
Is It Still Relevant to Discover New ACE Inhibitors from Natural Products? YES, but Only with Comprehensive Approaches to Address the Patients’ Real Problems: Chronic Dry Cough and Angioedema
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
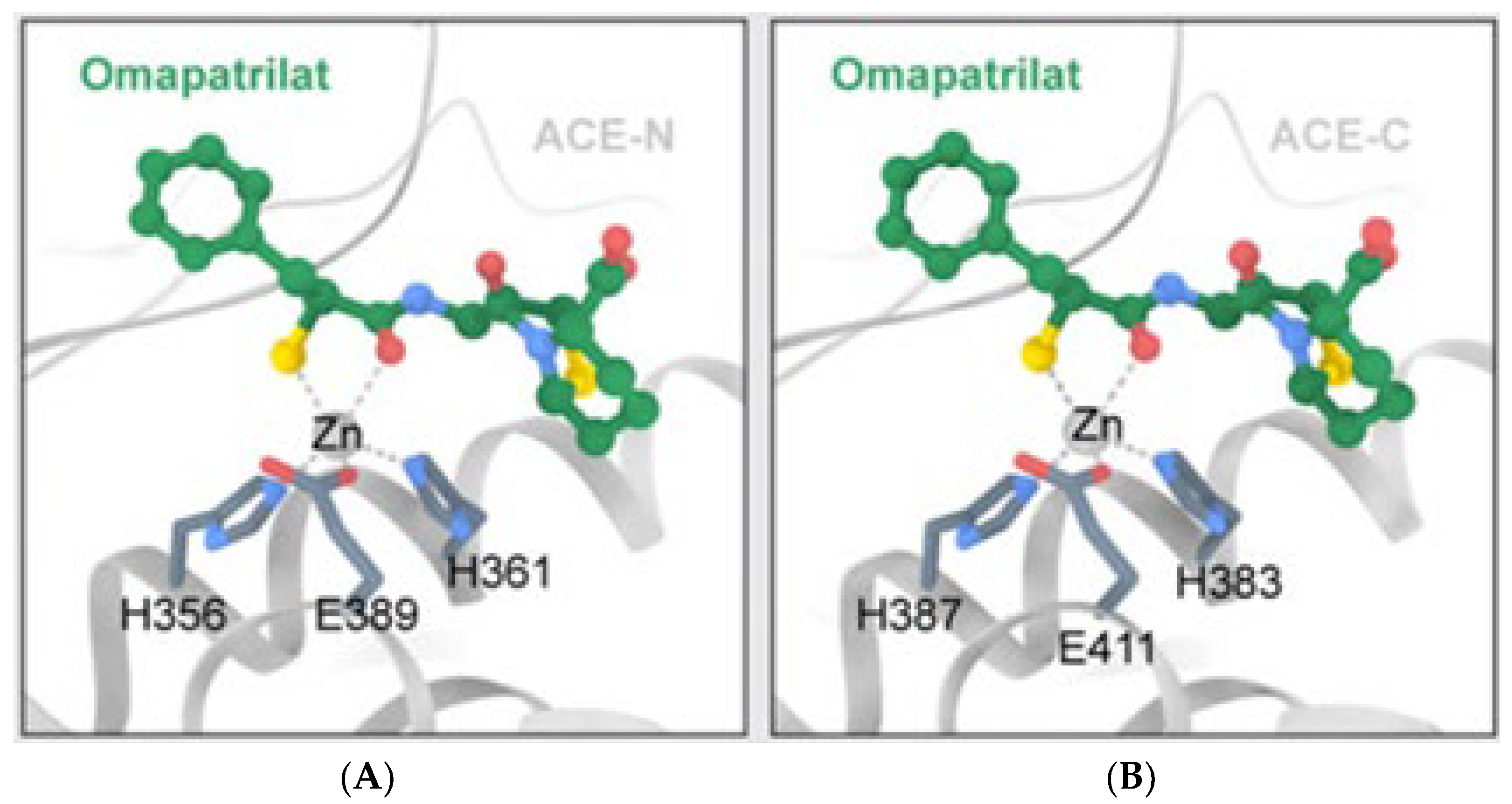
2. The ACE, Type of ACE, and Its Characteristics
3. ACE Inhibitors, Development of Dry Cough and Angioedema, and Possible Alternative Treatments to Address ACE Inhibitor-Induced Cough
4. Peptides’ Stability against Gastrointestinal Enzymes and Its ACE Domain-Specific Inhibition
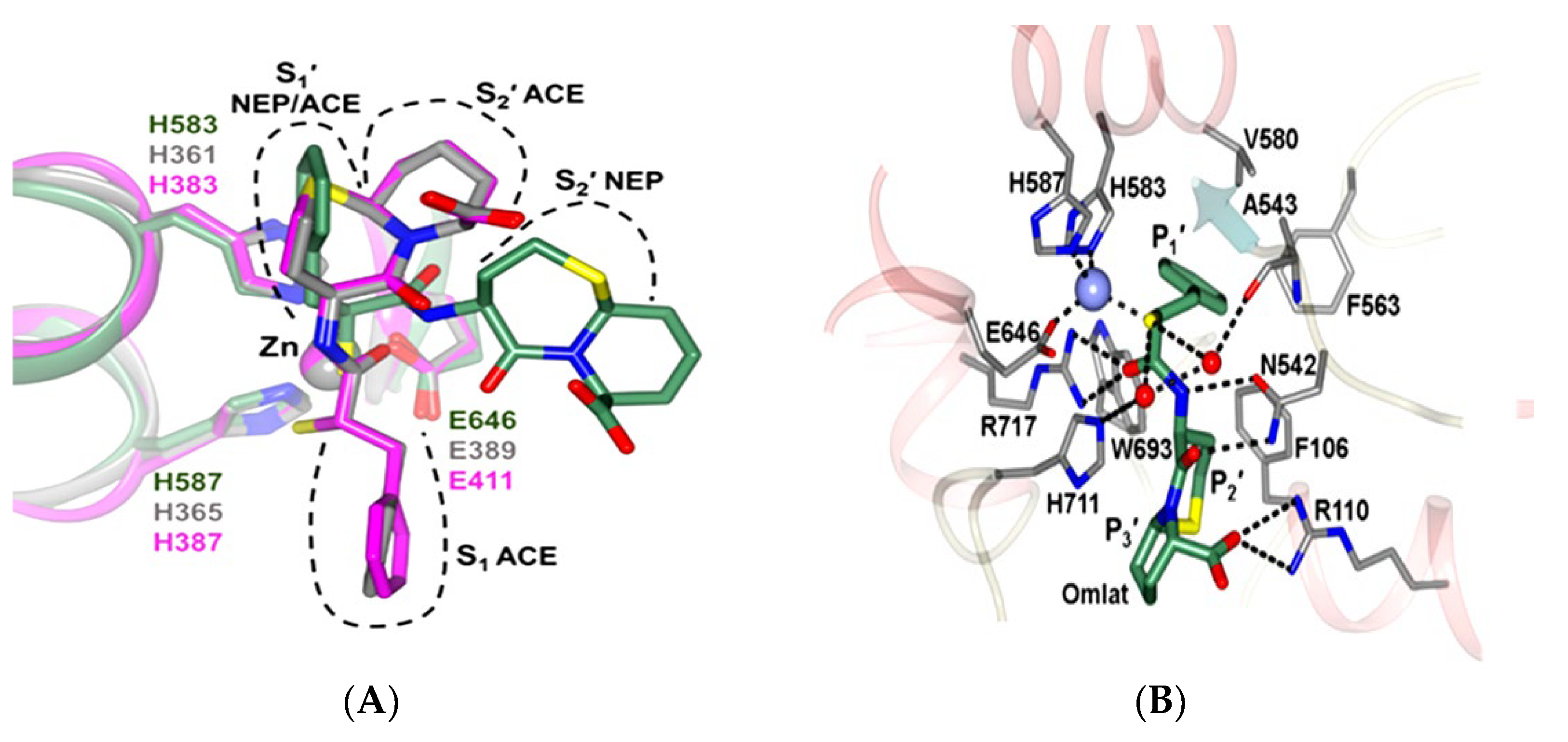
5. Function of Neprilysin and Chymase
6. Challenges Working with Naturally Derived Peptide-Based ACE Inhibitors
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anthony, C.S.; Masuyer, G.; Sturrock, E.D.; Acharya, K.R. Structure based drug design of angiotensin-I converting enzyme inhibitors. Curr. Med. Chem. 2012, 19, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.P.; Goh, P.S. Incidence of discontinuation of angiotensin-converting enzyme inhibitors due to cough, in a primary healthcare centre in Singapore. Singap. Med. J. 2014, 55, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.S.; Kwong, J.; Rezvani, F.; Coates, A.O. Angiotensin-converting enzyme related cough among Chinese-Americans. Am. J. Med. 2010, 123, 183.e11–183.e15. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Donald, S.; Howlett, J. Angiotensin-Converting Enzyme Inhibitors. Cardiology Advisor, 20 January 2019. Available online: https://www.thecardiologyadvisor.com/home/decision-support-in-medicine/cardiology/angiotensin-converting-enzyme-inhibitors/ (accessed on 26 February 2023).
- Gallo, G.; Volpe, M.; Rubattu, S. Angiotensin Receptor Blockers in the Management of Hypertension: A Real-World Perspective and Current Recommendations. Vasc. Health Risk Manag. 2022, 18, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Green, L.A. Hypertension: Which First-Line Medication Is Best? Medical News Today. 27 July 2021. Available online: https://www.medicalnewstoday.com/articles/hypertension-which-first-line-medication-is-best (accessed on 21 February 2023).
- Li, E.C.; Heran, B.S.; Wright, J.M. Angiotensin converting enzyme (ACE) inhibitors versus angiotensin receptor blockers for primary hypertension. Cochrane Database Syst. Rev. 2014, 2014, CD009096. [Google Scholar] [CrossRef]
- Naha, S.; Gardner, M.J.; Khangura, D.; Khangura, D.; Kurukulasuriya, R.L.; Sowers, J.R. Hypertension in Diabetes. In Endotext; Updated 2021; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279027 (accessed on 17 November 2022).
- Cheng, J.; Zhang, W.; Zhang, X.; Han, F.; Li, X.; He, X.; Li, Q.; Chen, J. Effect of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers on all-cause mortality, cardiovascular deaths, and cardiovascular events in patients with diabetes mellitus: A meta-analysis. JAMA Intern. Med. 2014, 174, 773–785. [Google Scholar] [CrossRef]
- Lv, X.; Zhang, Y.; Niu, Y.; Song, Q.; Zhao, Q. Comparison of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers on cardiovascular outcomes in hypertensive patients with type 2 diabetes mellitus: A PRISMA-compliant systematic review and meta-analysis. Medicine 2018, 97, e0256. [Google Scholar] [CrossRef]
- Yao, J.; Fan, S.; Shi, X.; Gong, X.; Zhao, J.; Fan, G. Angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers on insulin sensitivity in hypertensive patients: A meta-analysis of randomized controlled trials. PLoS ONE 2021, 16, e0253492. [Google Scholar] [CrossRef]
- Wang, R.; Yun, J.; Wu, S.; Bi, Y.; Zhao, F. Optimisation and Characterisation of Novel Angiotensin-Converting Enzyme Inhibitory Peptides Prepared by Double Enzymatic Hydrolysis from Agaricus bisporus Scraps. Foods 2022, 11, 394. [Google Scholar] [CrossRef]
- Zheng, W.; Tian, E.; Liu, Z.; Zhou, C.; Yang, P.; Tian, K.; Liao, W.; Li, J.; Ren, C. Small molecule angiotensin converting enzyme inhibitors: A medicinal chemistry perspective. Front. Pharmacol. 2022, 13, 968104. [Google Scholar] [CrossRef]
- Manoharan, S.; Shuib, A.S.; Abdullah, N. Structural characteristics and antihypertensive effects of angiotensin-I-converting enzyme inhibitory peptides in the renin-angiotensin and kallikrein kinin systems. Afr. J. Tradit. Complement. Altern. Med. 2017, 14, 383–406. [Google Scholar] [CrossRef]
- Wong, M.K.S. Angiotensin Converting Enzymes. In Handbook of Hormones; Academic Press: Cambridge, MA, USA, 2016; pp. 505–518. [Google Scholar]
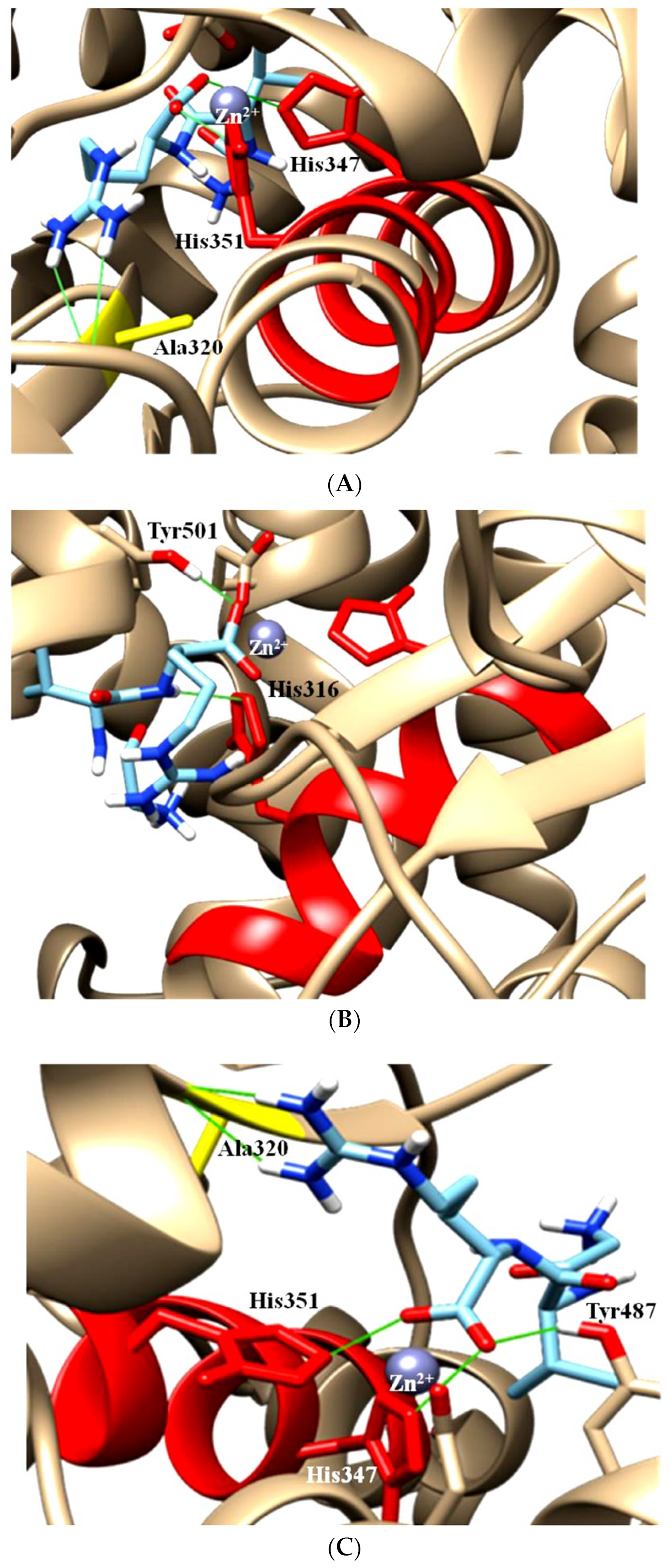
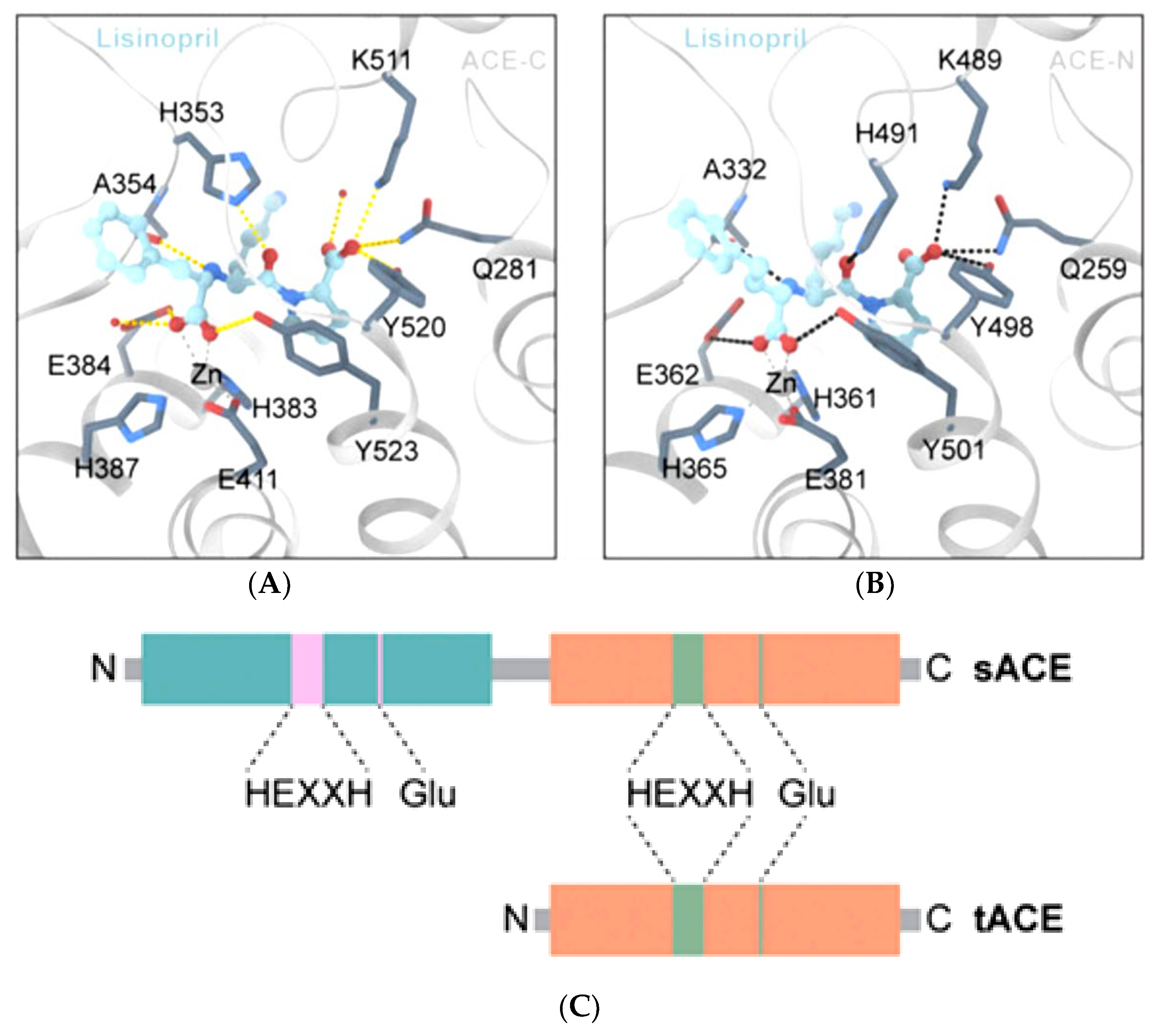
- Anthony, C.S.; Corradi, H.R.; Schwager, S.L.; Redelinghuys, P.; Georgiadis, D.; Dive, V.; Acharya, K.R.; Sturrock, E.D. The N domain of human angiotensin-I-converting enzyme: The role of N-glycosylation and the crystal structure in complex with an N domain-specific phosphinic inhibitor, RXP407. J. Biol. Chem. 2010, 285, 35685–35693. [Google Scholar] [CrossRef]
- Maluf-Meiken, L.C.; Fernandes, F.B.; Aragão, D.S.; Ronchi, F.A.; Andrade, M.C.; Franco, M.C.; Febba, A.C.; Plavnik, F.L.; Krieger, J.E.; Mill, J.G.; et al. N-domain isoform of Angiotensin I converting enzyme as a marker of hypertension: Populational study. Int. J. Hypertens. 2012, 2012, 581780. [Google Scholar] [CrossRef]
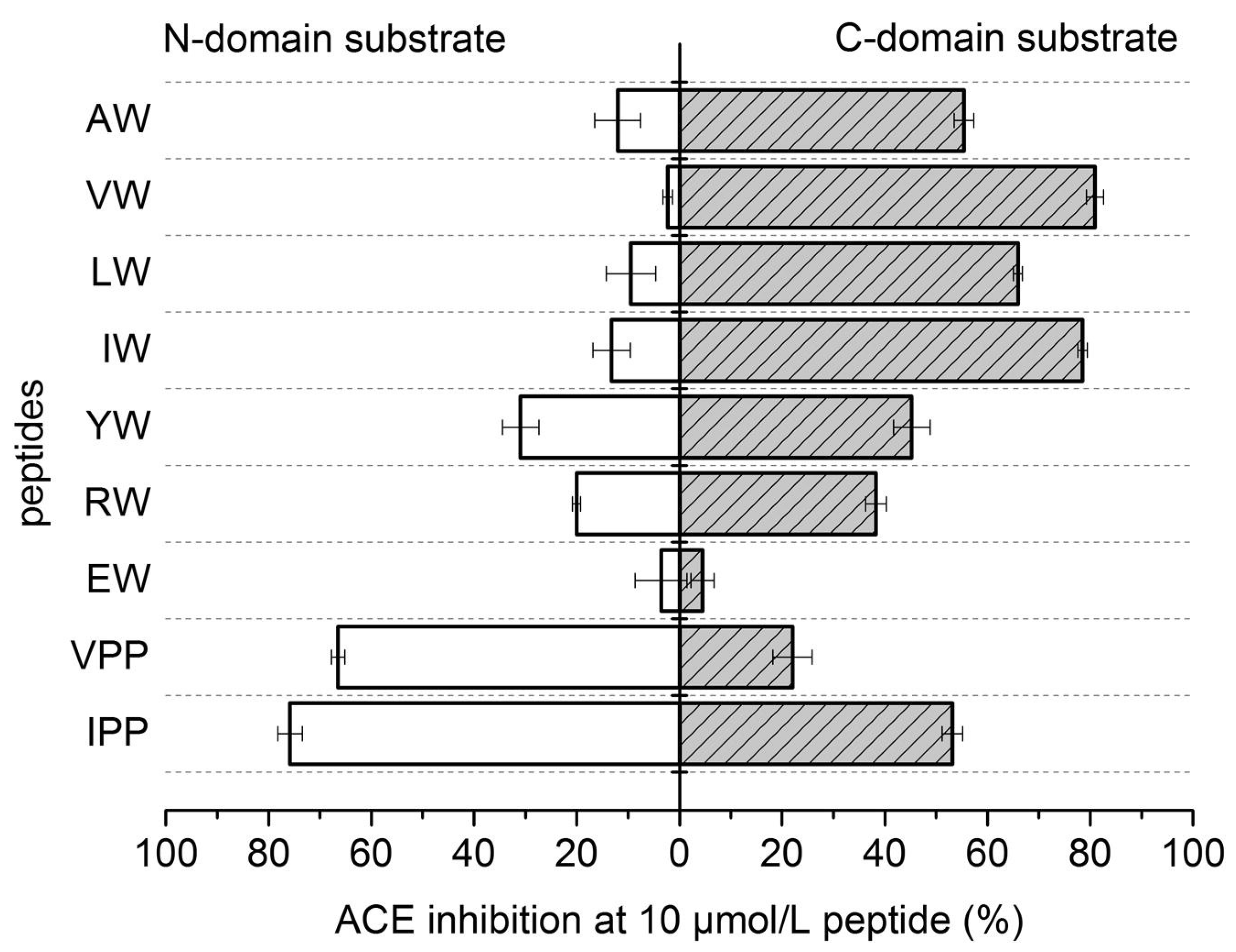
- Lunow, D.; Kaiser, S.; Rückriemen, J.; Pohl, C.; Henle, T. Tryptophan-containing dipeptides are C-domain selective inhibitors of angiotensin converting enzyme. Food Chem. 2015, 166, 596–602. [Google Scholar] [CrossRef]
- Manoharan, S.; Shuib, A.S.; Abdullah, N.; Ridzwan, N.F.W.; Mohamad, S.B. Molecular Docking Analysis of Human Somatic and Testicular Angiotensin Converting Enzyme Complexed with a Novel Compound Gly-Val-Arg. Adv. Pharmacol. Pharm. 2022, 10, 95–103. [Google Scholar] [CrossRef]
- Van Esch, J.H.; Tom, B.; Dive, V.; Batenburg, W.W.; Georgiadis, D.; Yiotakis, A.; van Gool, J.M.; de Bruijn, R.J.; de Vries, R.; Danser, A.H. Selective angiotensin-converting enzyme C-domain inhibition is sufficient to prevent angiotensin I-induced vasoconstriction. Hypertension 2005, 45, 120–125. [Google Scholar] [CrossRef]
- Georgiadis, D.; Beau, F.; Czarny, B.; Cotton, J.; Yiotakis, A.; Dive, V. Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin: Insights from selective inhibitors. Circ. Res. 2003, 93, 148–154. [Google Scholar] [CrossRef]
- Bersanetti, P.A.; Sabatini, R.A.; Matos, B.S.; Douglas, R.G.; Nchindam, A.; Juliano, M.A.; Pesquero, J.B.; Sturrock, E.D.; Carmona, A.K. Characterization of angiotensin I-converting enzyme N-domain selectivity using positional-scanning combinatorial libraries of fluorescence resonance energy transfer peptides. Biol. Chem. 2012, 393, 1547–1554. [Google Scholar] [CrossRef]
- ACE Inhibitors. Cleveland Clinic. 2021. Available online: https://my.clevelandclinic.org/health/treatments/21934-ace-inhibitors (accessed on 26 February 2023).
- Yılmaz, İ. Angiotensin-Converting Enzyme Inhibitors Induce Cough. Turk. Thorac. J. 2019, 20, 36–42. [Google Scholar] [CrossRef]
- Pinto, B.; Jadhav, U.; Singhai, P.; Sadhanandham, S.; Shah, N. ACEI-induced cough: A review of current evidence and its practical implications for optimal CV risk reduction. Indian Heart J. 2020, 72, 345–350. [Google Scholar] [CrossRef]
- Brown, T.; Gonzalez, J.; Monteleone, C. Angiotensin-converting enzyme inhibitor-induced angioedema: A review of the literature. J. Clin. Hypertens. 2017, 19, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Park, S.W.; Kim, D.K.; Lee, S.H.; Hong, K.P. Iron supplementation inhibits cough associated with ACE inhibitors. Hypertension 2001, 38, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Malini, P.L.; Strocchi, E.; Zanardi, M.; Milani, M.; Ambrosioni, E. Thromboxane antagonism and cough induced by angiotensin-converting-enzyme inhibitor. Lancet 1997, 350, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, E.; Reem, A. Fosinopril for Potential Resolution of Cough Associated with an Angiotensin-Converting Enzyme Inhibitor. Endocr. Pract. 2005, 11, 70. [Google Scholar]
- Sharif, M.N.; Evans, B.L.; Pylypchuk, G.B. Cough induced by quinapril with resolution after changing to fosinopril. Ann. Pharmacother. 1994, 28, 720–722. [Google Scholar] [CrossRef]
- David, D.; Jallad, N.; Germino, F.W.; Willett, M.S.; de Silva, J.; Weidner, S.M.; Mills, D.J. A Comparison of the Cough Profile of Fosinopril and Enalapril in Hypertensive Patients with a History of ACE Inhibitor-Associated Cough. Am. J. Ther. 1995, 2, 806–813. [Google Scholar] [CrossRef]
- Germino, F.W.; Lastra, J.; Pool, P.; Punzi, H.; Spinowitz, B.; Smith, W.; Wallace, S.; Willett, M.; Mills, D.; De Silva, J.; et al. Evaluation of the cough profile of fosinopril in hypertensive patients with ACE inhibitor associated cough-A pilot study. Curr. Ther. Res. 1993, 54, 469–475. [Google Scholar] [CrossRef]
- Manoharan, S.; Shuib, A.S.; Abdullah, N.; Mohamad, S.B.; Aminudin, N. Characterisation of novel angiotensin-I-converting enzyme inhibitory tripeptide, Gly-Val-Arg derived from mycelium of Pleurotus pulmonarius. Process Biochem. 2017, 62, 215. [Google Scholar] [CrossRef]
- Manoharan, S.; Shuib, A.S.; Abdullah, N.; Ashrafzadeh, A.; Kabir, N. Gly-Val-Arg, an angiotensin-I-converting enzyme inhibitory tripeptide ameliorates hypertension on spontaneously hypertensive rats. Process Biochem. 2018, 69, 224. [Google Scholar] [CrossRef]
- Abdou, M.M.; Dong, D.; O’Neill, P.M.; Amigues, E.; Matziari, M. Design, Synthesis, and Study of a Novel RXPA380-Proline Hybrid (RXPA380-P) as an Antihypertensive Agent. ACS Omega 2022, 7, 35035–35043. [Google Scholar] [CrossRef]
- Ningrum, S.; Sutrisno, A.; Hsu, J.L. An exploration of angiotensin-converting enzyme (ACE) inhibitory peptides derived from gastrointestinal protease hydrolysate of milk using a modified bioassay-guided fractionation approach coupled with in silico analysis. J. Dairy Sci. 2022, 105, 1913–1928. [Google Scholar] [CrossRef]
- Abdelhedi, O.; Nasri, R.; Mora, L.; Jridi, M.; Toldrá, F.; Nasri, M. In silico analysis and molecular docking study of angiotensin I-converting enzyme inhibitory peptides from smooth-hound viscera protein hydrolysates fractionated by ultrafiltration. Food Chem. 2018, 239, 453–463. [Google Scholar] [CrossRef]
- Iwaniak, A.; Minkiewicz, P.; Darewicz, M. Food-Originating ACE Inhibitors, Including Antihypertensive Peptides, as Preventive Food Components in Blood Pressure Reduction. Compr. Rev. Food Sci. Food Saf. 2014, 13, 114–134. [Google Scholar] [CrossRef]
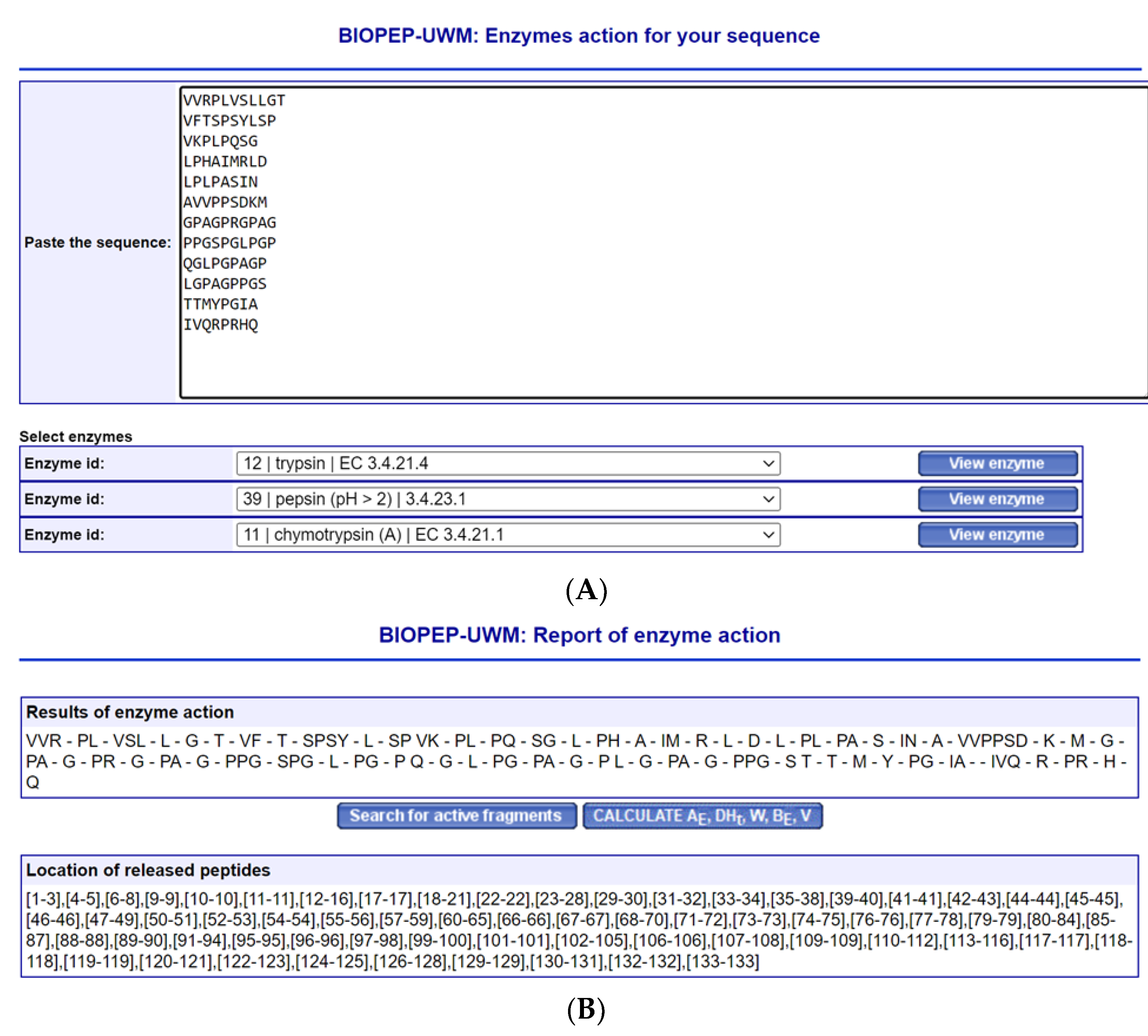
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM Virtual-A Novel Database of Food-Derived Peptides with In Silico-Predicted Biological Activity. Appl. Sci. 2022, 12, 7204. [Google Scholar] [CrossRef]
- Alves-Lopes, R.; Montezano, A.C.; Neves, K.B.; Harvey, A.; Rios, F.J.; Skiba, D.S.; Arendse, L.B.; Guzik, T.J.; Graham, D.; Poglitsch, M.; et al. Selective Inhibition of the C-Domain of ACE (Angiotensin-Converting Enzyme) Combined with Inhibition of NEP (Neprilysin): A Potential New Therapy for Hypertension. Hypertension 2021, 78, 604–616. [Google Scholar] [CrossRef]
- Sharma, U.; Cozier, G.E.; Sturrock, E.D.; Acharya, K.R. Molecular Basis for Omapatrilat and Sampatrilat Binding to Neprilysin-Implications for Dual Inhibitor Design with Angiotensin-Converting Enzyme. J. Med. Chem. 2020, 63, 5488–5500. [Google Scholar] [CrossRef]
- Lau, C.C.; Abdullah, N.; Shuib, A.S. Novel angiotensin I-converting enzyme inhibitory peptides derived from an edible mushroom, Pleurotus cystidiosus O.K. Miller identified by LC-MS/MS. BMC Complement. Altern. Med. 2013, 13, 313. [Google Scholar] [CrossRef]
- Ahmad, S.; Varagic, J.; VonCannon, J.L.; Groban, L.; Collawn, J.F.; Dell’Italia, L.J.; Ferrario, C.M. Primacy of cardiac chymase over angiotensin converting enzyme as an angiotensin-(1-12) metabolizing enzyme. Biochem. Biophys. Res. Commun. 2016, 478, 559–564. [Google Scholar] [CrossRef]
- Pitt, B. Clinical trials of angiotensin receptor blockers in heart failure: What do we know and what will we learn? Am. J. Hypertens. 2002, 15, 22S–27S. [Google Scholar] [CrossRef]
- Yao, S.; Agyei, D.; Udenigwe, C.C. Structural Basis of Bioactivity of Food Peptides in Promoting Metabolic Health. Adv. Food Nutr. Res. 2018, 84, 145–181. [Google Scholar]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal. Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manoharan, S. Is It Still Relevant to Discover New ACE Inhibitors from Natural Products? YES, but Only with Comprehensive Approaches to Address the Patients’ Real Problems: Chronic Dry Cough and Angioedema. Molecules 2023, 28, 4532. https://doi.org/10.3390/molecules28114532
Manoharan S. Is It Still Relevant to Discover New ACE Inhibitors from Natural Products? YES, but Only with Comprehensive Approaches to Address the Patients’ Real Problems: Chronic Dry Cough and Angioedema. Molecules. 2023; 28(11):4532. https://doi.org/10.3390/molecules28114532
Chicago/Turabian StyleManoharan, Sivananthan. 2023. "Is It Still Relevant to Discover New ACE Inhibitors from Natural Products? YES, but Only with Comprehensive Approaches to Address the Patients’ Real Problems: Chronic Dry Cough and Angioedema" Molecules 28, no. 11: 4532. https://doi.org/10.3390/molecules28114532
APA StyleManoharan, S. (2023). Is It Still Relevant to Discover New ACE Inhibitors from Natural Products? YES, but Only with Comprehensive Approaches to Address the Patients’ Real Problems: Chronic Dry Cough and Angioedema. Molecules, 28(11), 4532. https://doi.org/10.3390/molecules28114532