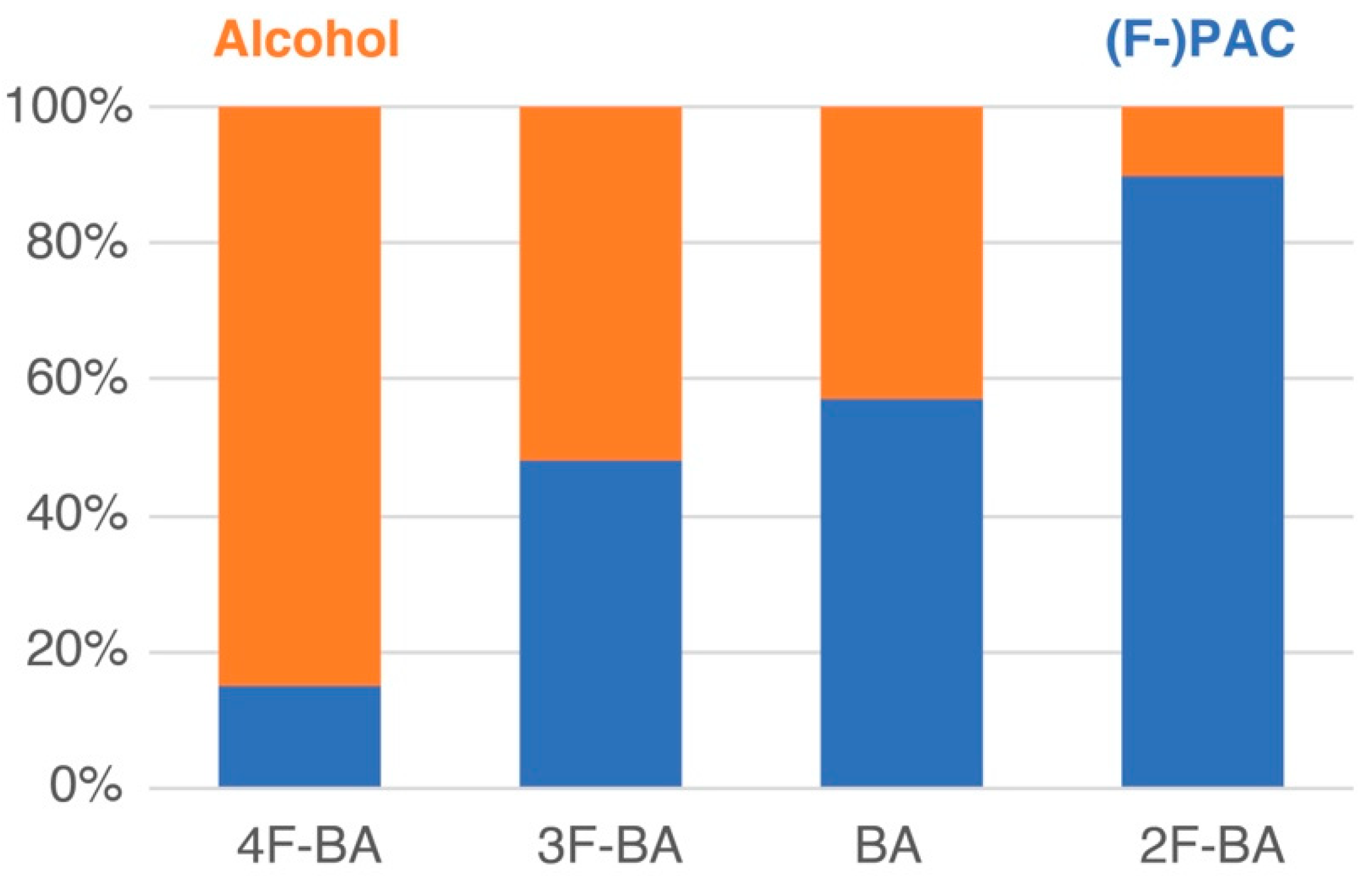
2.3. Product Composition from Whole-Cell Catalysis Exploiting Competition
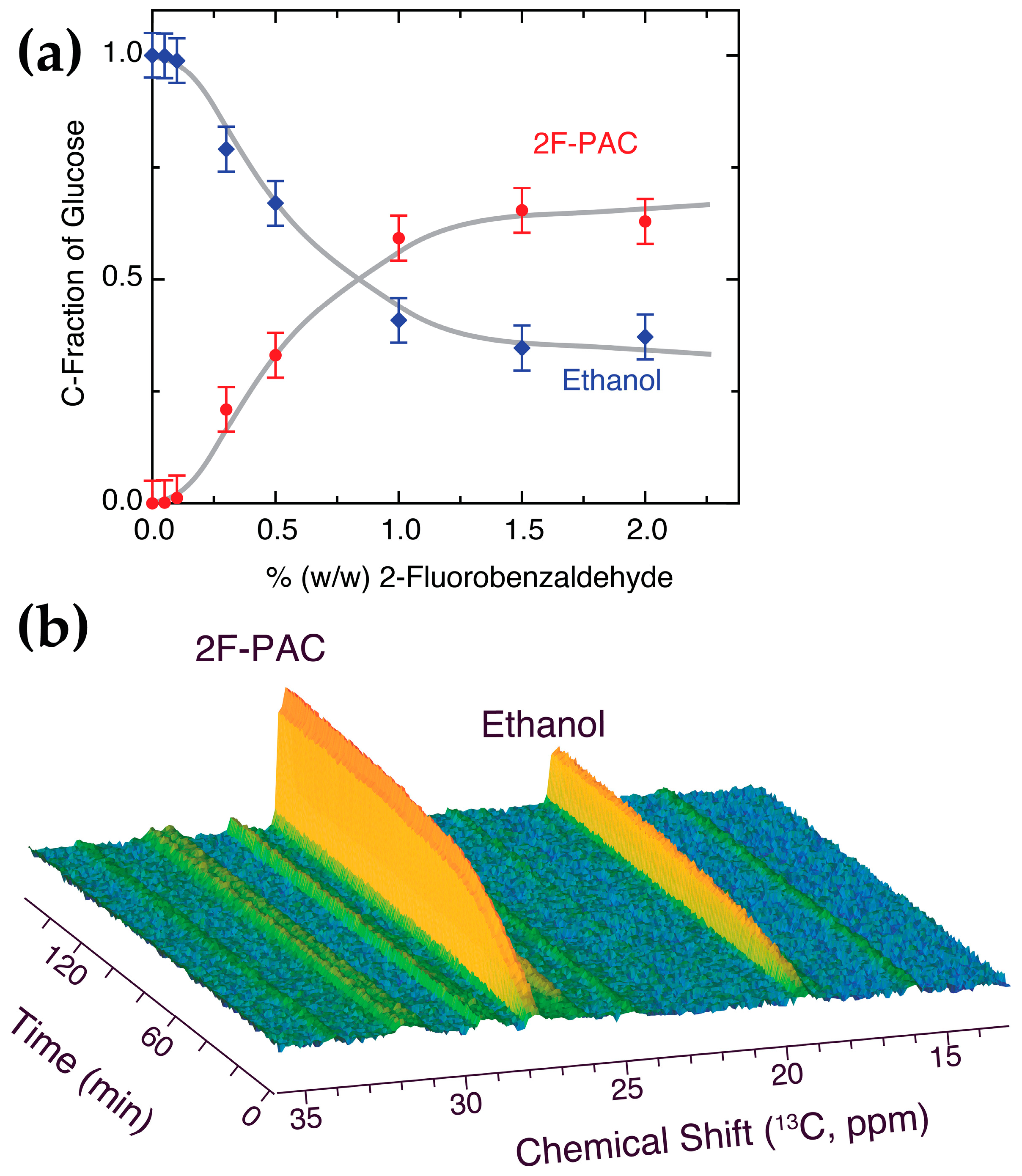
2.4. Competition with Natural Acetaldehyde Clearance and NADH Oxidation
2.5. Mechanistic Effect of Xenochemicals on Natural Biochemistry Using D-DNP NMR
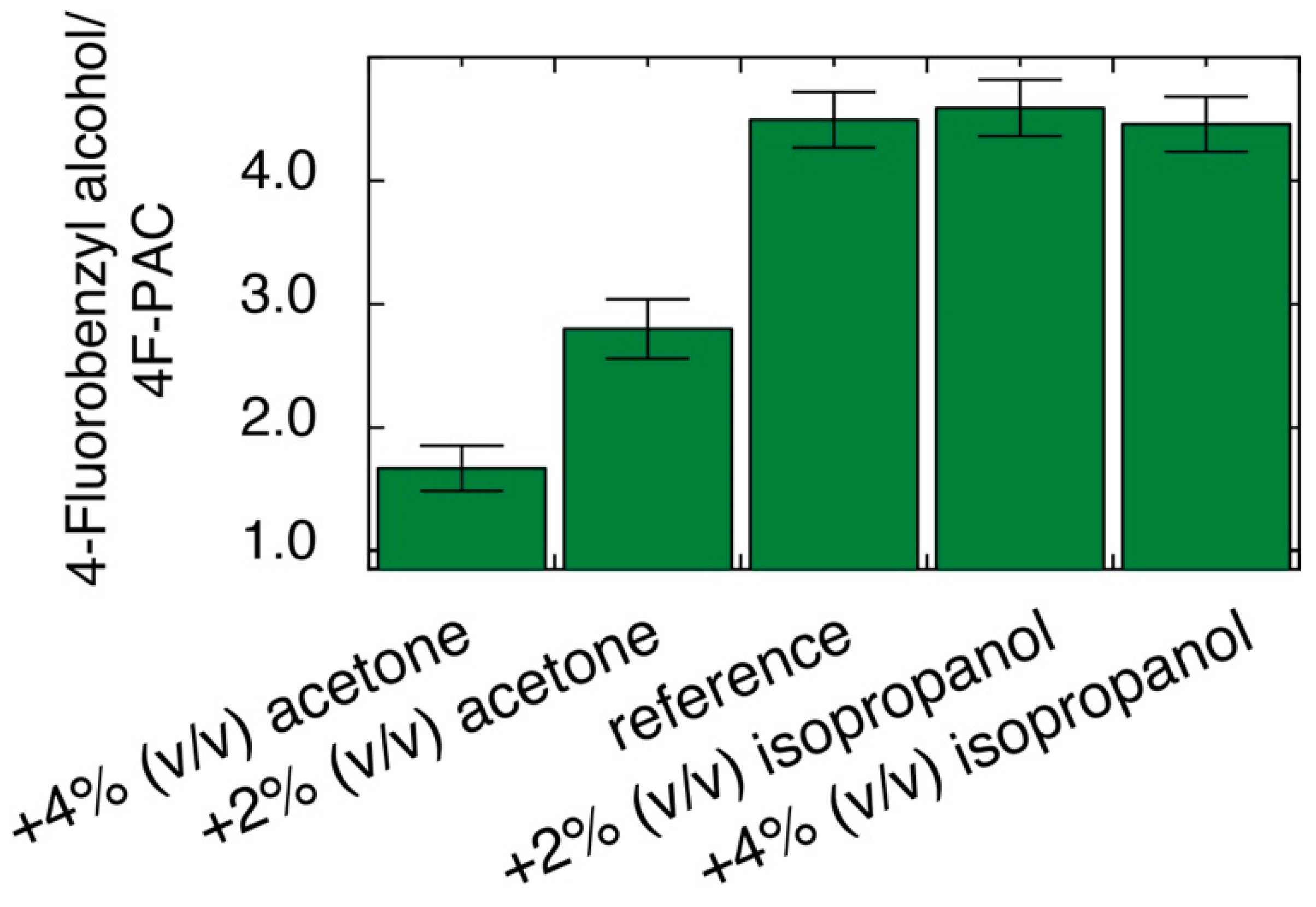
2.6. Effect of Oxidized Additive and Substrate
3. Materials and Methods
3.1. Chemicals
3.2. Distillation
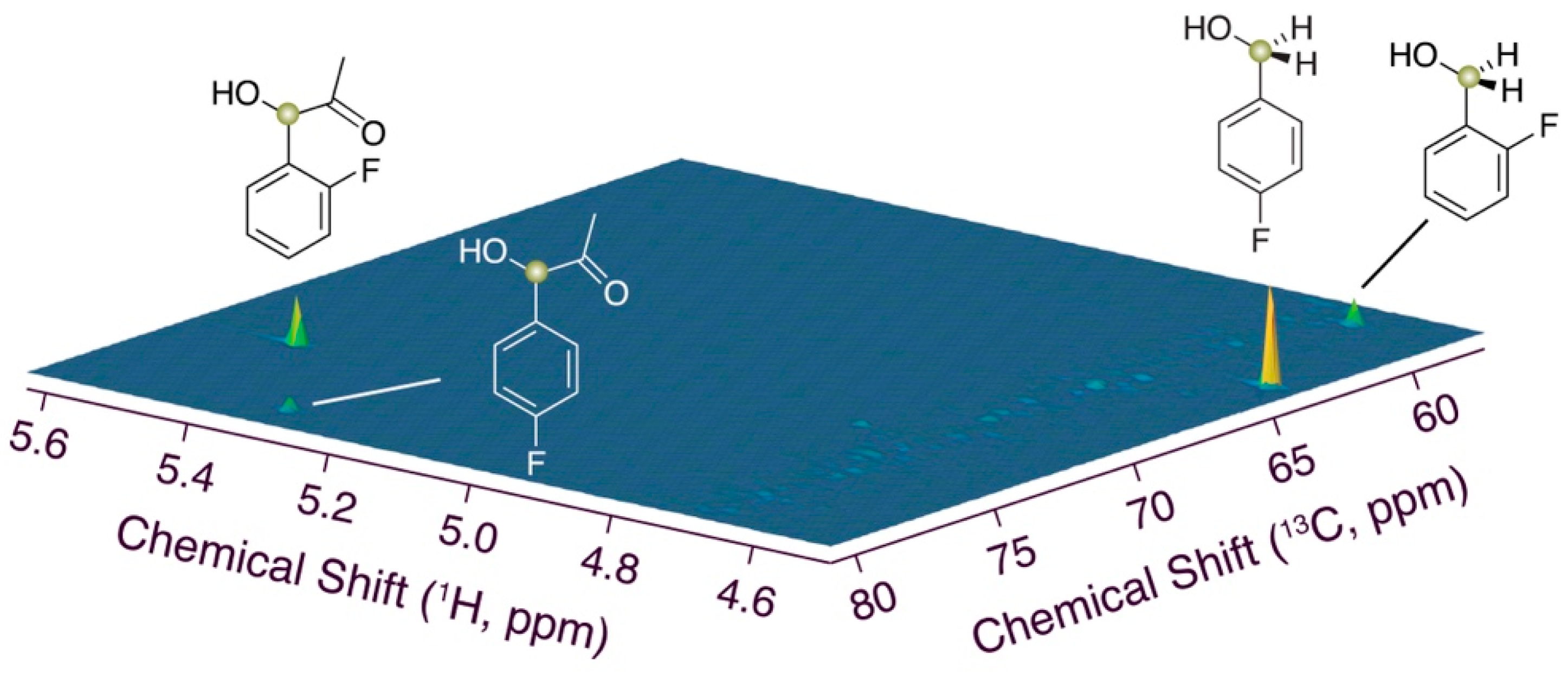
3.3. Assignment of Phenylacetyl Carbinol, including Monofluorinated Forms
3.4. Competition Assay
3.5. 1H-13C Assay of Distribution between Benzyl Alcohol and Phenylacetyl Carbinol Formation
3.6. Effect of Growth Phase and of Pyruvate As a Carbon Source
3.7. Hyperpolarization
3.8. D-DNP NMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Botti, L.; Kondrat, S.A.; Navar, R.; Padovan, D.; Martinez-Espin, J.S.; Meier, S.; Hammond, C. Solvent-Activated Hafnium-Containing Zeolites Enable Selective and Continuous Glucose–Fructose Isomerisation. Angew. Chem. 2020, 132, 20192–20198. [Google Scholar] [CrossRef]
- Rass-Hansen, J.; Falsig, H.; Jørgensen, B.; Christensen, C.H. Bioethanol: Fuel or Feedstock? J. Chem. Technol. Biotechnol. 2007, 82, 329–333. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef]
- Leresche, J.E.; Meyer, H.-P. Chemocatalysis and Biocatalysis (Biotransformation): Some Thoughts of a Chemist and of a Biotechnologist. Org. Process Res. Dev. 2006, 10, 572–580. [Google Scholar] [CrossRef]
- Cao, Y.; Li, X.; Ge, J. Enzyme Catalyst Engineering toward the Integration of Biocatalysis and Chemocatalysis. Trends Biotechnol. 2021, 39, 1173–1183. [Google Scholar] [CrossRef]
- Gröger, H.; Gallou, F.; Lipshutz, B.H. Where Chemocatalysis Meets Biocatalysis: In Water. Chem. Rev. 2022, 123, 5262–5296. [Google Scholar] [CrossRef]
- Yang, T.-X.; Zhao, L.-Q.; Wang, J.; Song, G.-L.; Liu, H.-M.; Cheng, H.; Yang, Z. Improving Whole-Cell Biocatalysis by Addition of Deep Eutectic Solvents and Natural Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2017, 5, 5713–5722. [Google Scholar] [CrossRef]
- Gröger, H.; Hummel, W. Combining the ‘Two Worlds’ of Chemocatalysis and Biocatalysis towards Multi-Step One-Pot Processes in Aqueous Media. Curr. Opin. Chem. Biol. 2014, 19, 171–179. [Google Scholar] [CrossRef]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and Challenges for Combining Chemo- and Biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- de Carvalho, C.C.C.R. Enzymatic and Whole Cell Catalysis: Finding New Strategies for Old Processes. Biotechnol. Adv. 2011, 29, 75–83. [Google Scholar] [CrossRef]
- Pinto, A.; Contente, M.L.; Tamborini, L. Advances on Whole-Cell Biocatalysis in Flow. Curr. Opin. Green Sustain. Chem. 2020, 25, 100343. [Google Scholar] [CrossRef]
- Nagy-Győr, L.; Lăcătuş, M.; Balogh-Weiser, D.; Csuka, P.; Bódai, V.; Erdélyi, B.; Molnár, Z.; Hornyánszky, G.; Paizs, C.; Poppe, L. How to Turn Yeast Cells into a Sustainable and Switchable Biocatalyst? On-Demand Catalysis of Ketone Bioreduction or Acyloin Condensation. ACS Sustain. Chem. Eng. 2019, 7, 19375–19383. [Google Scholar] [CrossRef]
- Cros, A.; Alfaro-Espinoza, G.; De Maria, A.; Wirth, N.T.; Nikel, P.I. Synthetic Metabolism for Biohalogenation. Curr. Opin. Biotechnol. 2022, 74, 180–193. [Google Scholar] [CrossRef]
- Nieto-Domínguez, M.; Nikel, P.I. Intersecting Xenobiology and Neometabolism To Bring Novel Chemistries to Life. ChemBioChem 2020, 21, 2551–2571. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, X.; Xia, Y.; Guo, X.; Guo, J.; Zhang, J.; Zhao, W.; Wu, Y.; Wang, J.; Zhong, F. Whole-Cell-Catalyzed Hydrogenation/Deuteration of Aryl Halides with a Genetically Repurposed Photodehalogenase. Chem, 2023; in press. [Google Scholar] [CrossRef]
- Miao, Y.; Rahimi, M.; Geertsema, E.M.; Poelarends, G.J. Recent Developments in Enzyme Promiscuity for Carbon–Carbon Bond-Forming Reactions. Curr. Opin. Chem. Biol. 2015, 25, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T.; Kazlauskas, R.J. Catalytic Promiscuity in Biocatalysis: Using Old Enzymes to Form New Bonds and Follow New Pathways. Angew. Chem. Int. Ed. 2004, 43, 6032–6040. [Google Scholar] [CrossRef] [PubMed]
- Kheronsky, O.; Tawfik, D.S. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [CrossRef]
- Hu, B.; Yu, H.; Zhou, J.; Li, J.; Chen, J.; Du, G.; Lee, S.Y.; Zhao, X. Whole-Cell P450 Biocatalysis Using Engineered Escherichia Coli with Fine-Tuned Heme Biosynthesis. Adv. Sci. 2023, 10, 2205580. [Google Scholar] [CrossRef]
- Alvarado, O.; Lizana, I.; Jaña, G.; Tuñon, I.; Delgado, E. A DFT Study on the Chiral Synthesis of R-Phenylacetyl Carbinol within the Quantum Chemical Cluster Approach. Chem. Phys. Lett. 2017, 677, 30–34. [Google Scholar] [CrossRef]
- Kuo, Y.-M.; Henry, R.A.; Andrews, A.J. Measuring Specificity in Multi-Substrate/Product Systems as a Tool to Investigate Selectivity in Vivo. Biochim. Biophys. Acta BBA-Proteins Proteom. 2016, 1864, 70–76. [Google Scholar] [CrossRef]
- Fan, T.W.-M.; Lane, A.N. Applications of NMR Spectroscopy to Systems Biochemistry. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 92–93, 18–53. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.; Jensen, P.R.; Duus, J.Ø. Real-Time Detection of Central Carbon Metabolism in Living Escherichia Coli and Its Response to Perturbations. FEBS Lett. 2011, 585, 3133–3138. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.R.; Matos, M.R.A.; Sonnenschein, N.; Meier, S. Combined In-Cell NMR and Simulation Approach to Probe Redox-Dependent Pathway Control. Anal. Chem. 2019, 91, 5395–5402. [Google Scholar] [CrossRef] [PubMed]
- Sannelli, F.; Gao, S.; Jensen, P.R.; Meier, S. Glucose/Furfural Substrate Mixtures in Non-Engineered Yeast: Potential for Massive Rerouting of Fermentation to C−C Bond Formation on Furfural. ChemCatChem 2022, 14, e202200933. [Google Scholar] [CrossRef]
- Meier, S.; Solodovnikova, N.; Jensen, P.R.; Wendland, J. Sulfite Action in Glycolytic Inhibition: In Vivo Real-Time Observation by Hyperpolarized 13 C NMR Spectroscopy. ChemBioChem 2012, 13, 2265–2269. [Google Scholar] [CrossRef]
- Křen, V.; Grout, D.H.G.; Dalton, H.; Hutchinson, D.W.; König, W.; Turner, M.M.; Dean, G.; Thomson, N. Pyruvate Decarboxylase: A New Enzyme for the Production of Acyloins by Biotransformation. J. Chem. Soc. Chem. Commun. 1993, 341–343. [Google Scholar] [CrossRef]
- Long, A.; Ward, O.P. Biotransformation of Aromatic Aldehydes BySaccharomyces Cerevisiae: Investigation of Reaction Rates. J. Ind. Microbiol. 1989, 4, 49–53. [Google Scholar] [CrossRef]
- Müller, M.; Gocke, D.; Pohl, M. Thiamin Diphosphate in Biological Chemistry: Exploitation of Diverse Thiamin Diphosphate-Dependent Enzymes for Asymmetric Chemoenzymatic Synthesis. FEBS J. 2009, 276, 2894–2904. [Google Scholar] [CrossRef]
- Attwood, P.V. The Structure and the Mechanism of Action of Pyruvate Carboxylase. Int. J. Biochem. Cell Biol. 1995, 27, 231–249. [Google Scholar] [CrossRef]
- Arjunan, P.; Umland, T.; Dyda, F.; Swaminathan, S.; Furey, W.; Sax, M.; Farrenkopf, B.; Gao, Y.; Zhang, D.; Jordan, F. Crystal Structure of the Thiamin Diphosphate-Dependent Enzyme Pyruvate Decarboxylase from the YeastSaccharomyces Cerevisiaeat 2.3 Å Resolution. J. Mol. Biol. 1996, 256, 590–600. [Google Scholar] [CrossRef]
- Lobell, M.; Crout, D.H.G. Pyruvate Decarboxylase: A Molecular Modeling Study of Pyruvate Decarboxylation and Acyloin Formation. J. Am. Chem. Soc. 1996, 118, 1867–1873. [Google Scholar] [CrossRef]
- Müller, M.; Sprenger, G.A.; Pohl, M. CC Bond Formation Using ThDP-Dependent Lyases. Curr. Opin. Chem. Biol. 2013, 17, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Crout, D.H.G.; Dalton, H.; Hutchinson, D.W.; Miyagoshi, M. Studies on Pyruvate Decarboxylase: Acyloin Formation from Aliphatic, Aromatic and Heterocyclic Aldehydes. J. Chem. Soc. Perkin 1991, 1, 1329–1334. [Google Scholar] [CrossRef]
- Long, A.; Ward, O.P. Biotransformation of Benzaldehyde BySaccharomyces Cerevisiae: Characterization of the Fermentation and Toxicity Effects of Substrates and Products. Biotechnol. Bioeng. 1989, 34, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Goličnik, M.; Masson, P. Time-Course of Enzyme-Catalyzed Competing Substrate Degradation for Michaelian Behavior and for Enzymes Showing Activation/Inhibition by Excess Substrate. Chem. Biol. Interact. 2019, 309, 108704. [Google Scholar] [CrossRef] [PubMed]
- Schnell, S. Time-Dependent Closed Form Solutions for Fully Competitive Enzyme Reactions. Bull. Math. Biol. 2000, 62, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Bøjstrup, M.; Petersen, B.O.; Beeren, S.R.; Hindsgaul, O.; Meier, S. Fast and Accurate Quantitation of Glucans in Complex Mixtures by Optimized Heteronuclear NMR Spectroscopy. Anal. Chem. 2013, 85, 8802–8808. [Google Scholar] [CrossRef]
- Petersen, B.O.; Hindsgaul, O.; Meier, S. Profiling of Carbohydrate Mixtures at Unprecedented Resolution Using High-Precision 1H-13C Chemical Shift Measurements and a Reference Library. Analyst 2013, 139, 401–406. [Google Scholar] [CrossRef]
- Jeannerat, D. Computer Optimized Spectral Aliasing in the Indirect Dimension of 1H–13C Heteronuclear 2D NMR Experiments. A New Algorithm and Examples of Applications to Small Molecules. J. Magn. Reson. 2007, 186, 112–122. [Google Scholar] [CrossRef]
- Jeannerat, D. High Resolution in Heteronuclear 1H–13C NMR Experiments by Optimizing Spectral Aliasing with One-Dimensional Carbon Data. Magn. Reson. Chem. 2003, 41, 3–17. [Google Scholar] [CrossRef]
- Marcó, N.; Fredi, A.; Parella, T. Ultra High-Resolution HSQC: Application to the Efficient and Accurate Measurement of Heteronuclear Coupling Constants. Chem. Commun. 2015, 51, 3262–3265. [Google Scholar] [CrossRef] [PubMed]
- Sannelli, F.; Jensen, P.R.; Meier, S. In-Cell NMR Approach for Real-Time Exploration of Pathway Versatility: Substrate Mixtures in Nonengineered Yeast. Anal. Chem. 2023, 95, 7262–7270. [Google Scholar] [CrossRef]
- Maugeri, Z.; de María, P.D. Whole-Cell Biocatalysis in Deep-Eutectic-Solvents/Aqueous Mixtures. ChemCatChem 2014, 6, 1535–1537. [Google Scholar] [CrossRef]
- Bode, H.B.; Bethe, B.; Höfs, R.; Zeeck, A. Big Effects from Small Changes: Possible Ways to Explore Nature’s Chemical Diversity. ChemBioChem 2002, 3, 619. [Google Scholar] [CrossRef] [PubMed]
- Ardenkjaer-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in Signal-to-Noise Ratio of > 10,000 Times in Liquid-State NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.; Karlsson, M.; Jensen, P.R.; Lerche, M.H.; Duus, J.Ø. Metabolic Pathway Visualization in Living Yeast by DNP-NMR. Mol. Biosyst. 2011, 7, 2834–2836. [Google Scholar] [CrossRef]
- Timm, K.N.; Hartl, J.; Keller, M.A.; Hu, D.-E.; Kettunen, M.I.; Rodrigues, T.B.; Ralser, M.; Brindle, K.M. Hyperpolarized [U- 2 H, U- 13 C]Glucose Reports on Glycolytic and Pentose Phosphate Pathway Activity in EL4 Tumors and Glycolytic Activity in Yeast Cells: Hyperpolarized [U-2H, U-13C]Glucose Metabolism. Magn. Reson. Med. 2015, 74, 1543–1547. [Google Scholar] [CrossRef]
- Harris, T.; Degani, H.; Frydman, L. Hyperpolarized 13C NMR Studies of Glucose Metabolism in Living Breast Cancer Cell Cultures. NMR Biomed. 2013, 26, 1831–1843. [Google Scholar] [CrossRef]
- Hansen, A.R.E.; Enemark-Rasmussen, K.; Mulder, F.A.A.; Jensen, P.R.; Meier, S. Versatile Procedures for Reliable NMR Quantification of CO 2 Electroreduction Products. J. Phys. Chem. C 2022, 126, 11026–11032. [Google Scholar] [CrossRef]
- Elliot, S.G.; Tosi, I.; Riisager, A.; Taarning, E.; Meier, S. Response Factors Enable Rapid Quantitative 2D NMR Analysis in Catalytic Biomass Conversion to Renewable Chemicals. Top. Catal. 2019, 62, 590–598. [Google Scholar] [CrossRef]
- Jensen, P.R.; Sannelli, F.; Stauning, L.T.; Meier, S. Enhanced 13C NMR Detects Extended Reaction Networks in Living Cells. Chem. Commun. 2021, 57, 10572–10575. [Google Scholar] [CrossRef] [PubMed]
- Macchiarulo, A.; Nobeli, I.; Thornton, J.M. Ligand Selectivity and Competition between Enzymes in Silico. Nat. Biotechnol. 2004, 22, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Nobeli, I.; Favia, A.D.; Thornton, J.M. Protein Promiscuity and Its Implications for Biotechnology. Nat. Biotechnol. 2009, 27, 157–167. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sannelli, F.; Sindahl, N.C.; Warthegau, S.S.; Jensen, P.R.; Meier, S. Conversion of Similar Xenochemicals to Dissimilar Products: Exploiting Competing Reactions in Whole-Cell Catalysis. Molecules 2023, 28, 5157. https://doi.org/10.3390/molecules28135157
Sannelli F, Sindahl NC, Warthegau SS, Jensen PR, Meier S. Conversion of Similar Xenochemicals to Dissimilar Products: Exploiting Competing Reactions in Whole-Cell Catalysis. Molecules. 2023; 28(13):5157. https://doi.org/10.3390/molecules28135157
Chicago/Turabian StyleSannelli, Francesca, Nikoline Corell Sindahl, Stefan S. Warthegau, Pernille Rose Jensen, and Sebastian Meier. 2023. "Conversion of Similar Xenochemicals to Dissimilar Products: Exploiting Competing Reactions in Whole-Cell Catalysis" Molecules 28, no. 13: 5157. https://doi.org/10.3390/molecules28135157
APA StyleSannelli, F., Sindahl, N. C., Warthegau, S. S., Jensen, P. R., & Meier, S. (2023). Conversion of Similar Xenochemicals to Dissimilar Products: Exploiting Competing Reactions in Whole-Cell Catalysis. Molecules, 28(13), 5157. https://doi.org/10.3390/molecules28135157