
The History of mARC


Abstract
1. From N-Oxygenation to N-Reduction
2. The Prodrug Principle: Amidoximes Instead of Amidines
3. Discovery of the mARC Enzyme System
4. Substrates of mARC
5. mARC Enzymes in Human Disease
6. Outlook
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clement, B.; Beckett, A.H. Metabolism of promethazine In vitro. Identificaton of N-oxidized products. Xenobiotica 1981, 11, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Clement, B. The N-oxidation of benzamidines in vitro. Xenobiotica 1983, 13, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Clement, B.; Zimmermann, M. Characteristics of the microsomal N-hydroxylation of benzamidine to benzamidoxime. Xenobiotica 1987, 17, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Clement, B. In vitro studies on the microsomal N-oxidation of N-substituted benzamidines. Arch. Pharm. 1984, 317, 925–933. [Google Scholar] [CrossRef]
- Gorrod, J.W. Differentiation of various types of biological oxidation of nitrogen in organic compounds. Chem. Biol. Interact. 1973, 289, 289–303. [Google Scholar] [CrossRef]
- Gorrod, J. The current status of the pKa concept in the differentiation of enzymic N-oxidation. In Biological Oxidation of Nitrogen; Elsevier: Amsterdam, The Netherlands, 1978; pp. 201–210. [Google Scholar]
- Clement, B.; Immel, M. Untersuchungen zur in vitro N-Oxygenierung N-tert.alkylsubstituierter Benzamidine. Arch. Pharm. 1987, 320, 660–665. [Google Scholar] [CrossRef]
- Clement, B.; Zimmermann, M. Hepatic microsomal N-demethylation of N-methylbenzamidine: N-dealkylation vs. N-oxygenation of amidines. Biochem. Pharmacol. 1987, 36, 3127–3133. [Google Scholar] [CrossRef]
- Clement, B.; Zimmermann, M. Mechanism of the microsomal N-hydroxylation of para-substituted benzamidines. Biochem. Pharmacol. 1988, 37, 4747–4752. [Google Scholar] [CrossRef]
- Clement, B. In-vitro-Untersuchungen zur mikrosomalen N-Oxidation einiger Guanidine. Arch. Pharm. 1986, 319, 961–968. [Google Scholar] [CrossRef]
- Clement, B.; Kunze, T. In vitro oxygenation of N,N’-diphenylguanidines. Xenobiotica 1993, 23, 155–167. [Google Scholar] [CrossRef]
- Clement, B.; Schultze-Mosgau, M.H.; Richter, P.H.; Besch, A. Cytochrome P450-dependent N-hydroxylation of an aminoguanidine (amidinohydrazone) and microsomal retroreduction of the N-hydroxylated product. Xenobiotica 1994, 24, 671–688. [Google Scholar] [CrossRef]
- Clement, B.; Kunze, T. Microsomal N-oxygenation of adenine to adenine 1-N-oxide. Arch. Pharm. 1993, 326, 25–27. [Google Scholar] [CrossRef]
- Clement, B. Structural requirements of microsomal N-oxygenations derived from studies on amidines. Drug Metab. Drug Interact. 1989, 7, 87–108. [Google Scholar] [CrossRef]
- Najmi, A.A.; Bischoff, R.; Permentier, H.P. N-Dealkylation of amines. Molecules 2022, 27, 3293. [Google Scholar] [CrossRef]
- Ott, G.; Reichmann, D.; Boerger, C.; Cascorbi, I.; Bittner, F.; Mendel, R.R.; Kunze, T.; Clement, B.; Havemeyer, A. Functional characterization of protein variants encoded by nonsynonymous single nucleotide polymorphisms in MARC1 and MARC2 in healthy caucasians. Drug Metab. Dispos. 2014, 42, 718–725. [Google Scholar] [CrossRef]
- Grant, D.M. Detoxification pathways in the liver. J. Inherit. Metab. Dis. 1991, 14, 421–430. [Google Scholar] [CrossRef]
- Clement, B.; Christiansen, K.; Girreser, U. Phase 2 metabolites of N-hydroxylated amidines (amidoximes): synthesis, In vitro formation by pig hepatocytes, and mutagenicity testing. Chem. Res. Toxicol. 2001, 14, 319–326. [Google Scholar] [CrossRef]
- Froehlich, A.K.; Girreser, U.; Clement, B. Metabolism of N-hydroxyguanidines (N-hydroxydebrisoquine) in human and porcine hepatocytes: Reduction and formation of glucuronides. Drug Metab. Dispos. 2005, 33, 1532–1537. [Google Scholar] [CrossRef]
- Clement, B.; Zimmermann, M.; Schmitt, S. Biotransformation des Benzamidins und des Benzamidoxims durch mikrosomale Enzyme vom Kaninchen. Arch. Pharm. 1989, 322, 431–435. [Google Scholar] [CrossRef]
- Clement, B.; Schmitt, S.; Zimmermann, M. Enzymatic reduction of benzamidoxime to benzamidoxine. Arch. Pharm. 1988, 321, 955–956. [Google Scholar] [CrossRef]
- Hauptmann, J.; Paintz, M.; Kaiser, B.; Richter, M. Reduction of a benzamidoxime derivative to the corresponding benzamidine in vivo and in vitro. Pharmazie 1988, 43, 559–560. [Google Scholar] [PubMed]
- Clement, B.; Immel, M.; Schmitt, S.; Steinmann, U. Biotransformations of benzamidine and benzamidoxime in vivo. Arch. Pharm. 1993, 326, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Berger, B.J.; Lombardy, R.J.; Marbury, G.D.; Bell, C.A.; Dykstra, C.C.; Hall, J.E.; Tidwell, R.R. Metabolic N-hydroxylation of pentamidine in vitro. Antimicrob. Agents Chemother. 1990, 34, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Clement, B.; Raether, W. Amidoximes of pentamidine: Synthesis, trypanocidal and leishmanicidal activity. Arzneimittelforschung 1985, 35, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Clement, B.; Immel, M.; Terlinden, R.; Wingen, F.-J. Reduction of amidoxime derivatives to pentamidine in vivo. Arch. Pharm. 1992, 325, 61–62. [Google Scholar] [CrossRef]
- Cohrs, B.; Zhao, Y.; Lützen, U.; Culman, J.; Clement, B.; Zuhayra, M. In vivo SPECT imaging of [123I]-labeled pentamidine prodrugs for the treatment of human African trypanosomiasis, pharmacokinetics, and bioavailability studies in rats. Int. J. Pharm. 2014, 477, 167–175. [Google Scholar] [CrossRef]
- Clement, B. Reduction of N-hydroxylated compounds: Amidoximes (N-hydroxyamidines) as pro-drugs of amidines. Drug Metab. Rev. 2002, 34, 565–579. [Google Scholar] [CrossRef]
- Reeh, C.; Wundt, J.; Clement, B. N,N′-Dihydroxyamidines: A new prodrug principle to improve the oral bioavailability of amidines. J. Med. Chem. 2007, 50, 6730–6734. [Google Scholar] [CrossRef]
- Kotthaus, J.; Kotthaus, J.; Schade, D.; Schwering, U.; Hungeling, H.; Müller-Fielitz, H.; Raasch, W.; Clement, B. New prodrugs of the antiprotozoal drug pentamidine. ChemMedChem 2011, 6, 2233–2242. [Google Scholar] [CrossRef]
- Schade, D.; Kotthaus, J.; Hungeling, H.; Kotthaus, J.; Clement, B. The peptidylglycine alpha-amidating monooxygenase (PAM): A novel prodrug strategy for amidoximes and N-hydroxyguanidines? ChemMedChem 2009, 4, 1595–1599. [Google Scholar] [CrossRef]
- Zikria, J.; Ansell, J. Oral anticoagulation with Factor Xa and thrombin inhibitors: Is there an alternative to warfarin? Discov. Med. 2009, 8, 196–203. [Google Scholar]
- Gresele, P.; Agnelli, G. Novel approaches to the treatment of thrombosis. Trends Pharmacol. Sci. 2002, 23, 25–32. [Google Scholar] [CrossRef]
- Stürzebecher, J.; Vieweg, H.; Wikström, P.; Turk, D.; Bode, W. Interactions of thrombin with benzamidine-based inhibitors. Biol. Chem. Hoppe Seyler 1992, 373, 491–496. [Google Scholar] [CrossRef]
- Tanizawa, K.; Ishii, S.-I.; Hamaguchi, K.; Kanaoka, Y. Proteolytic Enzymes: VI. aromatic amidines as competitive inhibitors of trypsin. J. Biochem. 1971, 69, 893–899. [Google Scholar] [CrossRef]
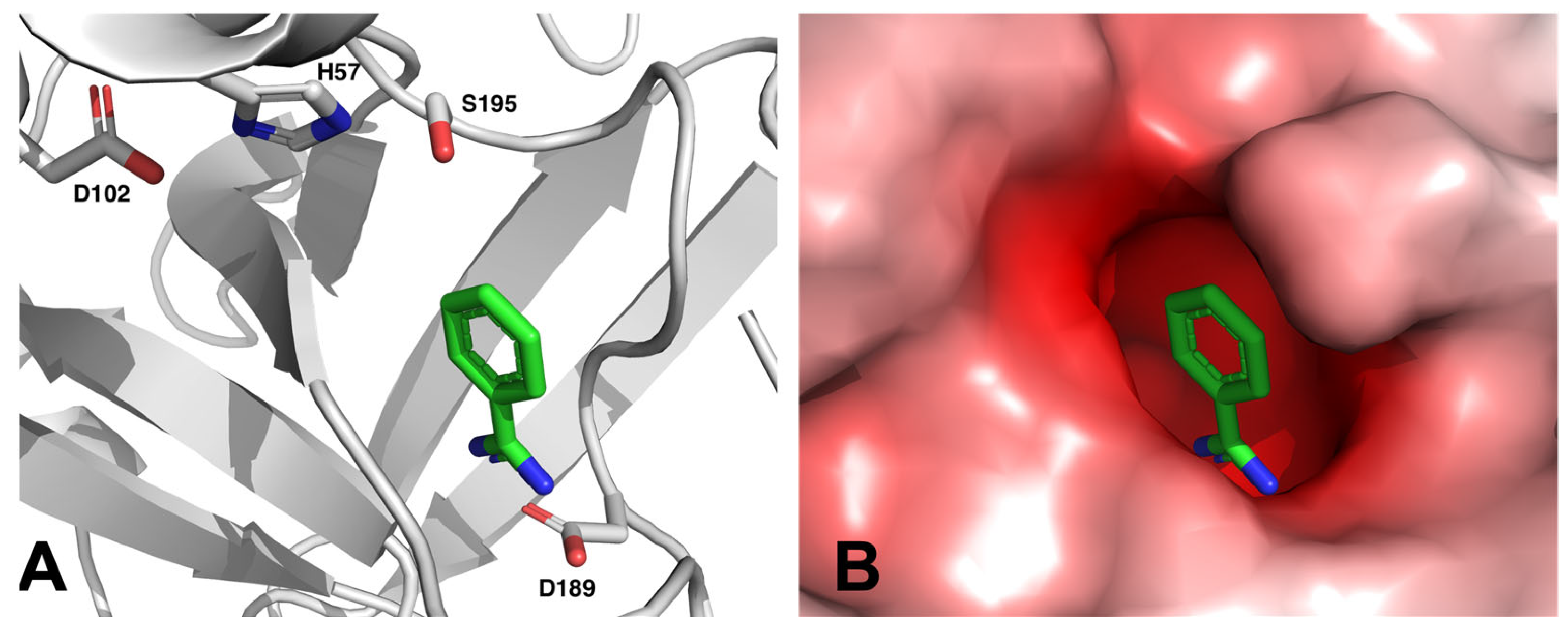
- Rühmann, E.; Betz, M.; Heine, A.; Klebe, G. Fragment binding ban be either more enthalpy-driven or entropy-driven: Crystal structures and residual hydration patterns suggest why. J. Med. Chem. 2015, 58, 6960–6971. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Weller, T.; Alig, L.; Beresini, M.; Blackburn, B.; Bunting, S.; Hadváry, P.; Hürzeler Müller, M.; Knopp, D.; Levet-Trafit, B.; Lipari, M.T.; et al. Orally active fibrinogen receptor antagonists. 2. Amidoximes as prodrugs of amidines. J. Med. Chem. 1996, 39, 3139–3147. [Google Scholar] [CrossRef]
- Wittke, B.; Mackie, I.J.; Machin, S.J.; Timm, U.; Zell, M.; Goggin, T. Pharmacokinetics and pharmacodynamics of Ro 44-3888 after single ascending oral doses of sibrafiban, an oral platelet aggregation inhibitor, in healthy male volunteers. Br. J. Clin. Pharmacol. 1999, 47, 521–530. [Google Scholar] [CrossRef]
- Song, Y.; Clizbe, L.; Bhakta, C.; Teng, W.; Wong, P.; Huang, B.; Tran, K.; Sinha, U.; Park, G.; Reed, A.; et al. Design and synthesis of factor Xa inhibitors and their prodrugs. Bioorg. Med. Chem. Lett. 2003, 13, 297–300. [Google Scholar] [CrossRef]
- Uchida, M.; Okazaki, K.; Mukaiyama, H.; Isawa, H.; Kobayashi, H.; Shiohara, H.; Muranaka, H.; Kai, Y.; Kikuchi, N.; Takeuchi, H.; et al. Orally active factor Xa inhibitors: Investigation of a novel series of 3-amidinophenylsulfonamide derivatives using an amidoxime prodrug strategy. Bioorg. Med. Chem. Lett. 2008, 18, 4682–4687. [Google Scholar] [CrossRef]
- Gustafsson, D.; Nyström, J.; Carlsson, S.; Bredberg, U.; Eriksson, U.; Gyzander, E.; Elg, M.; Antonsson, T.; Hoffmann, K.; Ungell, A.; et al. The direct thrombin inhibitor melagatran and its oral prodrug H 376/95: Intestinal absorption properties, biochemical and pharmacodynamic effects. Thromb. Res. 2001, 101, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Clement, B.; Lopian, K. Characterization of in vitro biotransformation of new, orally active, direct thrombin inhibitor ximelagatran, an amidoxime and ester prodrug. Drug Metab. Dispos. 2003, 31, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M.; Larrey, D.; Olsson, R.; Lewis, J.H.; Keisu, M.; Auclert, L.; Sheth, S. Hepatic findings in long-term clinical trials of ximelagatran. Drug Saf. 2005, 28, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Cully, M. Milestone 9: Ximelagatran sets the stage for NOACs. Nat. Rev. Cardiol. 2017. [Google Scholar] [CrossRef]
- Laizure, S.C.; Parker, R.B.; Herring, V.L.; Hu, Z.-Y. Identification of carboxylesterase-dependent dabigatran etexilate hydrolysis. Drug Metab. Dispos. 2014, 42, 201. [Google Scholar] [CrossRef]
- Blommel, M.L.; Blommel, A.L. Dabigatran etexilate: A novel oral direct thrombin inhibitor. Am. J. Health Syst. Pharm. 2011, 68, 1506–1519. [Google Scholar] [CrossRef]
- Stangier, J.; Stähle, H.; Rathgen, K.; Fuhr, R. Pharmacokinetics and pharmacodynamics of the direct oral thrombin inhibitor dabigatran in healthy elderly subjects. Clin. Pharmacokinet. 2008, 47, 47–59. [Google Scholar] [CrossRef]
- Clement, B.; Kotthaus, J.; Kotthaus, J.; Schade, D. Dabigatran-Amidoximester als Prodrugs und ihre Verwendung als Arzneimittel (EP 2550966 B1). EU Patent EP 2773347 B1, 13 September 2017. [Google Scholar]
- Froriep, D.; Clement, B.; Bittner, F.; Mendel, R.R.; Reichmann, D.; Schmalix, W.; Havemeyer, A. Activation of the anti-cancer agent upamostat by the mARC enzyme system. Xenobiotica 2013, 43, 780–784. [Google Scholar] [CrossRef]
- Plasse, T.F.; Delgado, B.; Potts, J.; Abramson, D.; Fehrmann, C.; Fathi, R.; McComsey, G.A. A randomized, placebo-controlled pilot study of upamostat, a host-directed serine protease inhibitor, for outpatient treatment of COVID-19. Int. J. Infect. Dis. 2023, 128, 148–156. [Google Scholar] [CrossRef]
- Schade, D.; Kotthaus, J.; Riebling, L.; Kotthaus, J.; Muller-Fielitz, H.; Raasch, W.; Hoffmann, A.; Schmidtke, M.; Clement, B. Zanamivir amidoxime- and N-hydroxyguanidine-based prodrug approaches to tackle poor oral bioavailability. J. Pharm. Sci. 2015, 104, 3208–3219. [Google Scholar] [CrossRef]
- Schade, D.; Kotthaus, J.; Riebling, L.; Kotthaus, J.; Muller-Fielitz, H.; Raasch, W.; Koch, O.; Seidel, N.; Schmidtke, M.; Clement, B. Development of novel potent orally bioavailable oseltamivir derivatives active against resistant influenza A. J. Med. Chem. 2014, 57, 759–769. [Google Scholar] [CrossRef]
- Maccallini, C.; Marinelli, L.; Indorf, P.; Cacciatore, I.; Fantacuzzi, M.; Clement, B.; Di Stefano, A.; Amoroso, R. A novel prodrug of a nNOS inhibitor with improved pharmacokinetic potential. ChemMedChem 2020, 15, 2157–2163. [Google Scholar] [CrossRef]
- Kadlubar, F.F.; Ziegler, D.M. Properties of a NADH-dependent N-hydroxy amine reductase isolated from pig liver microsomes. Arch. Biochem. Biophys. 1974, 162, 83–92. [Google Scholar] [CrossRef]
- Andersson, S.; Hofmann, Y.; Nordling, A.; Li, X.Q.; Nivelius, S.; Andersson, T.B.; Ingelman-Sundberg, M.; Johansson, I. Characterization and partial purification of the rat and human enzyme systems active in the reduction of N-hydroxymelagatran and benzamidoxime. Drug Metab. Dispos. 2005, 33, 570–578. [Google Scholar] [CrossRef]
- Bernheim, M.L.C.; Hochstein, P. Reduction of hydroxylamine by rat liver mitochondria. Arch. Biochem. Biophys. 1968, 124, 436–442. [Google Scholar] [CrossRef]
- Bernheim, M.L. The hydroxylamine reductase of mitochondria. Arch. Biochem. Biophys. 1969, 134, 408–413. [Google Scholar] [CrossRef]
- Clement, B.; Lomb, R.; Möller, W. Isolation and characterization of the protein components of the liver microsomal O2-insensitive NADH-benzamidoxime reductase. J. Biol. Chem. 1997, 272, 19615–19620. [Google Scholar] [CrossRef]
- Johansson, I.; Thelin, A.; Hofmann, Y.; Andersson, S.; Nordling, A.; Li, Q.; Carlsson, S.; Andersson, T.; Ingelman-Sundberg, M. Identification of stearoyl CoA desaturase as the enzyme responsible for the reduction of ximelagatran/N-hydroxymelagatran and benzamidoxime in adipocytes. Drug Metab. Rev. 2005, 37, 48–49. [Google Scholar]
- Reh, R.; Ozols, J.; Clement, B. Involvement of stearoyl-CoA desaturase in the reduction of amidoxime prodrugs. Xenobiotica 2008, 38, 1177–1190. [Google Scholar] [CrossRef]
- Havemeyer, A.; Bittner, F.; Wollers, S.; Mendel, R.; Kunze, T.; Clement, B. Identification of the missing component in the mitochondrial benzamidoxime prodrug-converting system as a novel molybdenum enzyme. J. Biol. Chem. 2006, 281, 34796–34802. [Google Scholar] [CrossRef]
- Anantharaman, V.; Aravind, L. MOSC domains: Ancient, predicted sulfur-carrier domains, present in diverse metal-sulfur cluster biosynthesis proteins including molybdenum cofactor sulfurases. FEMS Microbiol. Lett. 2002, 207, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.W.T.; Ooi, J.S.G.; Joly, G.L.C.; Chan, K.R. Gene Updater: A web tool that autocorrects and updates for Excel misidentified gene names. Sci. Rep. 2022, 12, 12743. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R. The history of the molybdenum cofactor—A personal view. Molecules 2022, 27, 4934. [Google Scholar] [CrossRef] [PubMed]
- Gruenewald, S.; Wahl, B.; Bittner, F.; Hungeling, H.; Kanzow, S.; Kotthaus, J.; Schwering, U.; Mendel, R.R.; Clement, B. The fourth molybdenum containing enzyme mARC: Cloning and involvement in the activation of N-hydroxylated prodrugs. J. Med. Chem. 2008, 51, 8173–8177. [Google Scholar] [CrossRef] [PubMed]
- Wahl, B.; Reichmann, D.; Niks, D.; Krompholz, N.; Havemeyer, A.; Clement, B.; Messerschmidt, T.; Rothkegel, M.; Biester, H.; Hille, R.; et al. Biochemical and spectroscopic characterization of the human Mitochondrial Amidoxime Reducing Components hmARC-1 and hmARC-2 suggests the existence of a new molybdenum enzyme family in eukaryotes. J. Biol. Chem. 2010, 285, 37847–37859. [Google Scholar] [CrossRef]
- Indorf, P.; Kubitza, C.; Scheidig, A.; Kunze, T.; Clement, B. Drug metabolism by the Mitochondrial Amidoxime Reducing Component (mARC): Rapid assay and identification of new substrates. J. Med. Chem. 2019, 63, 6538–6546. [Google Scholar] [CrossRef]
- Kalimuthu, P.; Havemeyer, A.; Clement, B.; Kubitza, C.; Scheidig, A.J.; Bernhardt, P.V. Human mitochondrial amidoxime reducing component (mARC): An electrochemical method for identifying new substrates and inhibitors. Electrochem. Commun. 2017, 84, 90–93. [Google Scholar] [CrossRef]
- Zapiter, J.; Harmer, J.R.; Struwe, M.; Scheidig, A.; Clement, B.; Bernhardt, P.V. Enzyme electrode biosensors for N-hydroxylated prodrugs incorporating the mitochondrial Amidoxime Reducing Component. Anal. Chem. 2022, 94, 9208–9215. [Google Scholar] [CrossRef]
- Klein, J.M.; Busch, J.D.; Potting, C.; Baker, M.J.; Langer, T.; Schwarz, G. The Mitochondrial Amidoxime-Reducing Component (mARC1) is a novel signal-anchored protein of the outer mitochondrial membrane. J. Biol. Chem. 2012, 287, 42795–42803. [Google Scholar] [CrossRef]
- Plitzko, B.; Ott, G.; Reichmann, D.; Henderson, C.J.; Wolf, C.R.; Mendel, R.; Bittner, F.; Clement, B.; Havemeyer, A. The involvement of mitochondrial amidoxime reducing components 1 and 2 and mitochondrial cytochrome b5 in N-reductive metabolism in human cells. J. Biol. Chem. 2013, 288, 20228–20237. [Google Scholar] [CrossRef]
- Plitzko, B.; Havemeyer, A.; Bork, B.; Bittner, F.; Mendel, R.; Clement, B. Defining the role of the NADH-cytochrome-b5 reductase 3 in the Mitochondrial Amidoxime Reducing Component enzyme system. Drug Metab. Dispos. 2016, 44, 1617–1621. [Google Scholar] [CrossRef]
- Kubitza, C.; Bittner, F.; Ginsel, C.; Havemeyer, A.; Clement, B.; Scheidig, A.J. Crystal structure of human mARC1 reveals its exceptional position among eukaryotic molybdenum enzymes. Proc. Natl. Acad. Sci. USA 2018, 115, 11958–11963. [Google Scholar] [CrossRef]
- Kubitza, C.; Ginsel, C.; Bittner, F.; Havemeyer, A.; Clement, B.; Scheidig, A.J. T4 lysozyme-facilitated crystallization of the human molybdenum cofactor-dependent enzyme mARC. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 337–344. [Google Scholar] [CrossRef]
- Bauch, E.; Reichmann, D.; Mendel, R.R.; Bittner, F.; Manke, A.M.; Kurz, P.; Girreser, U.; Havemeyer, A.; Clement, B. Electrochemical and mARC-catalyzed enzymatic reduction of para-substituted benzamidoximes: Consequences for the prodrug concept “amidoximes instead of amidines”. ChemMedChem 2015, 10, 360–367. [Google Scholar] [CrossRef]
- Kotthaus, J.; Wahl, B.; Havemeyer, A.; Kotthaus, J.; Schade, D.; Garbe-Schonberg, D.; Mendel, R.; Bittner, F.; Clement, B. Reduction of N(omega)-hydroxy-L-arginine by the mitochondrial amidoxime reducing component (mARC). Biochem. J. 2011, 433, 383–391. [Google Scholar] [CrossRef]
- Jakobs, H.H.; Froriep, D.; Havemeyer, A.; Mendel, R.R.; Bittner, F.; Clement, B. The mitochondrial amidoxime reducing component (mARC): Involvement in metabolic reduction of N-oxides, oximes and N-hydroxyamidinohydrazones. ChemMedChem 2014, 9, 2381–2387. [Google Scholar] [CrossRef]
- Havemeyer, A.; Grünewald, S.; Wahl, B.; Bittner, F.; Mendel, R.; Erdélyi, P.; Fischer, J.; Clement, B. Reduction of N-hydroxy-sulfonamides, including N-hydroxy-valdecoxib, by the molybdenum-containing enzyme mARC. Drug Metab. Dispos. 2010, 28, 1917–1921. [Google Scholar] [CrossRef]
- Ginsel, C.; Plitzko, B.; Froriep, D.; Stolfa, D.A.; Jung, M.; Kubitza, C.; Scheidig, A.J.; Havemeyer, A.; Clement, B. The involvement of the Mitochondrial Amidoxime Reducing Component (mARC) in the reductive metabolism of hydroxamic acids. Drug Metab. Dispos. 2018, 46, 1396–1402. [Google Scholar] [CrossRef]
- Schneider, J.; Girreser, U.; Havemeyer, A.; Bittner, F.; Clement, B. Detoxification of trimethylamine N-oxide by the Mitochondrial Amidoxime Reducing Component mARC. Chem. Res. Toxicol. 2018, 46, 1396–1402. [Google Scholar] [CrossRef]
- Testa, B.; Clement, B. Chapter 24—Biotransformation reactions and their enzymes. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth, C.G., Aldous, D., Raboisson, P., Rognan, D., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 561–584. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Ware, R.E.; Despotovic, J.M.; Mortier, N.A.; Flanagan, J.M.; He, J.; Smeltzer, M.P.; Kimble, A.C.; Aygun, B.; Wu, S.; Howard, T.; et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood 2011, 118, 4985–4991. [Google Scholar] [CrossRef] [PubMed]
- Kozmin, S.G.; Leroy, P.; Pavlov, Y.I.; Schaaper, R.M. YcbX and yiiM, two novel determinants for resistance of Escherichia coli to N-hydroxylated base analogues. Mol. Microbiol. 2008, 68, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Krompholz, N.; Krischkowski, C.; Reichmann, D.; Garbe-Schönberg, D.; Mendel, R.; Bittner, F.; Clement, B.; Havemeyer, A. The Mitochondrial Amidoxime Reducing Component (mARC) is involved in detoxification of N-hydroxylated base analogues. Chem. Res. Toxicol. 2012, 25, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Plitzko, B.; Havemeyer, A.; Kunze, T.; Clement, B. The pivotal role of the mitochondrial amidoxime reducing component 2 in protecting human cells against apoptotic effects of the base analog N6-hydroxylaminopurine. J. Biol. Chem. 2015, 290, 10126–10135. [Google Scholar] [CrossRef]
- Ott, G.; Plitzko, B.; Krischkowski, C.; Reichmann, D.; Bittner, F.; Mendel, R.R.; Kunze, T.; Clement, B.; Havemeyer, A. Reduction of sulfamethoxazole hydroxylamine (SMX-HA) by the mitochondrial amidoxime reducing component (mARC). Chem. Res. Toxicol. 2014, 27, 1687–1695. [Google Scholar] [CrossRef]
- Peter, J.W.; Erik, D.; Christer Von, B.; Snorri, S.T. Mechanism of N-hydroxyacetylarylamine mutagenicity in the Salmonella test system: Metabolic activation of N-hydroxyphenacetin by liver and kidney fractions from rat, mouse, hamster, and man. Mol. Pharmacol. 1980, 18, 117. [Google Scholar]
- Sparacino-Watkins, C.E.; Tejero, J.; Sun, B.; Gauthier, M.C.; Thomas, J.; Ragireddy, V.; Merchant, B.A.; Wang, J.; Azarov, I.; Basu, P.; et al. Nitrite reductase and nitric-oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J. Biol. Chem. 2014, 289, 10345–10358. [Google Scholar] [CrossRef]
- Cecco, E.; Gladwin, M.T.; Sparacino-Watkins, C. Oxygen inhibits nitrite reduction to nitric oxide by the molybdenum-fependent mARC-2 enzyme. Free. Radic. Biol. Med. 2017, 112, 34. [Google Scholar] [CrossRef]
- Wu, D.; Wang, Y.; Yang, G.; Zhang, S.; Liu, Y.; Zhou, S.; Guo, H.; Liang, S.; Cui, Y.; Zhang, B.; et al. A novel mitochondrial amidoxime reducing component 2 is a favorable indicator of cancer and suppresses the progression of hepatocellular carcinoma by regulating the expression of p27. Oncogene 2020, 39, 6099–6112. [Google Scholar] [CrossRef]
- Llamas, A.; Chamizo-Ampudia, A.; Tejada-Jimenez, M.; Galvan, A.; Fernandez, E. The molybdenum cofactor enzyme mARC: Moonlighting or promiscuous enzyme? Biofactors 2017, 43, 486–494. [Google Scholar] [CrossRef]
- Wu, D.; Liang, S.; Guo, H.; Zhang, S.; Yang, G.; Yuan, Y.; Liu, L. Downregulation of MARC2 promotes immune escape and is associated with immunosuppression of hepatocellular carcinoma. Front. Genet. 2021, 12, 790093. [Google Scholar] [CrossRef]
- Emdin, C.A.; Haas, M.E.; Khera, A.V.; Aragam, K.; Chaffin, M.; Klarin, D.; Hindy, G.; Jiang, L.; Wei, W.Q.; Feng, Q.; et al. A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 2020, 16, e1008629. [Google Scholar] [CrossRef]
- Neve, E.P.; Nordling, A.; Andersson, T.B.; Hellman, U.; Diczfalusy, U.; Johansson, I.; Ingelman-Sundberg, M. Amidoxime reductase system containing cytochrome b5 type B (CYB5B) and MOSC2 is of importance for lipid synthesis in adipocyte mitochondria. J. Biol. Chem. 2012, 287, 6307–6317. [Google Scholar] [CrossRef]
- Neve, E.P.; Kofeler, H.; Hendriks, D.F.; Nordling, A.; Gogvadze, V.; Mkrtchian, S.; Naslund, E.; Ingelman-Sundberg, M. Expression and function of mARC: Roles in lipogenesis and metabolic activation of ximelagatran. PLoS ONE 2015, 10, e0138487. [Google Scholar] [CrossRef]
- Rixen, S.; Havemeyer, A.; Tyl-Bielicka, A.; Pysniak, K.; Gajewska, M.; Kulecka, M.; Ostrowski, J.; Mikula, M.; Clement, B. Mitochondrial amidoxime-reducing component 2 (mARC2) has a significant role in N-reductive activity and energy metabolism. J. Biol. Chem. 2019, 294, 17593–17602. [Google Scholar] [CrossRef]
- Gladwin, M.; Sparacino-Watkins, C.E.; Jurczak, M. Method of Treating Insulin Resistance. U.S. Patent US20190160154, 30 May 2019. [Google Scholar]
- Hudert, C.A.; Adams, L.A.; Alisi, A.; Anstee, Q.M.; Crudele, A.; Draijer, L.G.; EU-PNAFLD Investigators; Furse, S.; Hengstler, J.G.; Jenkins, B.; et al. Variants in mitochondrial amidoxime reducing component 1 and hydroxysteroid 17-beta dehydrogenase 13 reduce severity of nonalcoholic fatty liver disease in children and suppress fibrotic pathways through distinct mechanisms. Hepatol. Commun. 2022, 8, 1934–1948. [Google Scholar] [CrossRef]
- Struwe, M.A.; Clement, B.; Scheidig, A. Letter to the editor: The clinically relevant MTARC1 p.Ala165Thr variant impacts neither the fold nor active site architecture of the human mARC1 protein. Hepatol. Commun. 2022, 6, 3277–3278. [Google Scholar] [CrossRef]
- Hudert, C.A.; Mann, J.P. Reply. Hepatol. Commun. 2022, 6, 3279. [Google Scholar] [CrossRef]
- Schneider, C.V.; Schneider, K.M.; Conlon, D.M.; Park, J.; Vujkovic, M.; Zandvakili, I.; Ko, Y.A.; Trautwein, C.; Center, R.; Carr, R.M.; et al. A genome-first approach to mortality and metabolic phenotypes in MTARC1 p.Ala165Thr (rs2642438) heterozygotes and homozygotes. Med 2021, 2, 851–863. [Google Scholar] [CrossRef]
- Sveinbjornsson, G.; Ulfarsson, M.O.; Thorolfsdottir, R.B.; Jonsson, B.A.; Einarsson, E.; Gunnlaugsson, G.; Rognvaldsson, S.; Arnar, D.O.; Baldvinsson, M.; Bjarnason, R.G.; et al. Multiomics study of nonalcoholic fatty liver disease. Nat. Genet. 2022, 54, 1652–1663. [Google Scholar] [CrossRef]
- Gao, C.; Marcketta, A.; Backman, J.D.; O’Dushlaine, C.; Staples, J.; Ferreira, M.A.R.; Lotta, L.A.; Overton, J.D.; Reid, J.G.; Mirshahi, T.; et al. Genome-wide association analysis of serum alanine and aspartate aminotransferase, and the modifying effects of BMI in 388k European individuals. Genet. Epidemiol. 2021, 54, 664–681. [Google Scholar] [CrossRef] [PubMed]
- Innes, H.; Buch, S.; Hutchinson, S.; Guha, I.N.; Morling, J.R.; Barnes, E.; Irving, W.; Forrest, E.; Pedergnan, V.; Goldberg, D.; et al. Genome-wide association study for alcohol-related cirrhosis identifies risk loci in MARC1 and HNRNPUL1. Gastroenterology 2020, 159, 1276–1289.e1277. [Google Scholar] [CrossRef] [PubMed]
- Fairfield, C.J.; Drake, T.M.; Pius, R.; Bretherick, A.D.; Campbell, A.; Clark, D.W.; Fallowfield, J.A.; Hayward, C.; Henderson, N.C.; Joshi, P.K.; et al. Genome-wide association study of NAFLD using electronic health records. Hepatol. Commun. 2022, 6, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.C.; Chen, L.; Hameed, L.S.; Kitchen, R.R.; Maroteau, C.; Nagarajan, S.R.; Norlin, J.; Daly, C.E.; Szczerbinska, I.; Hjuler, S.T.; et al. Hepatocyte mARC1 promotes fatty liver disease. JHEP Rep. 2023, 5, 100693. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Vaughan, C. Ximelagatran (Exanta): Alternative to warfarin? Bayl. Univ. Med Cent. Proc. 2005, 18, 76–80. [Google Scholar] [CrossRef][Green Version]
- Clement, B.; Mau, S.; Deters, S.; Havemeyer, A. Hepatic, extrahepatic, microsomal, and mitochondrial activation of the N-hydroxylated prodrugs benzamidoxime, guanoxabenz, and Ro 48-3656 ([[1-[(2S)-2-[[4-[(hydroxyamino)iminomethyl]benzoyl]amino]-1-oxopropyl]-4-piperid inyl]oxy]-acetic acid). Drug Metab. Dispos. 2005, 33, 1740–1747. [Google Scholar] [CrossRef]
- Tejada-Jimenez, M.; Chamizo-Ampudia, A.; Calatrava, V.; Galvan, A.; Fernandez, E.; Llamas, A. From the eukaryotic molybdenum cofactor biosynthesis to the moonlighting enzyme mARC. Molecules 2018, 23, 3287. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patent Number | Company | Title |
|---|---|---|
| WO2023282704 | OliX Pharmaceuticals | Asymmetric siRNA targeting MARC1 gene, and use thereof |
| WO2022248665 | Novo Nordisk | Compositions and methods for inhibiting mitochondria amidoxime-reducing component 1 (MARC1) expression |
| WO2022183065 | Ionis Pharmaceuticals | Modulation of MARC1 expression using antisense oligonucleotides or other inhibitors to treat liver disease |
| WO2022159158 | Alnylam Pharmaceuticals | Modified double-stranded oligonucleotides |
| WO2022036126 | Amgen | RNAi constructs and methods for inhibiting mARC1 expression |
| WO2021237097 | Alnylam Pharmaceuticals | Compositions and methods for inhibiting MARC1 gene expression in human using double-stranded RNA |
| US20210262022 | The General Hospital Corporation | Mitochondrial amidoxime-reducing component (MARC) gene variants associated with liver diseases and methods of protection against liver diseases or symptoms thereof |
| WO2020154567 | Viscient Biosciences | Compositions and methods for the diagnosis and treatment of diseases of the liver |
| Year | Discovery | Citation |
|---|---|---|
| 1983 | BA is oxidized to BAO in vitro | [2] |
| 1988 | N-reduction of BAO to BA is demonstrated in vitro | [21] |
| 1993 | In vivo studies show that N-reduction of BAO dominates physiologically | [23] |
| 2003 | The mARC-activated prodrug Ximelagatran is submitted to the FDA for approval | [110] |
| 2005 | N-reducing activity is highest in mitochondria | [111] |
| 2006 | mARC proteins were first isolated from porcine liver | [62] |
| 2010 | Detailed description of recombinant human mARC proteins | [67] |
| 2017 | Initial development of an electrochemical assay for mARC activity | [69] |
| 2018 | Crystal structure of human mARC1 | [74,75] |
| 2019 | In vivo murine MTARC2 knockout model underscores mARC’s role in lipid metabolism | [98] |
| 2019 | Development of a fast spectrophotometric mARC activity assay | [68] |
| 2020 | The mARC1 p.A165T variant is first shown to protect against liver disease | [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clement, B.; Struwe, M.A. The History of mARC. Molecules 2023, 28, 4713. https://doi.org/10.3390/molecules28124713
Clement B, Struwe MA. The History of mARC. Molecules. 2023; 28(12):4713. https://doi.org/10.3390/molecules28124713
Chicago/Turabian StyleClement, Bernd, and Michel A. Struwe. 2023. "The History of mARC" Molecules 28, no. 12: 4713. https://doi.org/10.3390/molecules28124713
APA StyleClement, B., & Struwe, M. A. (2023). The History of mARC. Molecules, 28(12), 4713. https://doi.org/10.3390/molecules28124713