
Recent Progress in the Integration of CO2 Capture and Utilization

Abstract
1. Introduction
2. Integration of CO2 Capture and Utilization
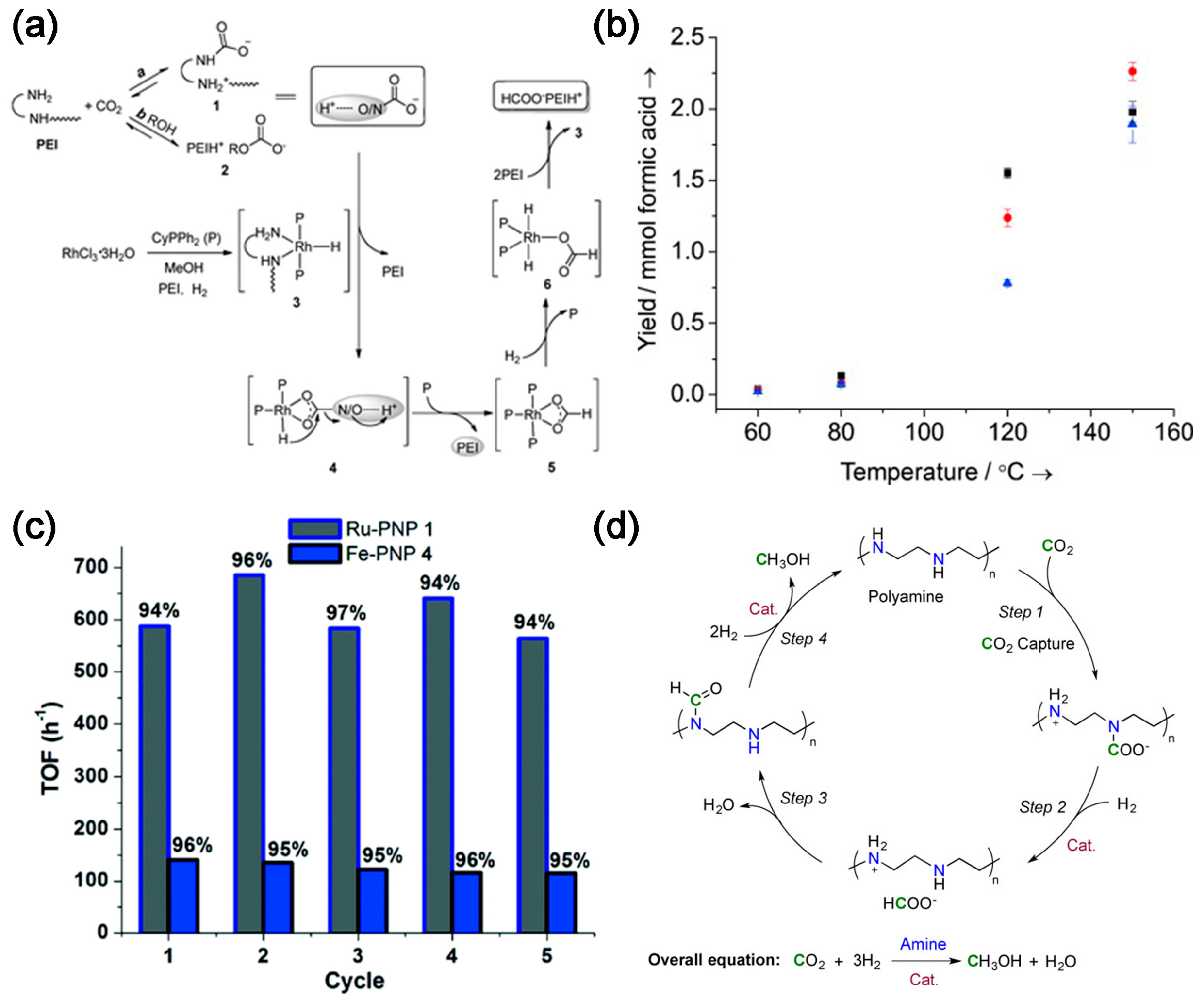
2.1. Integration of CO2 Absorption and Conversion
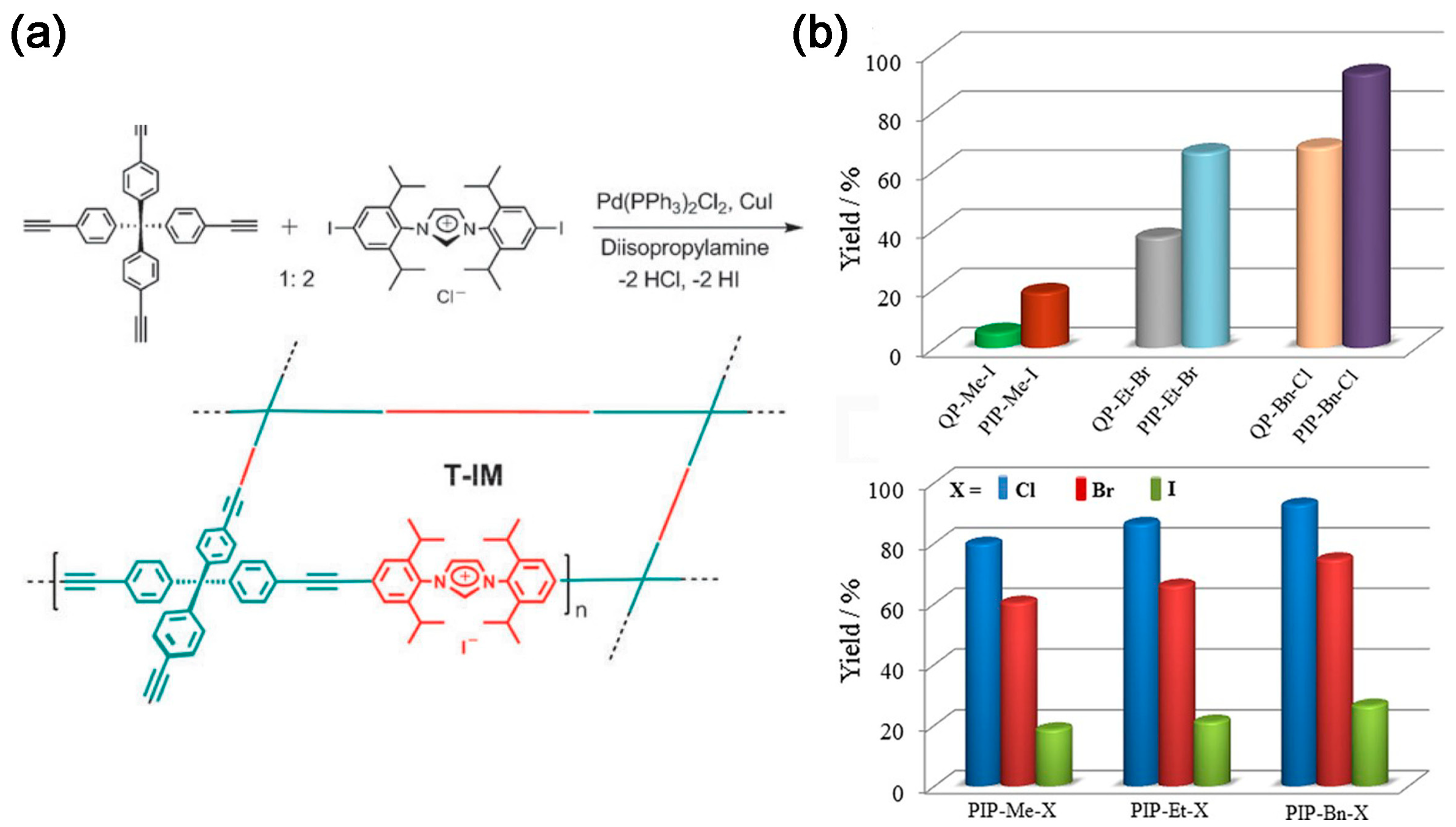
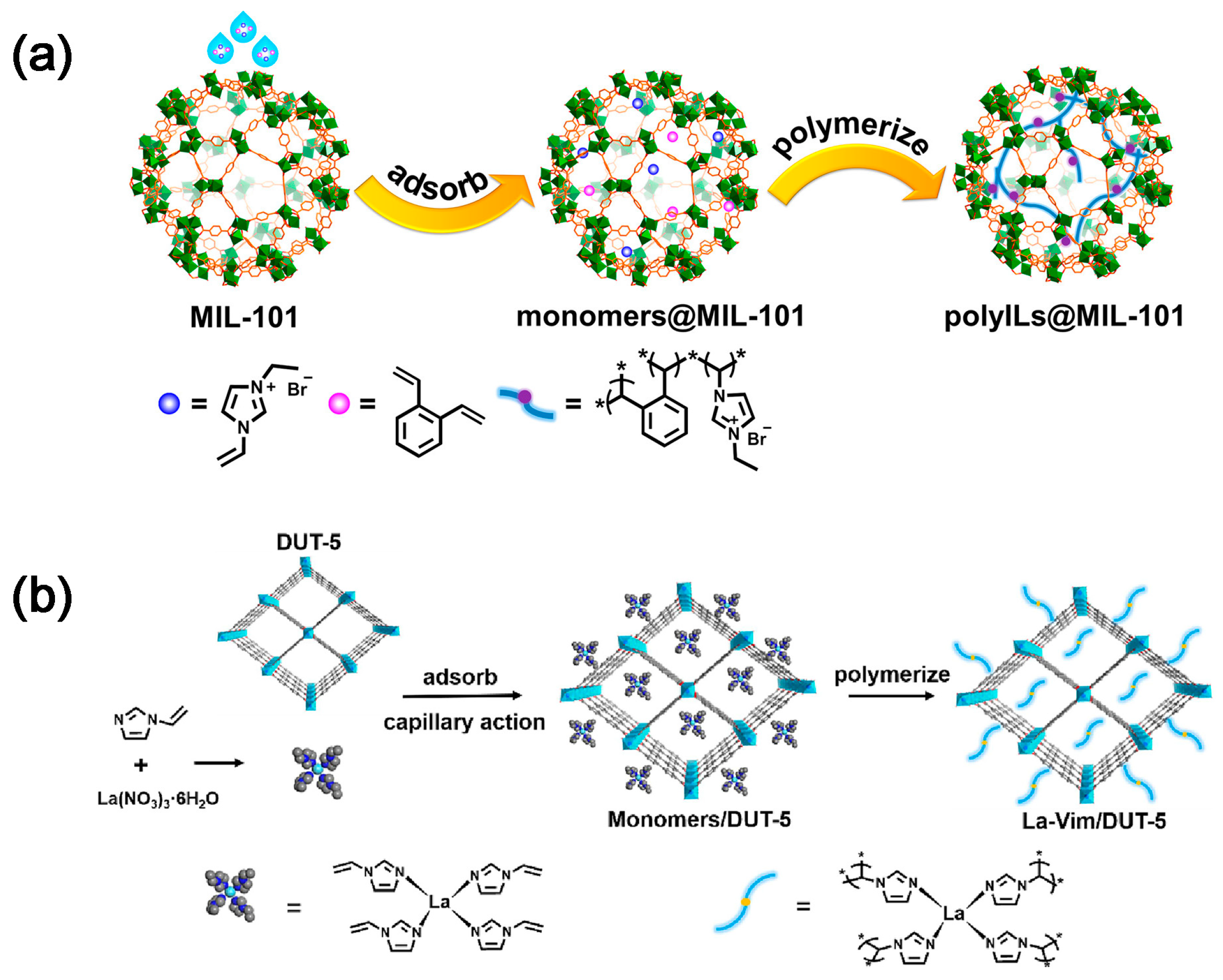
2.2. CO2 Adsorption and Conversion Integration
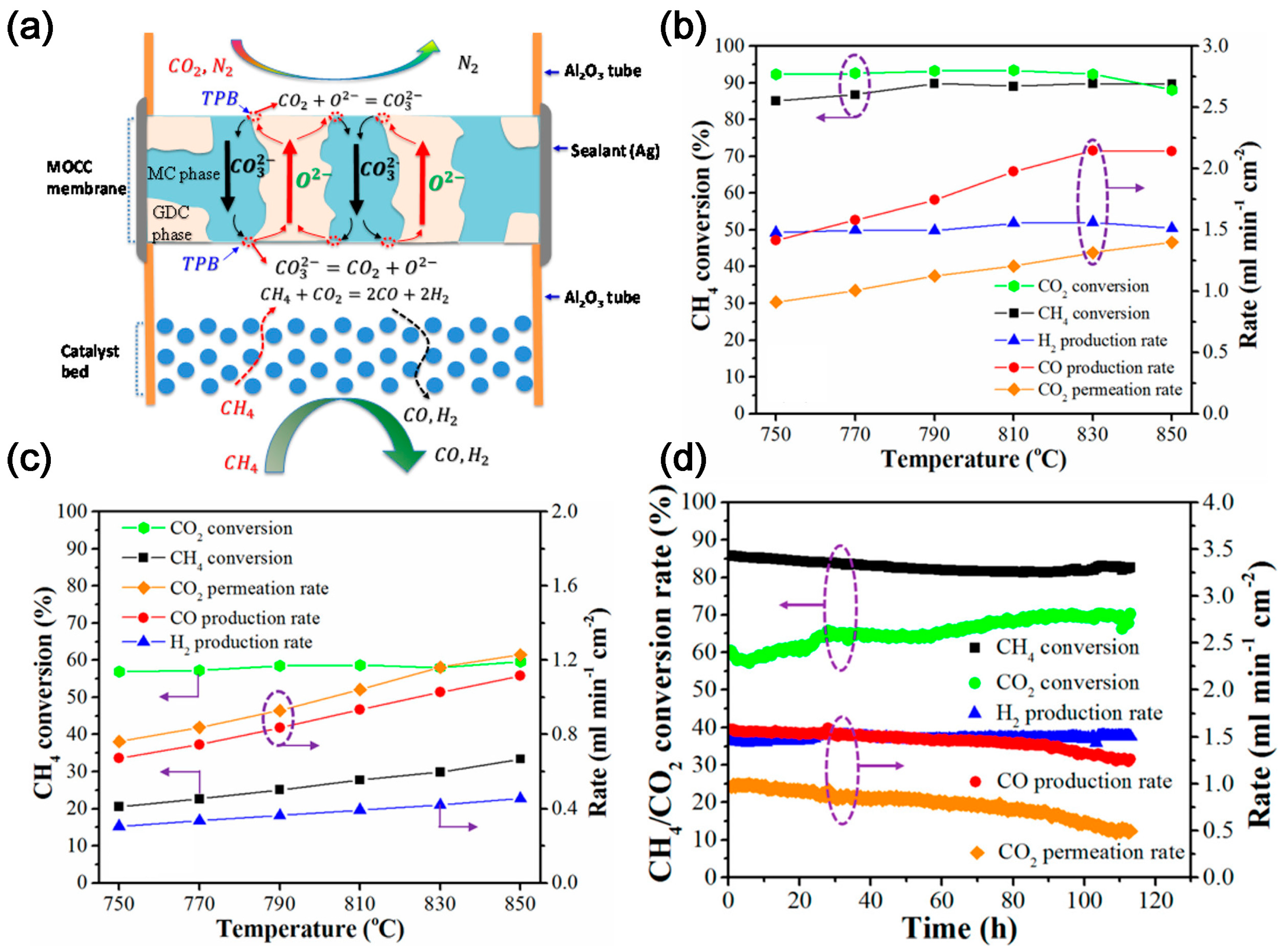
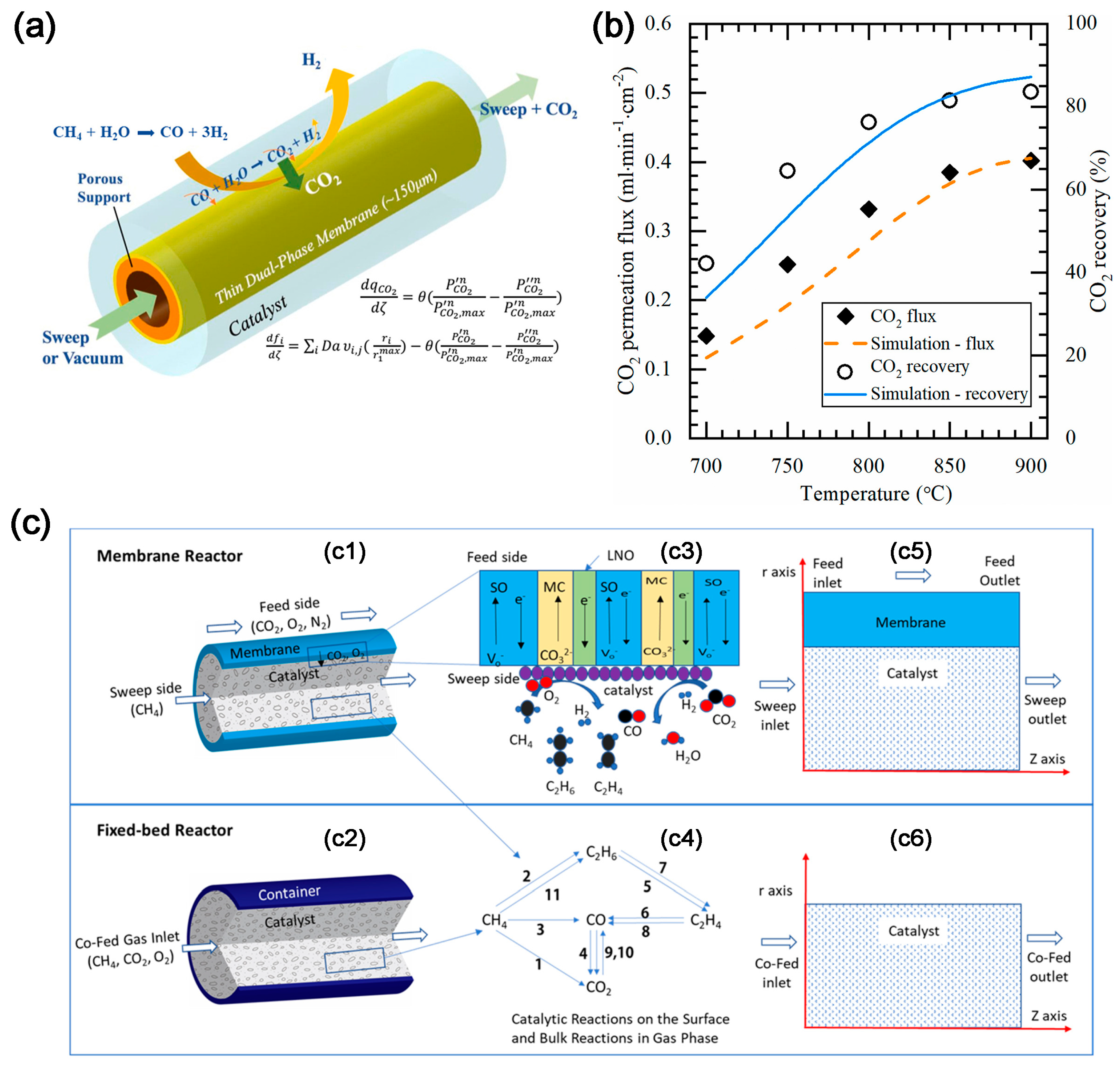
2.3. CO2 Electrochemical Membrane Separation and Conversion Integration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane | Catalyst | Reaction | Ref. |
|---|---|---|---|
| La0.6Sr0.4Co0.8Fe0.2O3-δ Li-Na-K | 10 wt%Ni-/γ-Al2O3 | DMR | [29] |
| Ce0.8Gd0.2O1.9 Li-Na | Ni-MgO-1 wt% Pt LaNi0.6Fe0.4O3-δ | DMR | [31] |
| Ag Li-Na | Ni-MgO-1 wt% Pt | DOMR | [32] |
| NiO-SDC Li-Na | Ni-MgO-1 wt% Pt | DOMR | [40] |
| Ce0.8Gd0.2O1.9 Li-Na | 5 wt% Cr2O3- ZSM-5 | Ethane-to-Ethylene | [33] |
| Ce0.9Pr0.1O2-δ-Pr0.6Sr0.4Fe0.5Co0.5O3-δ Li-Na-K | 10 wt%Ni-/γ-Al2O3 | DOMR | [41] |
| LNO/SDC Li-Na | LNO/LCNO | RWGS | [34] |
| BYS-SDC Li-Na-K | Ni-based catalyst (HiFUEL R110) | SMR | [36] |
| γ-LiAlO2-Ag Li-Na-K | γ-LiAlO2-Ag | Syngas production | [42] |
| NiO-SDC Li-Na | 2%Mn-5%Na2WO4/SiO2 | OCM | [39] |
2.4. CO2 Capture and Conversion over Dual-Function Materials (DFMs)
3. Conclusions, Challenges and Opportunities
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, K.S.; Fritz, P.W.; Coskun, A. Porous organic polymers for CO2 capture, separation and conversion. Chem. Soc. Rev. 2022, 51, 9831–9852. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, J.; Huang, K.; Zhu, X.; Yang, W. The current status of high temperature electrochemistry-based CO2 transport membranes and reactors for direct CO2 capture and conversion. Prog. Energy Combust. Sci. 2021, 82, 100888. [Google Scholar] [CrossRef]
- Yamada, H. Amine-based capture of CO2 for utilization and storage. Polym. J. 2021, 53, 93–102. [Google Scholar] [CrossRef]
- Maina, J.W.; Pringle, J.M.; Razal, J.M.; Nunes, S.; Vega, L.; Gallucci, F.; Dumée, L.F. Strategies for integrated capture and conversion of CO2 from dilute flue gases and the atmosphere. ChemSusChem 2021, 14, 1805–1820. [Google Scholar] [CrossRef] [PubMed]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.S. Conversion of CO2 from air into methanol using a polyamine and a homogeneous ruthenium catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Goeppert, A.; Prakash, G.S. Combined CO2 capture and hydrogenation to methanol: Amine immobilization enables easy recycling of active elements. ChemSusChem 2019, 12, 3172–3177. [Google Scholar] [CrossRef]
- Hanusch, J.M.; Kerschgens, I.P.; Huber, F.; Neuburger, M.; Gademann, K. Pyrrolizidines for direct air capture and CO2 conversion. Chem. Commun. 2019, 55, 949–952. [Google Scholar] [CrossRef]
- Guan, C.; Pan, Y.; Ang, E.P.L.; Hu, J.; Yao, C.; Huang, M.-H.; Li, H.; Lai, Z.; Huang, K.-W. Conversion of CO2 from air into formate using amines and phosphorus-nitrogen PN 3P-Ru (ii) pincer complexes. Green Chem. 2018, 20, 4201–4205. [Google Scholar] [CrossRef]
- Tailor, R.; Abboud, M.; Sayari, A. Supported polytertiary amines: Highly efficient and selective SO2 adsorbents. Environ. Sci. Technol. 2014, 48, 2025–2034. [Google Scholar] [CrossRef]
- Yang, S.; Zhan, L.; Xu, X.; Wang, Y.; Ling, L.; Feng, X. Graphene-based porous silica sheets impregnated with polyethyleneimine for superior CO2 capture. Adv. Mater. 2013, 25, 2130–2134. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; May, R.B.; Prakash, G.S.; Olah, G.A.; Narayanan, S. Carbon dioxide capture from the air using a polyamine based regenerable solid adsorbent. J. Am. Chem. Soc. 2011, 133, 20164–20167. [Google Scholar] [CrossRef]
- Li, Y.-N.; He, L.-N.; Liu, A.-H.; Lang, X.-D.; Yang, Z.-Z.; Yu, B.; Luan, C.-R. In situ hydrogenation of captured CO2 to formate with polyethyleneimine and Rh/monophosphine system. Green Chem. 2013, 15, 2825–2829. [Google Scholar] [CrossRef]
- McNamara, N.D.; Hicks, J.C. CO2 capture and conversion with a multifunctional polyethyleneimine-tethered iminophosphine iridium catalyst/adsorbent. ChemSusChem 2014, 7, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.S. CO2 capture by amines in aqueous media and its subsequent conversion to formate with reusable ruthenium and iron catalysts. Green Chem. 2016, 18, 5831–5838. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Huff, C.A.; Sanford, M.S. Tandem amine and ruthenium-catalyzed hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xing, G.; Chen, W.; Chen, L. Porous organic polymers: A promising platform for efficient photocatalysis. Mater. Chem. Front. 2020, 4, 332–353. [Google Scholar] [CrossRef]
- Cho, H.C.; Lee, H.S.; Chun, J.; Lee, S.M.; Kim, H.J.; Son, S.U. Tubular microporous organic networks bearing imidazolium salts and their catalytic CO2 conversion to cyclic carbonates. Chem. Commun. 2011, 47, 917–919. [Google Scholar] [CrossRef]
- Wang, J.; Sng, W.; Yi, G.; Zhang, Y. Imidazolium salt-modified porous hypercrosslinked polymers for synergistic CO2 capture and conversion. Chem. Commun. 2015, 51, 12076–12079. [Google Scholar] [CrossRef]
- Sun, Q.; Jin, Y.; Aguila, B.; Meng, X.; Ma, S.; Xiao, F.S. Porous ionic polymers as a robust and efficient platform for capture and chemical fixation of atmospheric CO2. ChemSusChem 2017, 10, 1160–1165. [Google Scholar]
- Smith, J.G. Organic Chemistry, 3rd ed.; McGraw-Hill: Singapore, 2011. [Google Scholar]
- Dai, Z.; Sun, Q.; Liu, X.; Bian, C.; Wu, Q.; Pan, S.; Wang, L.; Meng, X.; Deng, F.; Xiao, F.-S. Metalated porous porphyrin polymers as efficient heterogeneous catalysts for cycloaddition of epoxides with CO2 under ambient conditions. J. Catal. 2016, 338, 202–209. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Xu, Q.; Zhang, W.; Zhou, X.; Ji, H. State-of-the-art aluminum porphyrin-based heterogeneous catalysts for the chemical fixation of CO2 into cyclic carbonates at ambient conditions. ChemCatChem 2017, 9, 767–773. [Google Scholar] [CrossRef]
- Zhai, G.; Liu, Y.; Lei, L.; Wang, J.; Wang, Z.; Zheng, Z.; Wang, P.; Cheng, H.; Dai, Y.; Huang, B. Light-promoted CO2 conversion from epoxides to cyclic carbonates at ambient conditions over a Bi-based metal–organic framework. ACS Catal. 2021, 11, 1988–1994. [Google Scholar] [CrossRef]
- Ding, M.; Jiang, H.-L. Incorporation of imidazolium-based poly (ionic liquid)s into a metal–organic framework for CO2 capture and conversion. ACS Catal. 2018, 8, 3194–3201. [Google Scholar] [CrossRef]
- Dai, W.; Li, Q.; Long, J.; Mao, P.; Xu, Y.; Yang, L.; Zou, J.; Luo, X. Hierarchically mesoporous imidazole-functionalized covalent triazine framework: An efficient metal-and halogen-free heterogeneous catalyst towards the cycloaddition of CO2 with epoxides. J. CO2 Util. 2022, 62, 102101. [Google Scholar] [CrossRef]
- Liu, F.; Duan, X.; Dai, X.; Du, S.; Ma, J.; Liu, F.; Liu, M. Metal-decorated porous organic frameworks with cross-linked pyridyl and triazinyl as efficient platforms for CO2 activation and conversion under mild conditions. Chem. Eng. J. 2022, 445, 136687. [Google Scholar] [CrossRef]
- Ma, P.; Ding, M.; Zhang, Y.; Rong, W.; Yao, J. Integration of lanthanide-imidazole containing polymer with metal-organic frameworks for efficient cycloaddition of CO2 with epoxides. Sep. Purif. Technol. 2023, 313, 123498. [Google Scholar] [CrossRef]
- Rui, Z.; Ji, H.; Lin, Y.S. Modeling and analysis of ceramic-carbonate dual-phase membrane reactor for carbon dioxide reforming with methane. Int. J. Hydrogen Energy 2011, 36, 8292–8300. [Google Scholar] [CrossRef]
- Anderson, M.; Lin, Y.S. Carbon dioxide separation and dry reforming of methane for synthesis of syngas by a dual-phase membrane reactor. AlChE J. 2013, 59, 2207–2218. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, J.; Huang, K. Combining electrochemical CO2 capture with catalytic dry methane reforming in a single reactor for low-cost syngas production. ACS Sustain. Chem. Eng. 2016, 4, 7056–7065. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, J.; Huang, K. Dry-oxy methane reforming with mixed e(-)/CO32- conducting membranes. ACS Sustain. Chem. Eng. 2017, 5, 5432–5439. [Google Scholar]
- Zhang, P.; Tong, J.; Huang, K. Role of CO2 in catalytic ethane-to-ethylene conversion using a high-temperature CO2 transport membrane reactor. ACS Sustain. Chem. Eng. 2019, 7, 6889–6897. [Google Scholar] [CrossRef]
- Chen, T.J.; Wang, Z.G.; Liu, L.N.; Pati, S.; Wai, M.H.; Kawi, S. Coupling CO2 separation with catalytic reverse water-gas shift reaction via ceramic-carbonate dual-phase membrane reactor. Chem. Eng. J. 2020, 379, 122182. [Google Scholar] [CrossRef]
- Xing, W.; Peters, T.; Fontaine, M.-L.; Evans, A.; Henriksen, P.P.; Norby, T.; Bredesen, R. Steam-promoted CO2 flux in dual-phase CO2 separation membranes. J. Membr. Sci. 2015, 482, 115–119. [Google Scholar] [CrossRef]
- Wu, H.C.; Rui, Z.B.; Lin, J.Y.S. Hydrogen production with carbon dioxide capture by dual-phase ceramic-carbonate membrane reactor via steam reforming of methane. J. Membr. Sci. 2020, 598, 117780. [Google Scholar] [CrossRef]
- Ovalle-Encinia, O.; Wu, H.C.; Chen, T.J.; Lin, J.Y.S. CO2-permselective membrane reactor for steam reforming of methane. J. Membr. Sci. 2022, 641, 119914. [Google Scholar] [CrossRef]
- Li, X.; Huang, K.; Van Dam, N.; Jin, X. Performance projection of a high-temperature CO2 transport membrane reactor for combined CO2 capture and methane-to-ethylene conversion. J. Electrochem. Soc. 2022, 169, 053501. [Google Scholar]
- Zhang, K.; Sun, S.; Huang, K. Oxidative coupling of methane (OCM) conversion into C2 products through a CO2/O2 co-transport membrane reactor. J. Membr. Sci. 2022, 661, 120915. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, J.; Huang, K. Self-formed, mixed-conducting, triple-phase membrane for efficient CO2/O2 capture from flue gas and in situ dry-oxy methane reforming. ACS Sustain. Chem. Eng. 2018, 6, 14162–14169. [Google Scholar] [CrossRef]
- Fabian-Anguiano, J.A.; Mendoza-Serrato, C.G.; Gomez-Yanez, C.; Zeifert, B.; Ma, X.; Ortiz-Landeros, J. Simultaneous CO2 and O2 separation coupled to oxy-dry reforming of CH4 by means of a ceramic-carbonate membrane reactor for in situ syngas production. Chem. Eng. Sci. 2019, 210, 115250. [Google Scholar]
- Fabian-Anguiano, J.A.; Ramirez-Moreno, M.J.; Balmori-Ramirez, H.; Romero-Serrano, J.A.; Romero-Ibarra, I.C.; Ma, X.; Ortiz-Landeros, J. Syngas production with CO2 utilization through the oxidative reforming of methane in a new cermet-carbonate packed-bed membrane reactor. J. Membr. Sci. 2021, 637, 119607. [Google Scholar] [CrossRef]
- Shao, B.; Zhang, Y.; Sun, Z.; Li, J.; Gao, Z.; Xie, Z.; Hu, J.; Liu, H. CO2 capture and in-situ conversion: Recent progresses and perspectives. Green Chem. Eng. 2022, 3, 189–198. [Google Scholar] [CrossRef]
- Sun, S.; Sun, H.; Williams, P.T.; Wu, C. Recent advances in integrated CO2 capture and utilization: A review. Sustain. Energy Fuels 2021, 5, 4546–4559. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, D.; Pérez-Ramírez, J.; Chen, Z. Recent progress in materials exploration for thermocatalytic, photocatalytic, and integrated photothermocatalytic CO2-to-fuel conversion. Adv. Energy Sustain. Res. 2022, 3, 2100169. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Broda, M.; Hosseini, D.; Copéret, C.; Müller, C. Integrated CO2 capture and conversion as an efficient process for fuels from greenhouse gases. ACS Catal. 2018, 8, 2815–2823. [Google Scholar] [CrossRef]
- Tian, S.; Yan, F.; Zhang, Z.; Jiang, J. Calcium-looping reforming of methane realizes in situ CO2 utilization with improved energy efficiency. Sci. Adv. 2019, 5, eaav5077. [Google Scholar] [CrossRef]
- Hu, J.; Hongmanorom, P.; Chirawatkul, P.; Kawi, S. Efficient integration of CO2 capture and conversion over a Ni supported CeO2-modified CaO microsphere at moderate temperature. Chem. Eng. J. 2021, 426, 130864. [Google Scholar] [CrossRef]
- Wang, G.; Guo, Y.; Yu, J.; Liu, F.; Sun, J.; Wang, X.; Wang, T.; Zhao, C. Ni-CaO dual function materials prepared by different synthetic modes for integrated CO2 capture and conversion. Chem. Eng. J. 2022, 428, 132110. [Google Scholar] [CrossRef]
- Sun, H.; Wang, J.; Zhao, J.; Shen, B.; Shi, J.; Huang, J.; Wu, C. Dual functional catalytic materials of Ni over Ce-modified CaO sorbents for integrated CO2 capture and conversion. Appl. Catal. B Environ. 2019, 244, 63–75. [Google Scholar] [CrossRef]
- Duyar, M.S.; Trevino, M.A.A.; Farrauto, R.J. Dual function materials for CO2 capture and conversion using renewable H2. Appl. Catal. B Environ. 2015, 168, 370–376. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; He, Z.; Libby, M.C.; Farrauto, R.J. Catalysts and adsorbents for CO2 capture and conversion with dual function materials: Limitations of Ni-containing DFMs for flue gas applications. J. CO2 Util. 2019, 31, 143–151. [Google Scholar] [CrossRef]
- Porta, A.; Visconti, C.G.; Castoldi, L.; Matarrese, R.; Jeong-Potter, C.; Farrauto, R.; Lietti, L. Ru-Ba synergistic effect in dual functioning materials for cyclic CO2 capture and methanation. Appl. Catal. B Environ. 2021, 283, 119654. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.; González-Velasco, J. Mechanism of the CO2 storage and in situ hydrogenation to CH4. Temperature and adsorbent loading effects over Ru-CaO/Al2O3 and Ru-Na2CO3/Al2O3 catalysts. Appl. Catal. B Environ. 2019, 256, 117845. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.; González-Velasco, J. Ni loading effects on dual function materials for capture and in-situ conversion of CO2 to CH4 using CaO or Na2CO3. J. CO2 Util. 2019, 34, 576–587. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Rownaghi, A.A.; Rezaei, F. Combined capture and utilization of CO2 for syngas production over dual-function materials. ACS Sustain. Chem. Eng. 2018, 6, 13551–13561. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Trevino, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrogen Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Wang, S.; Schrunk, E.T.; Mahajan, H.; Farrauto, R.J. The role of ruthenium in CO2 capture and catalytic conversion to fuel by dual function materials (DFM). Catalysts 2017, 7, 88. [Google Scholar] [CrossRef]
- Wang, S.; Farrauto, R.J.; Karp, S.; Jeon, J.H.; Schrunk, E.T. Parametric, cyclic aging and characterization studies for CO2 capture from flue gas and catalytic conversion to synthetic natural gas using a dual functional material (DFM). J. CO2 Util. 2018, 27, 390–397. [Google Scholar] [CrossRef]






| Entry | Catalysts | Additives | Time (h) | Conv. (%) a | Select. (%) a |
|---|---|---|---|---|---|
| 1 b | POP-TPP | n-Bu4NBr | 24 | 52.1 | >99.0 |
| 2 c | Co/POP-TPP | n-Bu4NBr | 24 | 95.6 | 99 |
| 3 c | Co/TPP | n-Bu4NBr | 24 | 97.5 | 99 |
| 4 | Co/POP-TPP | None | 24 | 9.7 | 99 |
| 5 | Co/TPP | None | 24 | 18.5 | 99 |
| 6 | None | n-Bu4NBr | 24 | 34 | 99 |
| 7 d | Co/POP-TPP | n-Bu4NBr | 96 | 96.1 | 99 |
| 8 e | Co/POP-TPP | n-Bu4NBr | 24 | 88.9 | 99 |
| 9 f | Zn/POP-TPP | n-Bu4NBr | 24 | 93.2 | >99.0 |
| 10 f | Zn/TPP | n-Bu4NBr | 24 | 93.5 | >99.0 |
| 11 g | Mg/POP-TPP | n-Bu4NBr | 24 | 80.5 | >99.0 |
| 12 g | Mg/TPP | n-Bu4NBr | 24 | 99.3 | >99.0 |
| 13 h | Co/POP-TPP | n-Bu4NBr | 24 | 93.6 | 99 |
| DFM | Condition (°C) | Ref. | |
|---|---|---|---|
| Absorption | Reaction | ||
| Ni-CaO/Al2O3 | 280–520 10% CO2/Ar | 280–520 10% H2/Ar | [55] |
| Ni-Na2CO3/Al2O3 | 280–520 10% CO2/Ar | 280–520 10% H2/Ar | |
| Ni-Na2O/Al2O3 | 320 7.5% CO2/N2 and 7.5% CO2, 4.5% O2, 15% H2O/N2 | 320 15% H2/N2 | [52] |
| Ru-Na2O/γ-Al2O3 | 320 15% CO2/N2 | 320 20% H2/N2 | [57] |
| Rh-CaO/γ-Al2O3 | 320 10% CO2/N2 | 320 2% H2/N2 | |
| Ru-CaO/γ-Al2O3 | 280–400 1.4% CO2/Ar and 11% CO2/Ar | 280–400 10% H2/Ar | [54] |
| Ru-Na2CO3/γ-Al2O3 | 280–400 1.4% CO2/Ar and 11% CO2/A | 280–400 10% H2/Ar | |
| Ru-CaO/γ-Al2O3 | 320 10% CO2/air and 8% CO2/21% H2O/air | 320 5% H2/N2 | [51] |
| Ru-CaO/γ-Al2O3 | 320 10% CO2/N2 | 320 4% H2/N2 | [57] |
| Ru-CaO/γ-Al2O3 | 320 7.5% CO2, 4.5% O2, 15% H2O/N2 | 320 5% H2/N2 | [58] |
| Ru-Na2CO3/γ-Al2O3 | 320 7.5% CO2/N2 and 7.5% CO2, 4.5% O2, 15% H2O/N2 | 320 5% H2/N2 | [59] |
| Ru-Na2O/γ-Al2O3 | 250–350 7.5% CO2, 4.5% O2, 15% H2O/N2 | 250–350 15% H2/N2 | [60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, H.; Li, Y.; Zhang, C. Recent Progress in the Integration of CO2 Capture and Utilization. Molecules 2023, 28, 4500. https://doi.org/10.3390/molecules28114500
Ning H, Li Y, Zhang C. Recent Progress in the Integration of CO2 Capture and Utilization. Molecules. 2023; 28(11):4500. https://doi.org/10.3390/molecules28114500
Chicago/Turabian StyleNing, Huanghao, Yongdan Li, and Cuijuan Zhang. 2023. "Recent Progress in the Integration of CO2 Capture and Utilization" Molecules 28, no. 11: 4500. https://doi.org/10.3390/molecules28114500
APA StyleNing, H., Li, Y., & Zhang, C. (2023). Recent Progress in the Integration of CO2 Capture and Utilization. Molecules, 28(11), 4500. https://doi.org/10.3390/molecules28114500