

Ligand-Based Virtual Screening, Molecular Docking, and Molecular Dynamic Simulations of New β-Estrogen Receptor Activators with Potential for Pharmacological Obesity Treatment
 and
and 
Abstract
1. Introduction
2. Results
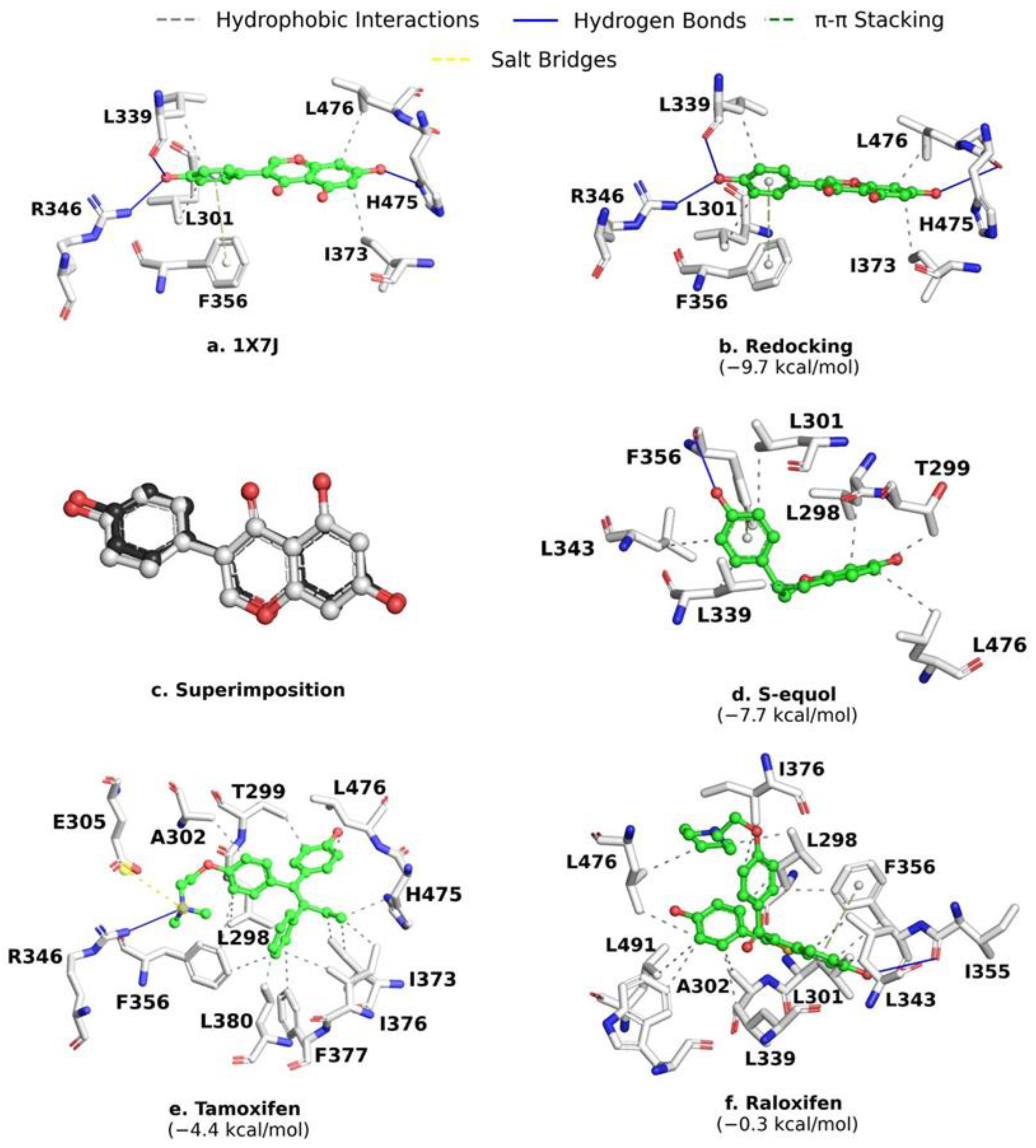
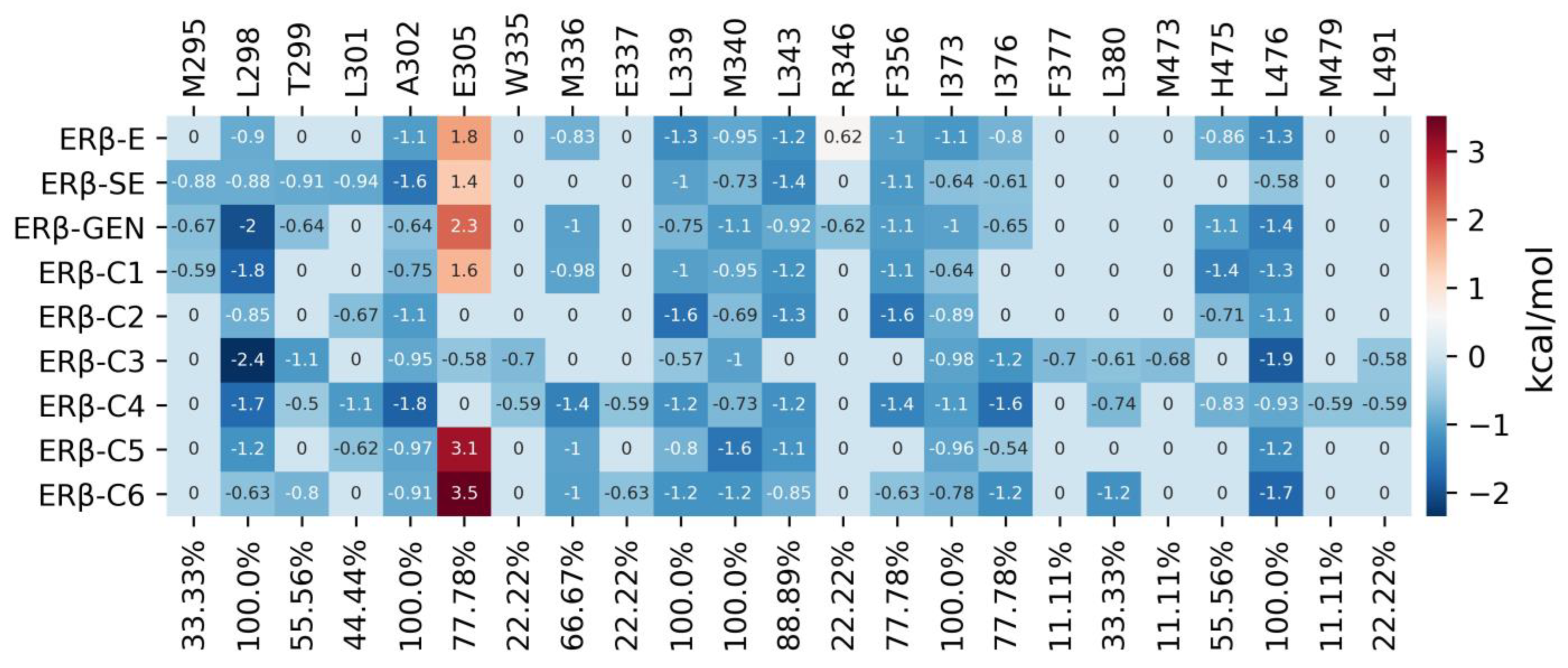
2.1. Analysis of ERβ-Ligand Complexes
2.2. MD Validation and Control Compounds Analysis
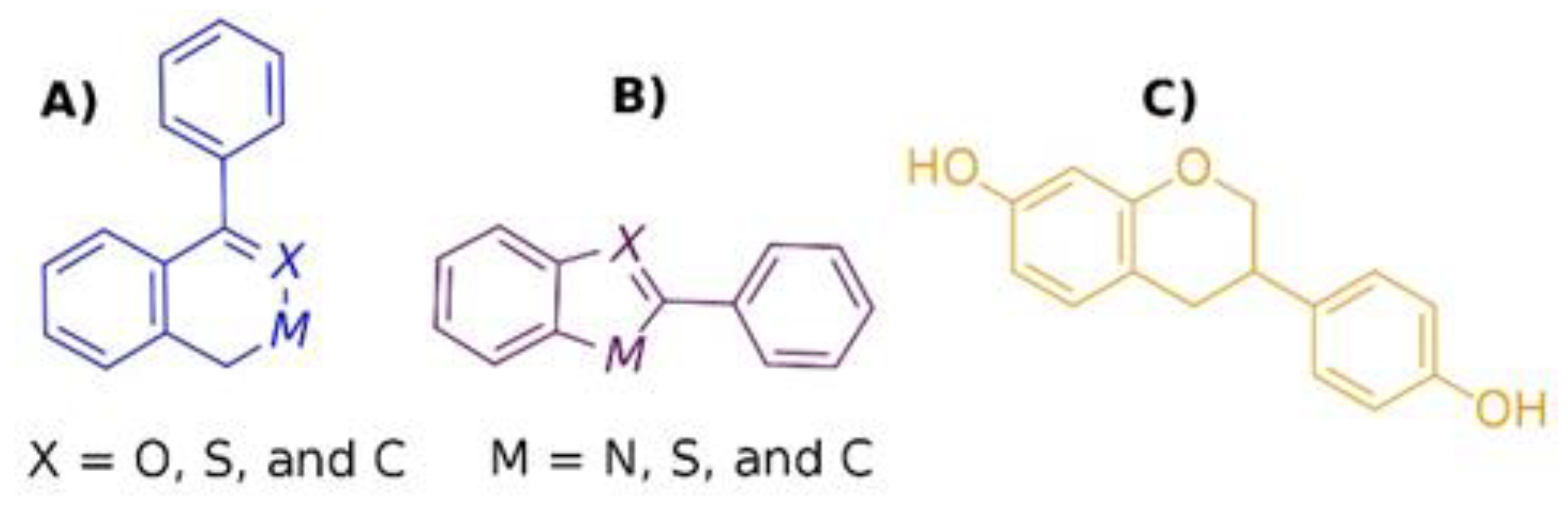
2.3. LBVS by Structure Similarity
2.4. LBVS by Substructure Similarity
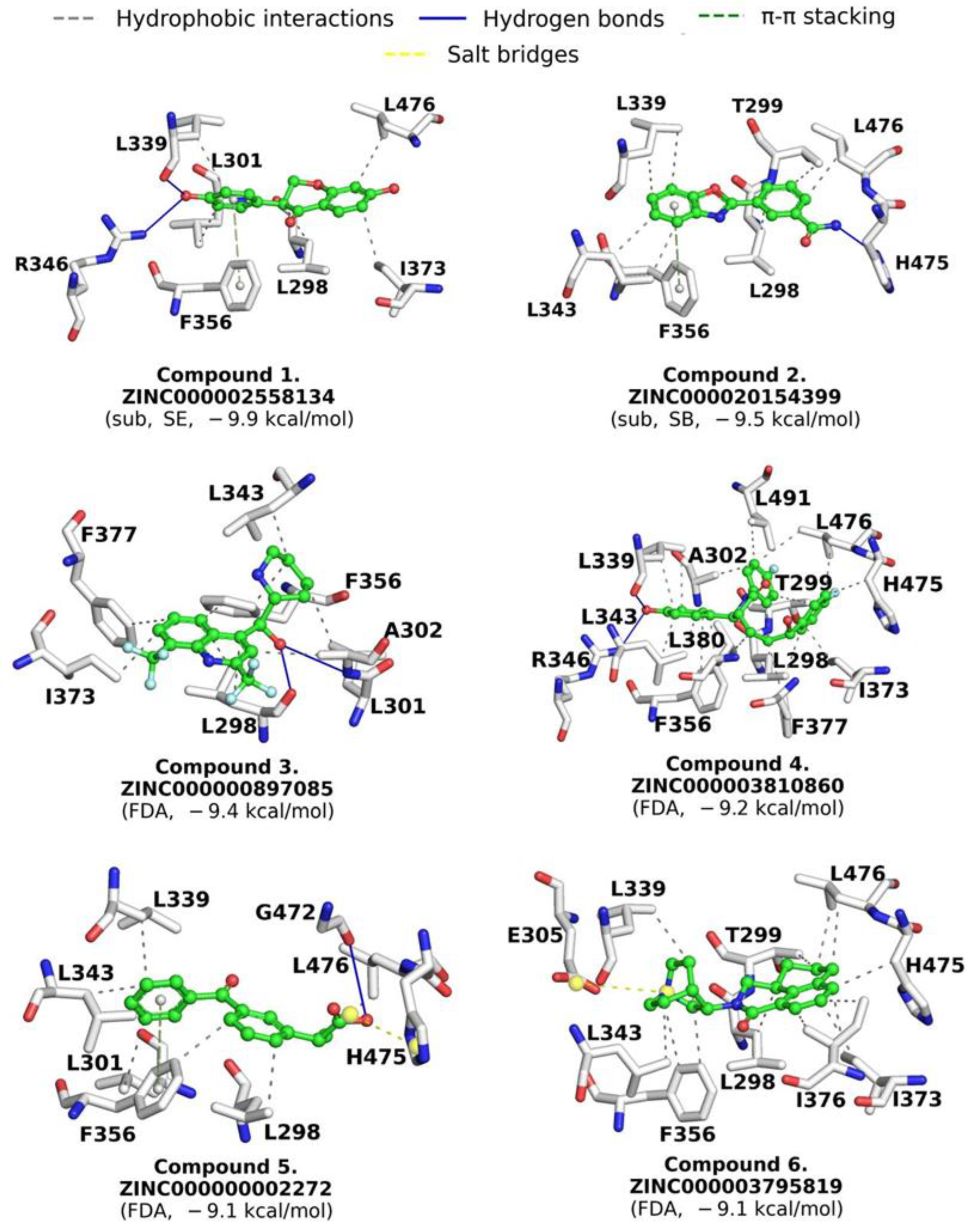
2.5. Drugs Repositioning
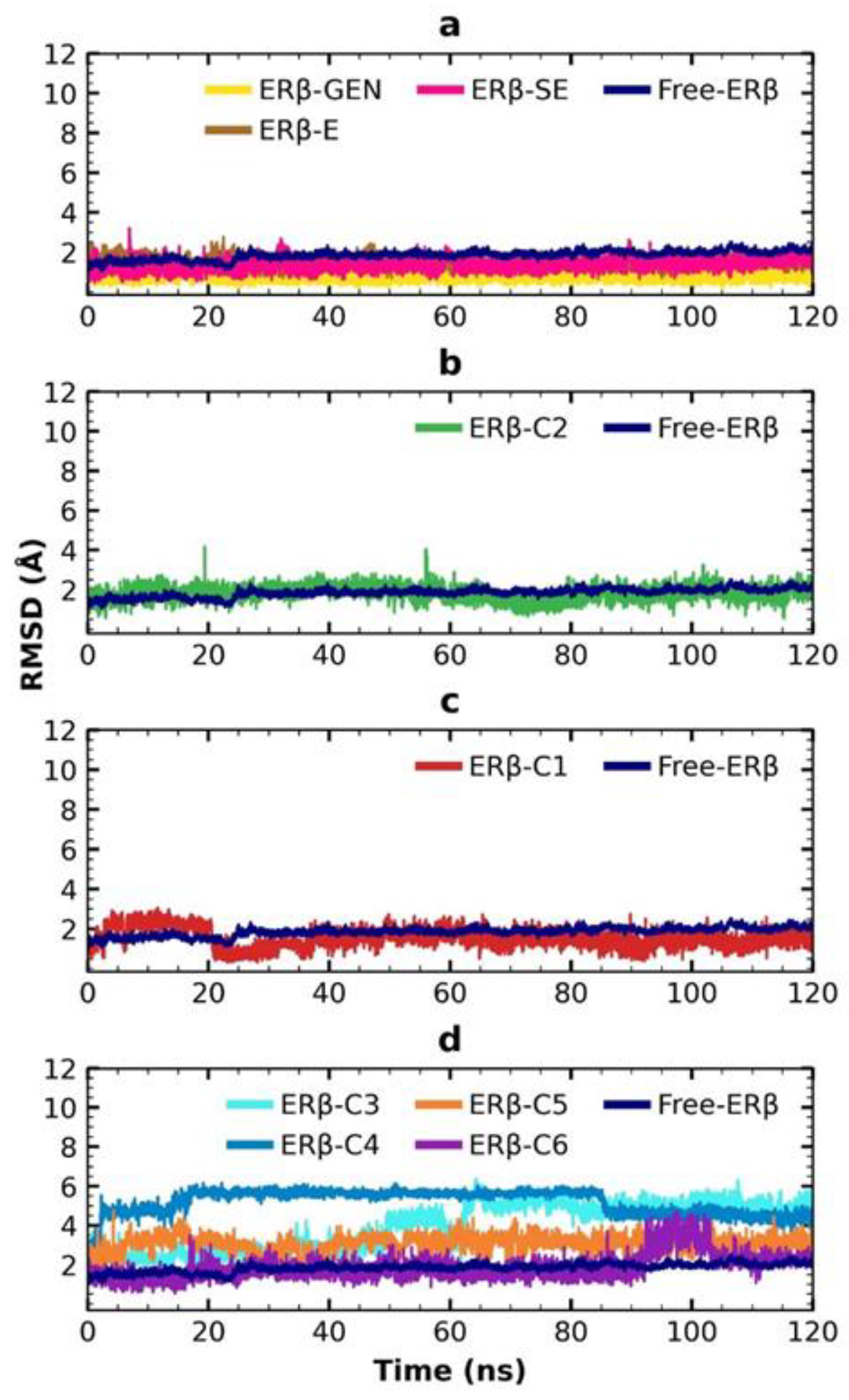
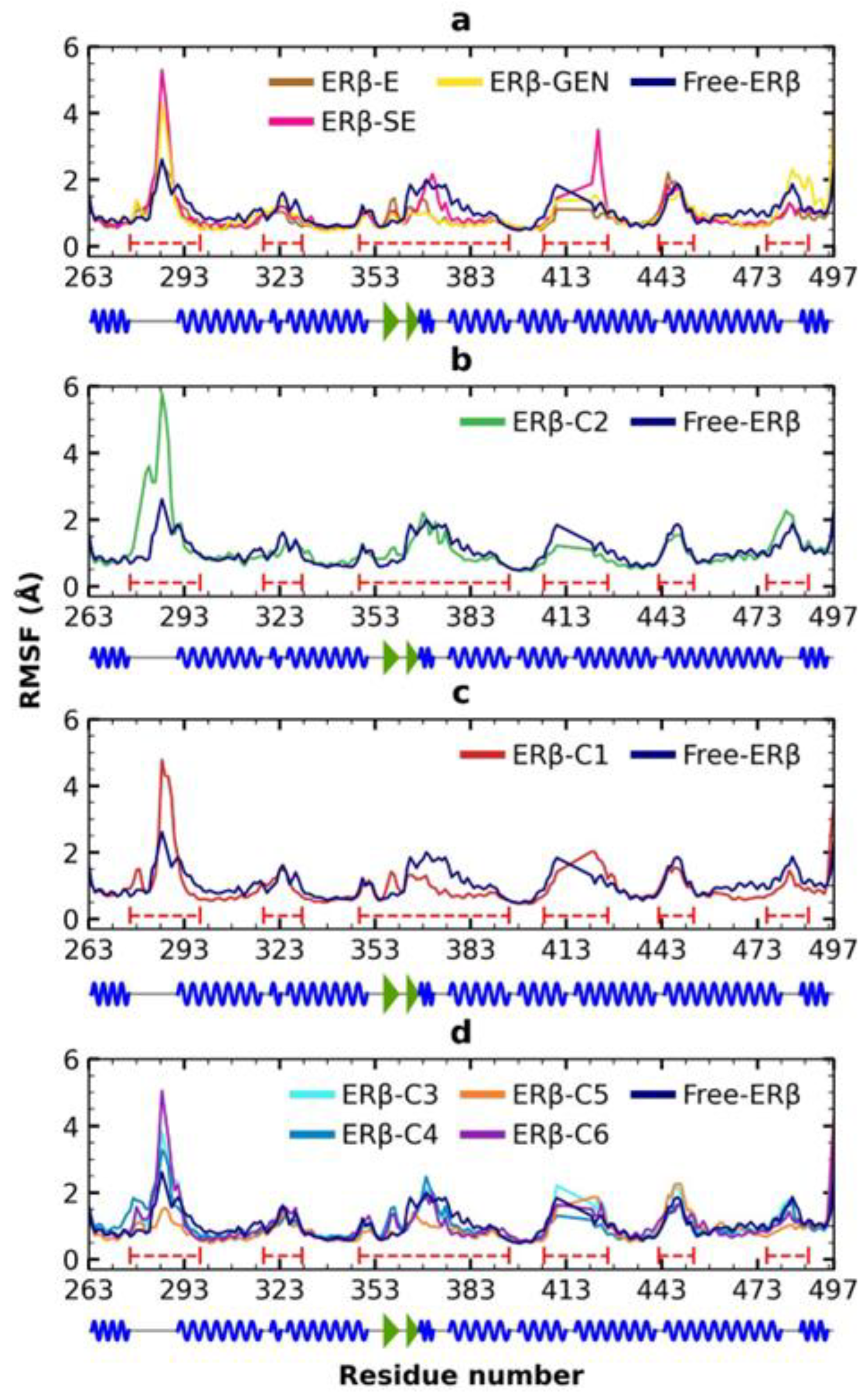
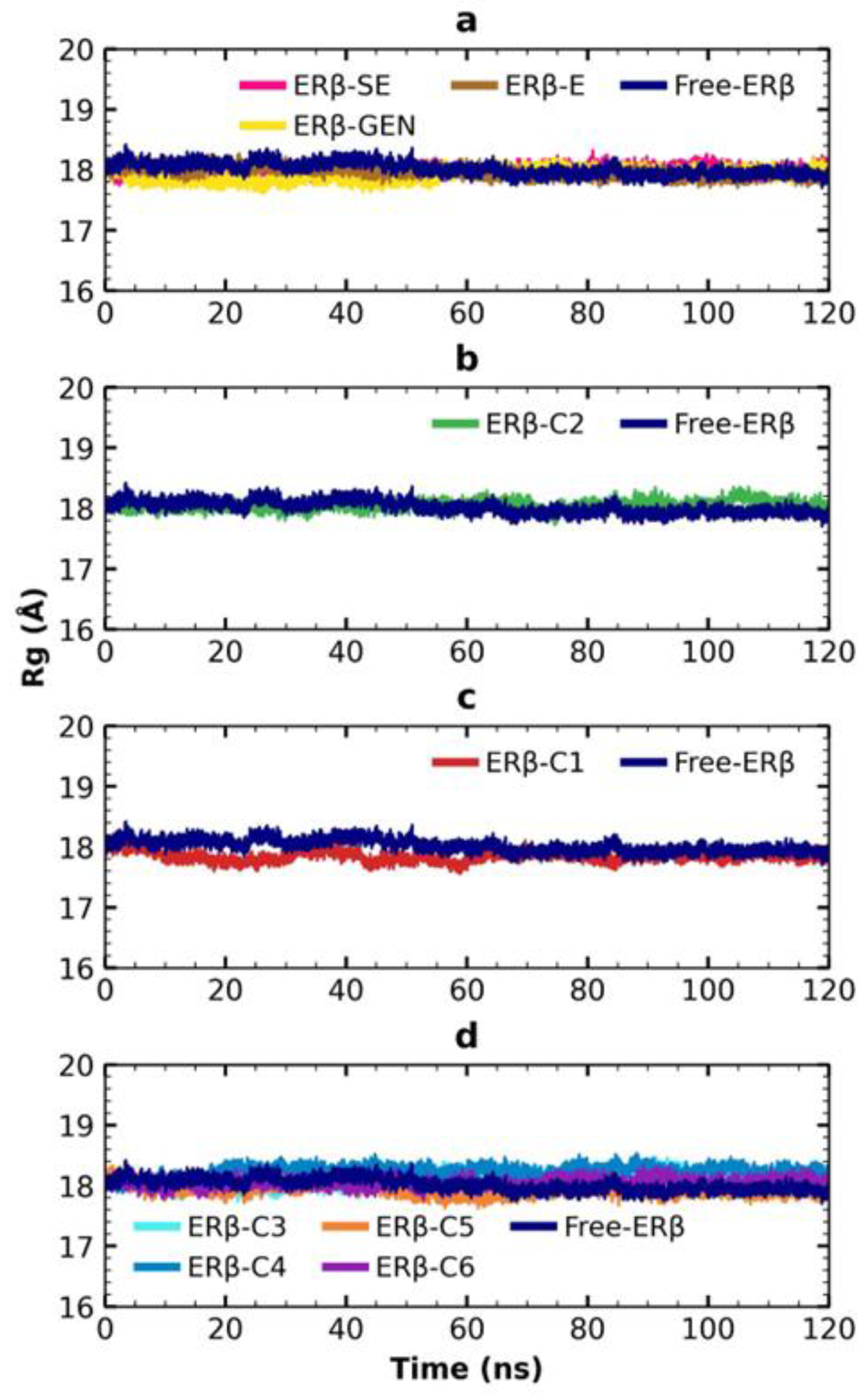
2.6. MDS Analysis
2.7. ADMET Analysis
3. Discussion
4. Materials and Methods
4.1. Analysis of ERβ and Its Ligands
4.2. Preparation of ERβ Tridimensional Structure
4.3. MD Validation and Analysis of Control Compounds
4.4. Identification of New Potential ERβ Ligands
4.4.1. LBVS
4.4.2. Drug Repositioning
4.4.3. Interaction Fingerprint Analysis
4.4.4. Molecular Dynamics Simulations (MDS)
4.4.5. MDS Trajectories Analysis
4.4.6. Prediction of ADMET Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- WHO. Obesity and Overweight. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 22 September 2021).
- National Heart, Lung and Blood Institute (NHLBI). Overweight and Obesity. 2021. Available online: https://www.nhlbi.nih.gov/health-topics/overweight-and-obesity (accessed on 22 September 2021).
- Tabarés Seisdedos, R. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar]
- May, M.; Schindler, C.; Engeli, S. Modern pharmacological treatment of obese patients. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018819897527. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Clemmensen, C.; Finan, B.; DiMarchi, R.; Tschöp, M. Anti-obesity therapy: From rainbow pills to polyagonists. Pharmacol. Rev. 2018, 70, 712–746. [Google Scholar] [CrossRef] [PubMed]
- Pilitsi, E.; Farr, O.M.; Polyzos, S.A.; Perakakis, N.; Nolen-Doerr, E.; Papathanasiou, A.-E.; Mantzoros, C.S. Pharmacotherapy of obesity: Available medications and drugs under investigation. Metabolism 2019, 92, 170–192. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Pelliccia, S.; Famiglini, V.; Stefanucci, A.; Macedonio, G.; Chiavaroli, A.; Orlando, G.; Brunetti, L.; Ferrante, C.; Pieretti, S.; et al. Exploring the first Rimonabant analog-opioid peptide hybrid compound, as bivalent ligand for CB1 and opioid receptors. J. Enzym. Inhib. Med. Chem. 2017, 32, 444–451. [Google Scholar] [CrossRef]
- Dvorácskó, S.; Dimmito, M.P.; Sebastiani, J.; La Regina, G.; Silvestri, R.; Pieretti, S.; Stefanucci, A.; Tömböly, C.; Mollica, A. Rimonabant-Based Compounds Bearing Hydrophobic Amino Acid Derivatives as Cannabinoid Receptor Subtype 1 Ligands. ACS Med. Chem. Lett. 2023, 14, 479–486. [Google Scholar] [CrossRef]
- Müller, T.D.; Blüher, M.; Tschöp, M.H.; DiMarchi, R.D. Anti-obesity drug discovery: Advances and challenges. Nat. Rev. Drug Discov. 2022, 21, 201–223. [Google Scholar] [CrossRef]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen–estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef]
- Yepuru, M.; Eswaraka, J.; Kearbey, J.D.; Barrett, C.M.; Raghow, S.; Veverka, K.A.; Miller, D.D.; Dalton, J.T.; Narayanan, R. Estrogen receptor-β-selective ligands alleviate high-fat diet-and ovariectomy-induced obesity in mice. J. Biol. Chem. 2010, 285, 31292–31303. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Tran, Q.T.; Harvey, I.; Smallwood, H.S.; Thiyagarajan, T.; Banerjee, S.; Johnson, D.L.; Dalton, J.T.; Sullivan, R.D.; Miller, D.D.; et al. Pharmacologic activation of estrogen receptor β increases mitochondrial function, energy expenditure, and brown adipose tissue. FASEB J. 2017, 31, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, D.; Sugiyama, N.; Yonekubo, S.; Pawlak, A.; Murasawa, H.; Nakamura, M.; Hayashi, M.; Ogawa, T.; Moro, M.; Washizuka, S.; et al. Novel oestrogen receptor β-selective ligand reduces obesity and depressive-like behaviour in ovariectomized mice. Sci. Rep. 2017, 7, 4663. [Google Scholar] [CrossRef]
- Barros, R.P.; Gustafsson, J.-Å. Estrogen receptors and the metabolic network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Delgado, B.J.; Lopez-Ojeda, W. Estrogen. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- González-Granillo, M.; Savva, C.; Li, X.; Fitch, M.; Pedrelli, M.; Hellerstein, M.; Parini, P.; Korach-André, M.; Gustafsson, J. ERβ activation in obesity improves whole body metabolism via adipose tissue function and enhanced mitochondria biogenesis. Mol. Cell. Endocrinol. 2019, 479, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.; Clerici, C.; Lephart, E.D.; Cole, S.J.; Heenan, C.; Castellani, D.; Wolfe, B.E.; Nechemias-Zimmer, L.; Brown, N.M.; Lund, T.D.; et al. S-equol, a potent ligand for estrogen receptor β, is the exclusive enantiomeric form of the soy isoflavone metabolite produced by human intestinal bacterial flora. Am. J. Clin. Nutr. 2005, 81, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Mabuchi, K.; Takano, A.; Hara, Y.; Negishi, H.; Morimoto, K.; Ueno, T.; Uchiyama, S.; Takamata, A. S-equol exerts estradiol-like anorectic action with minimal stimulation of estrogen receptor-α in ovariectomized rats. Front. Endocrinol. 2017, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Mandujano-Lázaro, G.; Galaviz-Hernández, C.; Reyes-López, C.A.; Almanza-Pérez, J.C.; Giacoman-Martínez, A.; López-Camarillo, C.; Huang, F.; Marchat, L.A. A short S-equol exposure has a long-term inhibitory effect on adipogenesis in mouse 3T3-L1 cells. Appl. Sci. 2021, 11, 9657. [Google Scholar] [CrossRef]
- Sneha, P.; Doss, C.G.P. Molecular dynamics: New frontier in personalized medicine. Adv. Protein Chem. Struct. Biol. 2016, 102, 181–224. [Google Scholar]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without con-sidering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Kuhl, H. Pharmacology of Estrogens and Progestogens: Influence of Different Routes of Administration. Climacteric 2005, 8 (Suppl. S1), 3–63. [Google Scholar] [CrossRef]
- PubChem. Raloxifene. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5035 (accessed on 2 October 2021).
- Zafar, A.; Ahmad, S.; Naseem, I. Insight into the structural stability of coumestrol with human estrogen receptor α and β subtypes: A combined approach involving docking and molecular dynamics simulation studies. RSC Adv. 2015, 5, 81295–81312. [Google Scholar] [CrossRef]
- Dow, G.; Bauman, R.; Caridha, D.; Cabezas, M.; Du, F.; Gomez-Lobo, R.; Park, M.; Smith, K.; Cannard, K. Mefloquine induces dose-related neurological effects in a rat model. Antimicrob. Agents Chemother. 2006, 50, 1045–1053. [Google Scholar] [CrossRef]
- Takase, H.; Dohi, Y.; Okado, T.; Hashimoto, T.; Goto, Y.; Kimura, G. Effects of ezetimibe on visceral fat in the metabolic syndrome: A randomised controlled study. Eur. J. Clin. Investig. 2012, 42, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Ohbu-Murayama, K.; Adachi, H.; Hirai, Y.; Enomoto, M.; Fukami, A.; Obuchi, A.; Yoshimura, A.; Nakamura, S.; Nohara, Y.; Nakao, E.; et al. Ezetimibe combined with standard diet and exercise therapy improves insulin resistance and atherosclerotic markers in patients with metabolic syndrome. J. Diabetes Investig. 2015, 6, 325–333. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, R.-H.; Park, H.; Wang, H.J.; Lee, H.; Kang, E.S. Effect of ezetimibe on glucose metabolism and inflammatory markers in adipose tissue. Biomedicines 2020, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.H.; Mukherjee, S.; Jang, M.H.; Pham, H.G.; Choi, M.; Yun, J.W. Ketoprofen alleviates diet-induced obesity and promotes white fat browning in mice via the activation of COX-2 through mTORC1-p38 signaling pathway. Pflügers Arch. -Eur. J. Physiol. 2020, 472, 583–596. [Google Scholar] [CrossRef]
- Gouveia de Araujo Ferreira, N.; Cavalcanti, I.L.; Assad, A.R.; Barrucand, L.; Braga, E.L.C.; Verçosa, N. A prospective, randomized, double-blind trial to compare body weight-adjusted and fixed doses of palonosetron for preventing postoperative nausea and vomiting in obese female patients. PLoS ONE 2020, 15, e0227490. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Loschwitz, J.; Jäckering, A.; Keutmann, M.; Olagunju, M.; Olubiyi, O.O.; Strodel, B. Dataset of AMBER force field parameters of drugs, natural products and steroids for simulations using GROMACS. Data Brief. 2021, 35, 106948. [Google Scholar] [CrossRef]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Lemkul, J. From proteins to perturbed Hamiltonians: A suite of tutorials for the GROMACS-2018 molecular simulation package [article v1. 0]. Living J. Comp. Mol. Sci. 2018, 1, 5068. [Google Scholar] [CrossRef]
- Polishchuk, P.; Kutlushina, A.; Bashirova, D.; Mokshyna, O.; Madzhidov, T. Virtual screening using pharmacophore models retrieved from molecular dynamic simulations. Int. J. Mol. Sci. 2019, 20, 5834. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔEvdw | ΔEele | ΔGpolar | ΔGSA | ΔGb |
|---|---|---|---|---|---|
| ERβ-E | −41.24 ± 0.30 | −8.15 ± 0.52 | 21.09 ± 0.25 | −3.86 ± 0.02 | −32.17 ± 0.37 |
| ERβ-SE | −35.77 ± 0.43 | −14.68 ± 0.26 | 26.87 ± 0.27 | −3.41 ± 0.02 | −27.01 ± 0.41 |
| ERβ-GEN | −37.72 ± 0.35 | −14.43 ± 0.22 | 28.65 ± 0.18 | −3.49 ± 0.02 | −27.01 ± 0.30 |
| ERβ-C1 | −34.08 ± 0.38 | −13.69 ± 0.18 | 27.02 ± 0.19 | −3.52 ± 0.02 | −24.27 ± 0.34 |
| ERβ-C2 | −37.72 ± 0.25 | −4.33 ± 0.14 | 22.1 ± 0.19 | −3.38 ± 0.02 | −23.33 ± 0.30 |
| ERβ-C3 | −44.71 ± 0.30 | −1.87 ± 0.18 | 17.94 ± 0.16 | −4.47 ± 0.02 | −33.11 ± 0.30 |
| ERβ-C4 | −56.91 ± 0.28 | −3.54 ± 0.4 | 28.89 ± 0.41 | −4.92 ± 0.02 | −36.5 ± 0.34 |
| ERβ-C5 | −38.96 ± 0.29 | −4.4 ± 0.14 | 24.4 ± 0.30 | −3.7 ± 0.02 | −22.65 ± 0.36 |
| ERβ-C6 | −43.79 ± 0.27 | −1.61 ± 0.11 | 20.07 ± 0.44 | −4.17 ± 0.02 | −29.55 ± 0.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méndez-Álvarez, D.; Torres-Rojas, M.F.; Lara-Ramirez, E.E.; Marchat, L.A.; Rivera, G. Ligand-Based Virtual Screening, Molecular Docking, and Molecular Dynamic Simulations of New β-Estrogen Receptor Activators with Potential for Pharmacological Obesity Treatment. Molecules 2023, 28, 4389. https://doi.org/10.3390/molecules28114389
Méndez-Álvarez D, Torres-Rojas MF, Lara-Ramirez EE, Marchat LA, Rivera G. Ligand-Based Virtual Screening, Molecular Docking, and Molecular Dynamic Simulations of New β-Estrogen Receptor Activators with Potential for Pharmacological Obesity Treatment. Molecules. 2023; 28(11):4389. https://doi.org/10.3390/molecules28114389
Chicago/Turabian StyleMéndez-Álvarez, Domingo, Maria F. Torres-Rojas, Edgar E. Lara-Ramirez, Laurence A. Marchat, and Gildardo Rivera. 2023. "Ligand-Based Virtual Screening, Molecular Docking, and Molecular Dynamic Simulations of New β-Estrogen Receptor Activators with Potential for Pharmacological Obesity Treatment" Molecules 28, no. 11: 4389. https://doi.org/10.3390/molecules28114389
APA StyleMéndez-Álvarez, D., Torres-Rojas, M. F., Lara-Ramirez, E. E., Marchat, L. A., & Rivera, G. (2023). Ligand-Based Virtual Screening, Molecular Docking, and Molecular Dynamic Simulations of New β-Estrogen Receptor Activators with Potential for Pharmacological Obesity Treatment. Molecules, 28(11), 4389. https://doi.org/10.3390/molecules28114389