
Finding the Right Solvent: A Novel Screening Protocol for Identifying Environmentally Friendly and Cost-Effective Options for Benzenesulfonamide




Abstract
1. Introduction
2. Results and Discussion
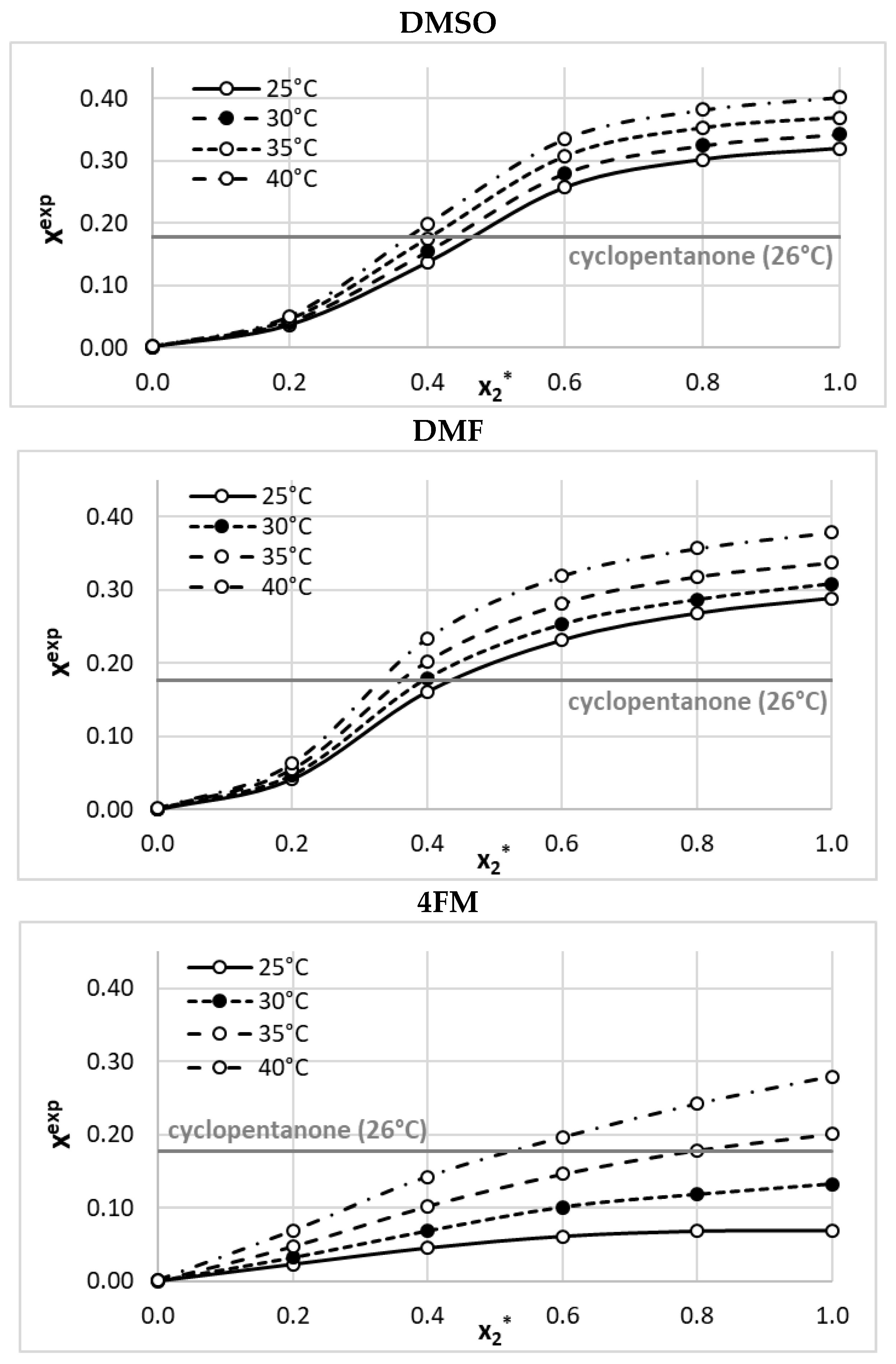
2.1. Solubility Measurements
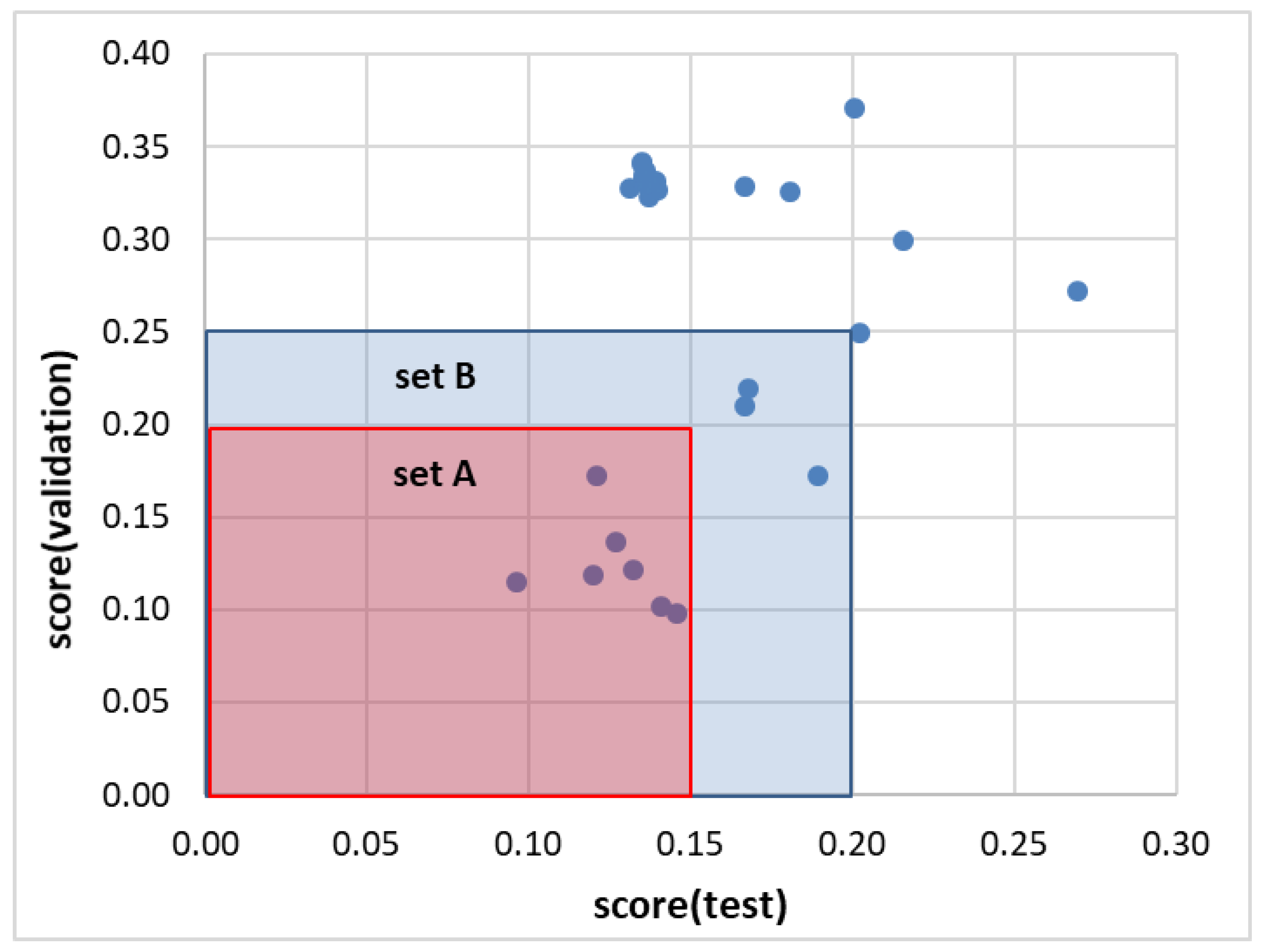
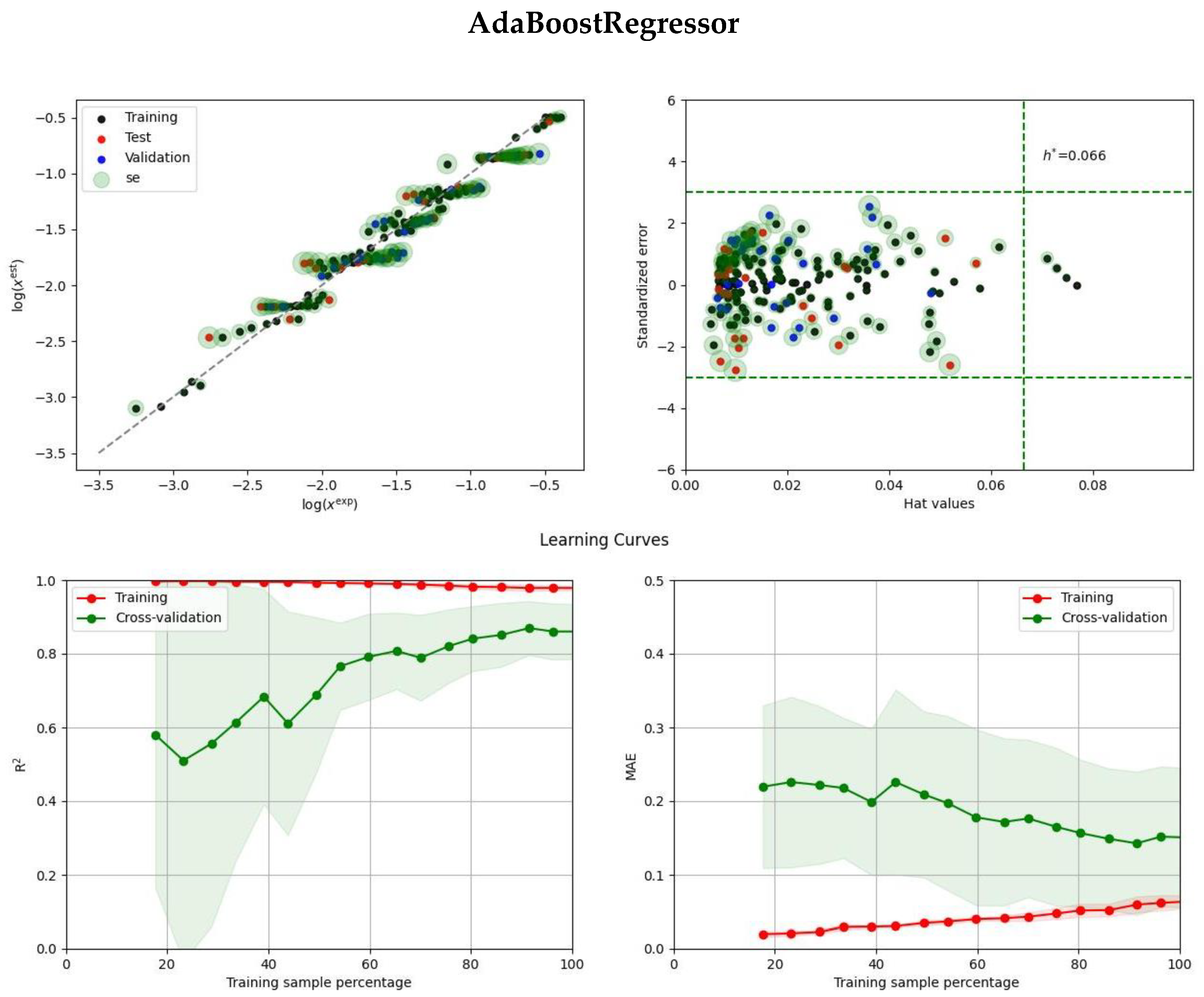
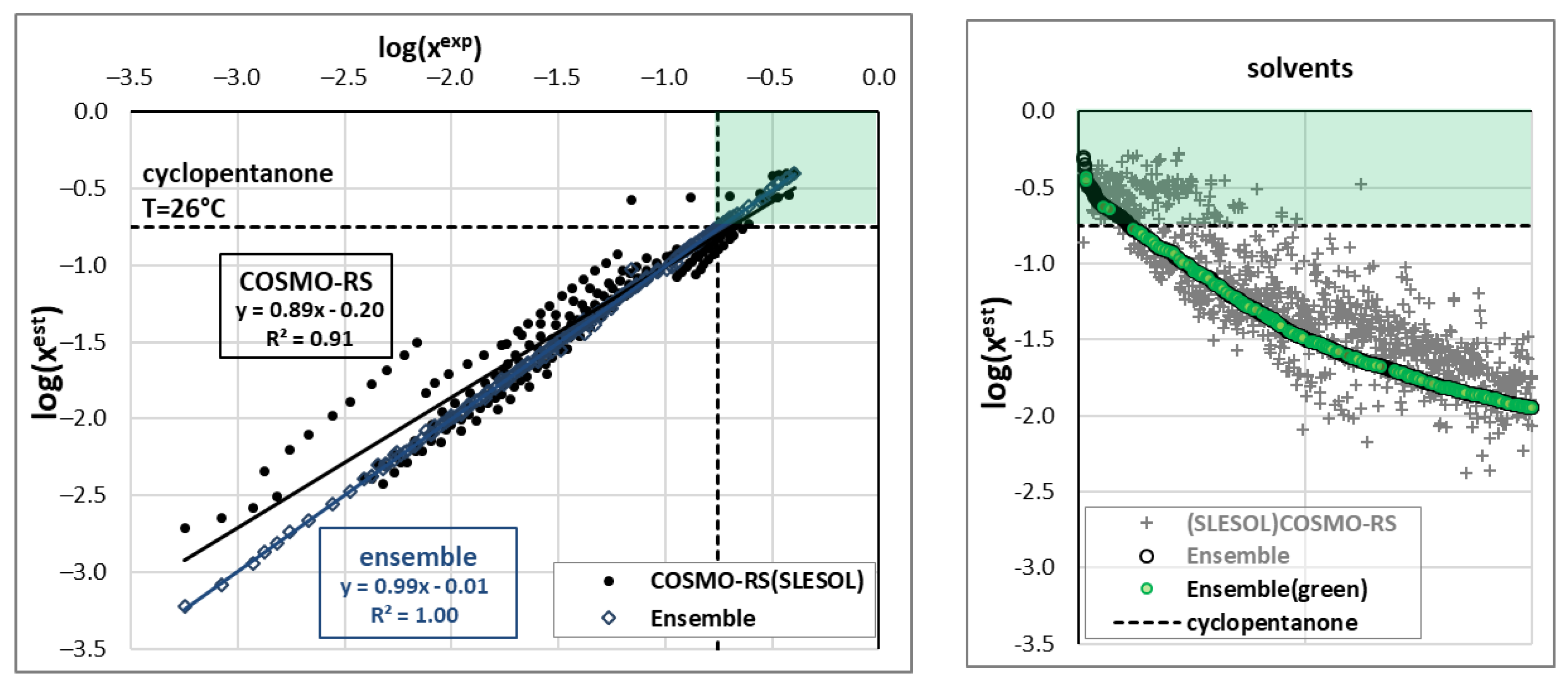
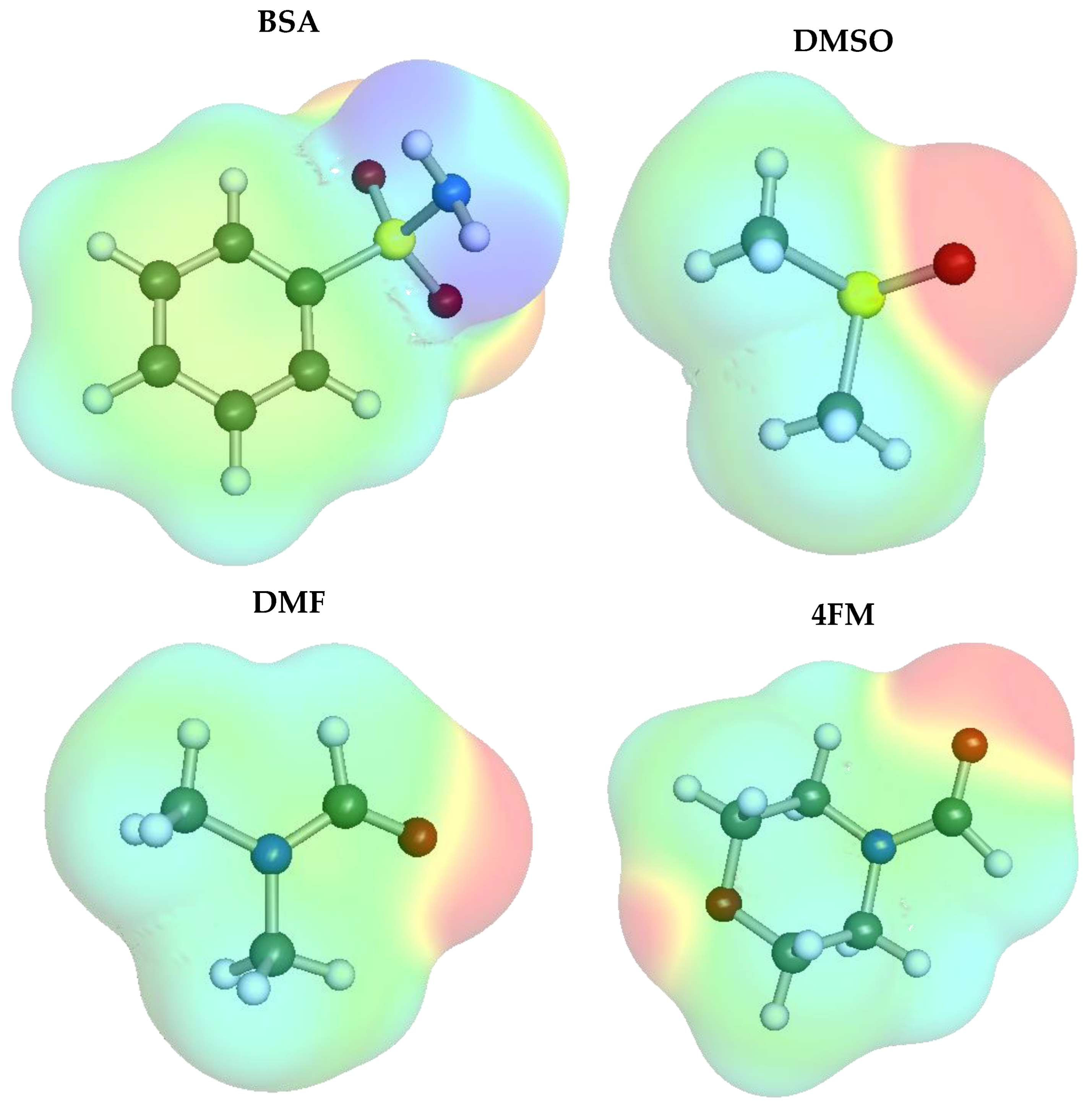
2.2. Ensemble Solubility Model
2.3. Screening of New Solvents
3. Materials and Methods
3.1. Materials
3.2. Solubility Measurements
3.3. Instrumental Analysis of Solid Residues
3.4. Solubility Dataset
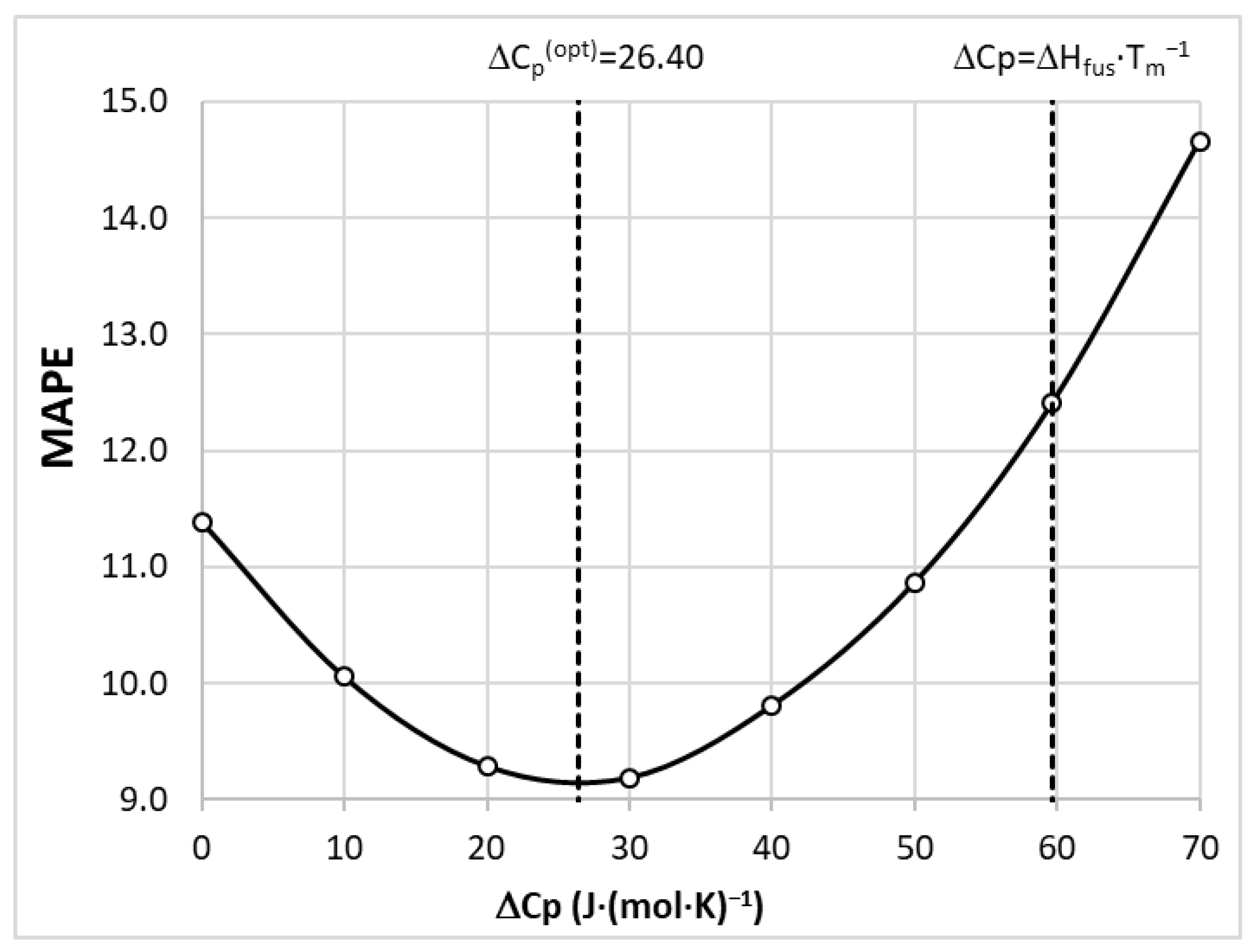
3.5. Model Development
3.6. Molecular Descriptors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- DeSimone, J.M. Practical approaches to green solvents. Science 2002, 297, 799–803. [Google Scholar] [CrossRef]
- Jessop, P.G. Searching for green solvents. Green Chem. 2011, 13, 1391–1398. [Google Scholar] [CrossRef]
- Cvjetko Bubalo, M.; Vidović, S.; Radojčić Redovniković, I.; Jokić, S. Green solvents for green technologies. J. Chem. Technol. Biotechnol. 2015, 90, 1631–1639. [Google Scholar] [CrossRef]
- Häckl, K.; Kunz, W. Some aspects of green solvents. Comptes Rendus Chim. 2018, 21, 572–580. [Google Scholar] [CrossRef]
- e Silva, A.P.S.; Pires, F.C.S.; Ferreira, M.C.R.; Silva, I.Q.; Aires, G.C.M.; Ribeiro, T.M.; Ortiz, E.G.; Martins, M.L.H.S.; de Carvalho, R.N. Case studies of green solvents in the pharmaceutical industry. In Green Sustainable Process for Chemical and Environmental Engineering and Science; Elsevier: Amsterdam, The Netherlands, 2021; pp. 151–159. [Google Scholar]
- Aissou, M.; Chemat-Djenni, Z.; Yara-Varón, E.; Fabiano-Tixier, A.S.; Chemat, F. Limonene as an agro-chemical building block for the synthesis and extraction of bioactive compounds. Comptes Rendus Chim. 2017, 20, 346–358. [Google Scholar] [CrossRef]
- Zhang, S.; Ye, L.; Zhang, H.; Hou, J. Green-solvent-processable organic solar cells. Mater. Today 2016, 19, 533–543. [Google Scholar] [CrossRef]
- Martínez, F.; Jouyban, A.; Acree, W.E. Pharmaceuticals solubility is still nowadays widely studied everywhere. Pharm. Sci. 2017, 23, 1–2. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.-C.; Kim, D.-H.; Lee, S.-E.; Kim, J.-K.; Park, J.-S. Overview of the Manufacturing Methods of Solid Dispersion Technology for Improving the Solubility of Poorly Water-Soluble Drugs and Application to Anticancer Drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Hancock, B.C.; York, P.; Rowe, R.C. The use of solubility parameters in pharmaceutical dosage form design. Int. J. Pharm. 1997, 148, 1–21. [Google Scholar] [CrossRef]
- Blagden, N.; de Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Grossmann, L.; McClements, D.J. Current insights into protein solubility: A review of its importance for alternative proteins. Food Hydrocoll. 2023, 137, 108416. [Google Scholar] [CrossRef]
- Sou, T.; Bergström, C.A.S. Automated assays for thermodynamic (equilibrium) solubility determination. Drug Discov. Today Technol. 2018, 27, 11–19. [Google Scholar] [CrossRef]
- Lu, W.; Chen, H. Application of deep eutectic solvents (DESs) as trace level drug extractants and drug solubility enhancers: State-of-the-art, prospects and challenges. J. Mol. Liq. 2022, 349, 118105. [Google Scholar] [CrossRef]
- Suwanwong, Y.; Boonpangrak, S. Molecularly imprinted polymers for the extraction and determination of water-soluble vitamins: A review from 2001 to 2020. Eur. Polym. J. 2021, 161, 110835. [Google Scholar] [CrossRef]
- George, R.F.; Bua, S.; Supuran, C.T.; Awadallah, F.M. Synthesis of some N-aroyl-2-oxindole benzenesulfonamide conjugates with carbonic anhydrase inhibitory activity. Bioorg. Chem. 2020, 96, 103635. [Google Scholar] [CrossRef] [PubMed]
- Buza, A.; Türkeş, C.; Arslan, M.; Demir, Y.; Dincer, B.; Nixha, A.R.; Beydemir, Ş. Discovery of novel benzenesulfonamides incorporating 1,2,3-triazole scaffold as carbonic anhydrase I, II, IX, and XII inhibitors. Int. J. Biol. Macromol. 2023, 239, 124232. [Google Scholar] [CrossRef]
- Nguyen, V.V.L.; Huynh, D.P. Synthesis poly(ethylene glycol)-poly(benzenesulfonamide serinol lactide urethane) copolymer for preparation pH sensitive hydrogel. Mater. Today Proc. 2022, 66, 2806–2810. [Google Scholar] [CrossRef]
- Kumar, N.; Solt, L.A.; Conkright, J.J.; Wang, Y.; Istrate, M.A.; Busby, S.A.; Garcia-Ordonez, R.D.; Burris, T.P.; Griffin, P.R. The Benzenesulfoamide T0901317 [N-(2,2,2-Trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide] Is a Novel Retinoic Acid Receptor-Related Orphan Receptor-α/γ Inverse Agonist. Mol. Pharmacol. 2010, 77, 228–236. [Google Scholar] [CrossRef]
- Prabhakaran, J.; Underwood, M.D.; Parsey, R.V.; Arango, V.; Majo, V.J.; Simpson, N.R.; Van Heertum, R.; Mann, J.J.; Kumar, J.S.D. Synthesis and in vivo evaluation of [18F]-4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide as a PET imaging probe for COX-2 expression. Bioorg. Med. Chem. 2007, 15, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Thilagavathi, G.; Jayachitra, R.; Kanagavalli, A.; Elangovan, N.; Sirajunnisa, A.; Sowrirajan, S.; Thomas, R. Synthesis, computational, molecular docking studies and photophysical properties of (Z)-N-(pyrimidin-2-yl)-4-(thiophen-2-ylmethylene)amino) benzenesulfonamide. J. Indian Chem. Soc. 2023, 100, 100835. [Google Scholar] [CrossRef]
- Castro, Ó.; Borrull, S.; Pocurull, E.; Borrull, F. Determination of benzothiazoles, benzotriazoles and benzenesulfonamides in seafood using quick, easy, cheap, effective, rugged and safe extraction followed by gas chromatography—Tandem mass spectrometry: Method development and risk assessment. J. Chromatogr. A 2023, 1691, 463841. [Google Scholar] [CrossRef]
- Talebi, M.R.; Nematollahi, D.; Massah, A.R. Comparative electrochemical study of N-(4-aminophenyl) and N-(4-hydroxyphenyl)benzenesulfonamide derivatives. Electrochim. Acta 2023, 457, 142499. [Google Scholar] [CrossRef]
- Du, J.; Liu, P.; Zhu, Y.; Wang, G.; Xing, S.; Liu, T.; Xia, J.; Dong, S.; Lv, N.; Li, Z. Novel tryptanthrin derivatives with benzenesulfonamide substituents: Design, synthesis, and anti-inflammatory evaluation. Eur. J. Med. Chem. 2023, 246, 114956. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.S.; Wang, Y.B.; Zhang, Z.T.; Liu, H.T. Solubilities of benzene sulfonamide in supercritical CO2 in the absence and presence of cosolvent. Thermochim. Acta 2012, 527, 165–171. [Google Scholar] [CrossRef]
- Li, Y.; Wu, K.; Liang, L. Solubility Determination, Modeling, and Thermodynamic Dissolution Properties of Benzenesulfonamide in 16 Neat Solvents from 273.15 to 324.45 K. J. Chem. Eng. Data 2019, 64, 3606–3616. [Google Scholar] [CrossRef]
- Duereh, A.; Sato, Y.; Smith, R.L.; Inomata, H. Methodology for replacing dipolar aprotic solvents used in API processing with safe hydrogen-bond donor and acceptor solvent-pair mixtures. Org. Process Res. Dev. 2017, 21, 114–124. [Google Scholar] [CrossRef]
- Wongsawa, T.; Hronec, M.; Lothongkum, A.W.; Pancharoen, U.; Phatanasri, S. Experiments and thermodynamic models for ternary (liquid–liquid) equilibrium systems of water + cyclopentanone + organic solvents at T = 298.2 K. J. Mol. Liq. 2014, 196, 98–106. [Google Scholar] [CrossRef]
- Chen, G.; Xia, M.; Lei, W.; Wang, F.; Gong, X. Prediction of crystal morphology of cyclotrimethylene trinitramine in the solvent medium by computer simulation: A case of cyclohexanone solvent. J. Phys. Chem. A 2014, 118, 11471–11478. [Google Scholar] [CrossRef]
- Tong, X.; Woods, D.; Acree, W.E.; Abraham, M.H. Updated Abraham model correlations for correlating solute transfer into dry butanone and dry cyclohexanone solvents. Phys. Chem. Liq. 2018, 56, 571–583. [Google Scholar] [CrossRef]
- Höckelmann, C.; Jüttner, F. Volatile organic compound (VOC) analysis and sources of limonene, cyclohexanone and straight chain aldehydes in axenic cultures of Calothrix and Plectonema. Water Sci. Technol. 2004, 49, 47–54. [Google Scholar] [CrossRef]
- Wang, N.; Shi, M.; Wu, S.; Guo, X.; Zhang, X.; Ni, N.; Sha, S.; Zhang, H. Study on Volatile Organic Compound (VOC) Emission Control and Reduction Potential in the Pesticide Industry in China. Atmosphere 2022, 13, 1241. [Google Scholar] [CrossRef]
- de Gouw, J.A.; Gilman, J.B.; Kim, S.-W.; Alvarez, S.L.; Dusanter, S.; Graus, M.; Griffith, S.M.; Isaacman-VanWertz, G.; Kuster, W.C.; Lefer, B.L.; et al. Chemistry of Volatile Organic Compounds in the Los Angeles Basin: Formation of Oxygenated Compounds and Determination of Emission Ratios. J. Geophys. Res. Atmos. 2018, 123, 2298–2319. [Google Scholar] [CrossRef]
- Lee, Y.H.; Chung, Y.H.; Kim, H.Y.; Shin, S.H.; Lee, S.B. Subacute Inhalation Toxicity of Cyclohexanone in B6C3F1 Mice. Toxicol. Res. 2018, 34, 49–53. [Google Scholar] [CrossRef]
- Scognamiglio, J.; Jones, L.; Letizia, C.S.; Api, A.M. Fragrance material review on cyclopentanone. Food Chem. Toxicol. 2012, 50, S608–S612. [Google Scholar] [CrossRef]
- Belsito, D.; Bickers, D.; Bruze, M.; Calow, P.; Dagli, M.L.; Dekant, W.; Fryer, A.D.; Greim, H.; Miyachi, Y.; Saurat, J.H.; et al. A toxicologic and dermatologic assessment of cyclopentanones and cyclopentenones when used as fragrance ingredients. Food Chem. Toxicol. 2012, 50, S517–S556. [Google Scholar] [CrossRef]
- Begum, M.Y. Advanced modeling based on machine learning for evaluation of drug nanoparticle preparation via green technology: Theoretical assessment of solubility variations. Case Stud. Therm. Eng. 2023, 45, 103029. [Google Scholar] [CrossRef]
- Abouzied, A.S.; Alshahrani, S.M.; Hani, U.; Obaidullah, A.J.; Al Awadh, A.A.; Lahiq, A.A.; Al-fanhrawi, H.J. Assessment of solid-dosage drug nanonization by theoretical advanced models: Modeling of solubility variations using hybrid machine learning models. Case Stud. Therm. Eng. 2023, 47, 103101. [Google Scholar] [CrossRef]
- Boobier, S.; Hose, D.R.J.; Blacker, A.J.; Nguyen, B.N. Machine learning with physicochemical relationships: Solubility prediction in organic solvents and water. Nat. Commun. 2020, 11, 5753. [Google Scholar] [CrossRef]
- Cysewski, P.; Przybyłek, M.; Rozalski, R. Experimental and theoretical screening for green solvents improving sulfamethizole solubility. Materials 2021, 14, 5915. [Google Scholar] [CrossRef] [PubMed]
- Cysewski, P.; Jeliński, T.; Przybyłek, M.; Nowak, W.; Olczak, M. Solubility Characteristics of Acetaminophen and Phenacetin in Binary Mixtures of Aqueous Organic Solvents: Experimental and Deep Machine Learning Screening of Green Dissolution Media. Pharmaceutics 2022, 14, 2828. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Jiao, Z.; Zhang, Z.; Wang, Q. Development of Solubility Prediction Models with Ensemble Learning. Ind. Eng. Chem. Res. 2021, 60, 11627–11635. [Google Scholar] [CrossRef]
- Mousavi, S.P.; Nakhaei-Kohani, R.; Atashrouz, S.; Hadavimoghaddam, F.; Abedi, A.; Hemmati-Sarapardeh, A.; Mohaddespour, A. Modeling of H2S solubility in ionic liquids: Comparison of white-box machine learning, deep learning and ensemble learning approaches. Sci. Rep. 2023, 13, 7946. [Google Scholar] [CrossRef] [PubMed]
- Cysewski, P.; Jeliński, T.; Cymerman, P.; Przybyłek, M. Solvent screening for solubility enhancement of theophylline in neat, binary and ternary NADES solvents: New measurements and ensemble machine learning. Int. J. Mol. Sci. 2021, 22, 7347. [Google Scholar] [CrossRef]
- Jeliński, T.; Bugalska, N.; Koszucka, K.; Przybyłek, M.; Cysewski, P. Solubility of sulfanilamide in binary solvents containing water: Measurements and prediction using Buchowski-Ksiazczak solubility model. J. Mol. Liq. 2020, 319, 114342. [Google Scholar] [CrossRef]
- Zhao, H.; Xia, S.; Ma, P. Use of ionic liquids as ‘green’ solvents for extractions. J. Chem. Technol. Biotechnol. 2005, 80, 1089–1096. [Google Scholar] [CrossRef]
- Ratti, R. Industrial applications of green chemistry: Status, Challenges and Prospects. SN Appl. Sci. 2020, 2, 263. [Google Scholar] [CrossRef]
- Wania, F.; MacKay, D. Tracking the Distribution of Persistent Organic Pollutants. Environ. Sci. Technol. 1996, 30, 390A–396A. [Google Scholar] [CrossRef]
- Pedregosa Fabianpedregosa, F.; Michel, V.; Grisel Oliviergrisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Vanderplas, J.; Cournapeau, D.; Pedregosa, F.; Varoquaux, G.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Dupeux, T.; Gaudin, T.; Marteau-Roussy, C.; Aubry, J.; Nardello-Rataj, V. COSMO-RS as an effective tool for predicting the physicochemical properties of fragrance raw materials. Flavour Fragr. J. 2022, 37, 106–120. [Google Scholar] [CrossRef]
- Eckert, F.; Klamt, A. Accurate prediction of basicity in aqueous solution with COSMO-RS. J. Comput. Chem. 2006, 27, 11–19. [Google Scholar] [CrossRef]
- Loschen, C.; Klamt, A. Solubility prediction, solvate and cocrystal screening as tools for rational crystal engineering. J. Pharm. Pharmacol. 2015, 67, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Harten, P.; Martin, T.; Gonzalez, M.; Young, D. The software tool to find greener solvent replacements, PARIS III. Environ. Prog. Sustain. Energy 2020, 39, e13331. [Google Scholar] [CrossRef] [PubMed]
- Misuri, L.; Cappiello, M.; Balestri, F.; Moschini, R.; Barracco, V.; Mura, U.; Del-Corso, A. The use of dimethylsulfoxide as a solvent in enzyme inhibition studies: The case of aldose reductase. J. Enzyme Inhib. Med. Chem. 2017, 32, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, C.; Lyzenga, W.; Shao, D.; Agu, R.U. Water-soluble organic solubilizers for in vitro drug delivery studies with respiratory epithelial cells: Selection based on various toxicity indicators. Drug Deliv. 2010, 17, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Timm, M.; Saaby, L.; Moesby, L.; Hansen, E.W. Considerations regarding use of solvents in in vitro cell based assays. Cytotechnology 2013, 65, 887–894. [Google Scholar] [CrossRef]
- Popa-Burke, I.; Russell, J. Compound Precipitation in High-Concentration DMSO Solutions. SLAS Discov. 2014, 19, 1302–1308. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Biological assay challenges from compound solubility: Strategies for bioassay optimization. Drug Discov. Today 2006, 11, 446–451. [Google Scholar] [CrossRef]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-generation Hyperparameter Optimization Framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 2623–2631. [Google Scholar]
- Engel, T. Representation of Chemical Compounds. In Chemoinformatics; Gasteiger, J., Engel, T., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; pp. 15–168. [Google Scholar]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799. [Google Scholar] [CrossRef]
- Dassault Systèmes. COSMOtherm, Version 22.0.0; Dassault Systèmes, Biovia: San Diego, CA, USA, 2022.
- Cramer, R.D.; Bunce, J.D.; Patterson, D.E.; Frank, I.E. Crossvalidation, Bootstrapping, and Partial Least Squares Compared with Multiple Regression in Conventional QSAR Studies. Quant. Struct. Relatsh. 1988, 7, 18–25. [Google Scholar] [CrossRef]
- Miranda, M.S.; Matos, M.A.R.; Morais, V.M.F.; Liebman, J.F. Combined experimental and computational study on the energetics of 1,2-benzisothiazol-3(2H)-one and 1,4-benzothiazin-3(2H, 4H)-one. J. Chem. Thermodyn. 2011, 43, 635–644. [Google Scholar] [CrossRef]
- Cysewski, P.; Przybyłek, M.; Kowalska, A.; Tymorek, N. Thermodynamics and Intermolecular Interactions of Nicotinamide in Neat and Binary Solutions: Experimental Measurements and COSMO-RS Concentration Dependent Reactions Investigations. Int. J. Mol. Sci. 2021, 22, 7365. [Google Scholar] [CrossRef] [PubMed]
- Przybyłek, M.; Kowalska, A.; Tymorek, N.; Dziaman, T.; Cysewski, P. Thermodynamic Characteristics of Phenacetin in Solid State and Saturated Solutions in Several Neat and Binary Solvents. Molecules 2021, 26, 4078. [Google Scholar] [CrossRef]
- Cysewski, P.; Jeliński, T.; Przybyłek, M. Intermolecular Interactions of Edaravone in Aqueous Solutions of Ethaline and Glyceline Inferred from Experiments and Quantum Chemistry Computations. Molecules 2023, 28, 629. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes. COSMOconf, Version 22.0.0; Dassault Systèmes, Biovia: San Diego, CA, USA, 2022.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent Name | CAS | EI | Relative Price | log(xBSApred) |
|---|---|---|---|---|
| Ethanamine | [109-85-3] | 0.81 | 6.9 | −0.43 |
| DMSO | [67-68-5] | 0.26 | 1.0 | −0.45 |
| 2,2-Dimethoxyethylmethylamine | [122-07-6] | 0.95 | 7.3 | −0.63 |
| N-Methyl-2-pyrrolidone | [872-50-4] | 0.97 | 0.3 | −0.67 |
| Delta-octanolactone | [698-76-0] | 0.61 | 434.1 | −0.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cysewski, P.; Jeliński, T.; Przybyłek, M. Finding the Right Solvent: A Novel Screening Protocol for Identifying Environmentally Friendly and Cost-Effective Options for Benzenesulfonamide. Molecules 2023, 28, 5008. https://doi.org/10.3390/molecules28135008
Cysewski P, Jeliński T, Przybyłek M. Finding the Right Solvent: A Novel Screening Protocol for Identifying Environmentally Friendly and Cost-Effective Options for Benzenesulfonamide. Molecules. 2023; 28(13):5008. https://doi.org/10.3390/molecules28135008
Chicago/Turabian StyleCysewski, Piotr, Tomasz Jeliński, and Maciej Przybyłek. 2023. "Finding the Right Solvent: A Novel Screening Protocol for Identifying Environmentally Friendly and Cost-Effective Options for Benzenesulfonamide" Molecules 28, no. 13: 5008. https://doi.org/10.3390/molecules28135008
APA StyleCysewski, P., Jeliński, T., & Przybyłek, M. (2023). Finding the Right Solvent: A Novel Screening Protocol for Identifying Environmentally Friendly and Cost-Effective Options for Benzenesulfonamide. Molecules, 28(13), 5008. https://doi.org/10.3390/molecules28135008