
Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides

Abstract
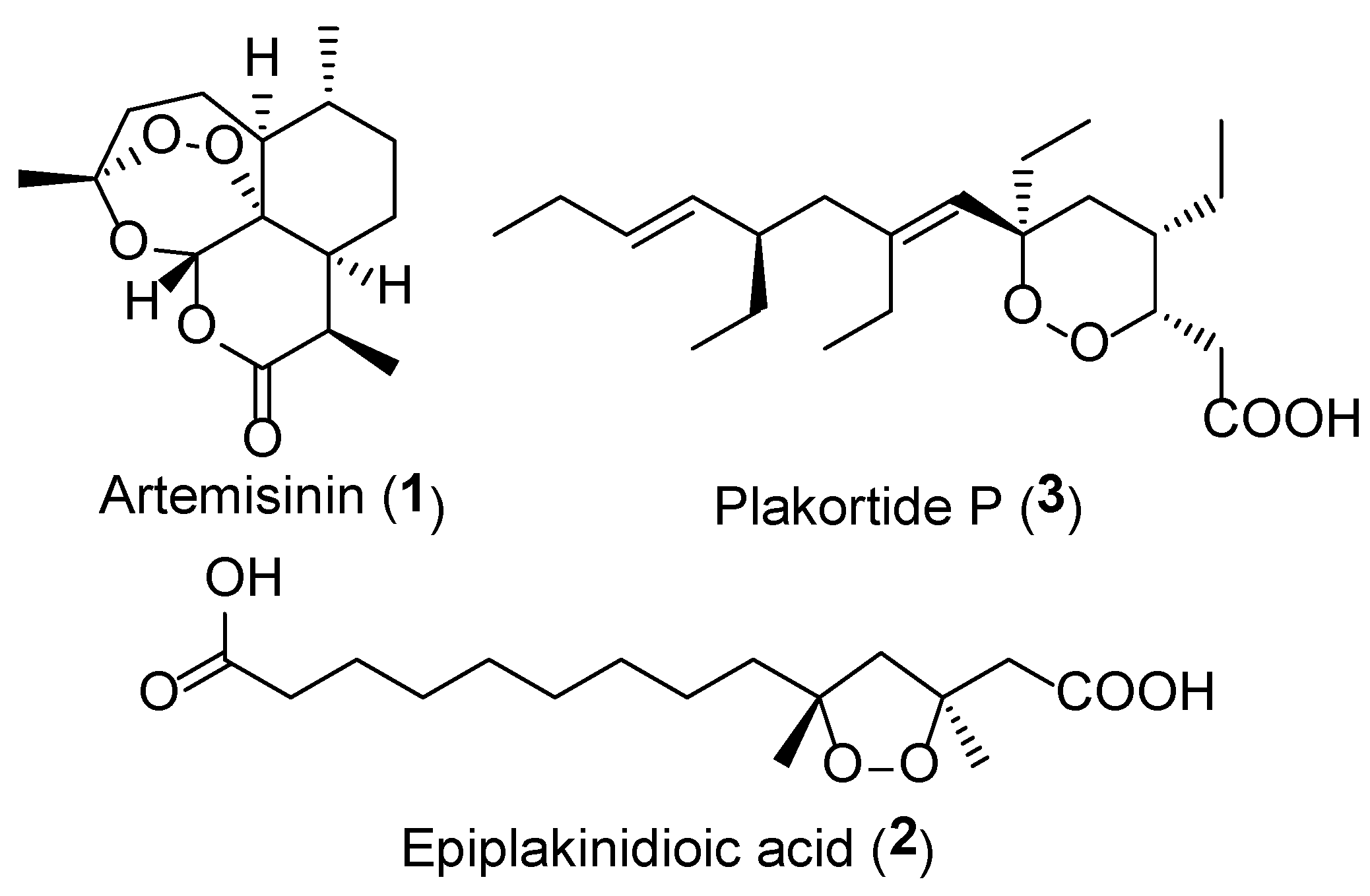
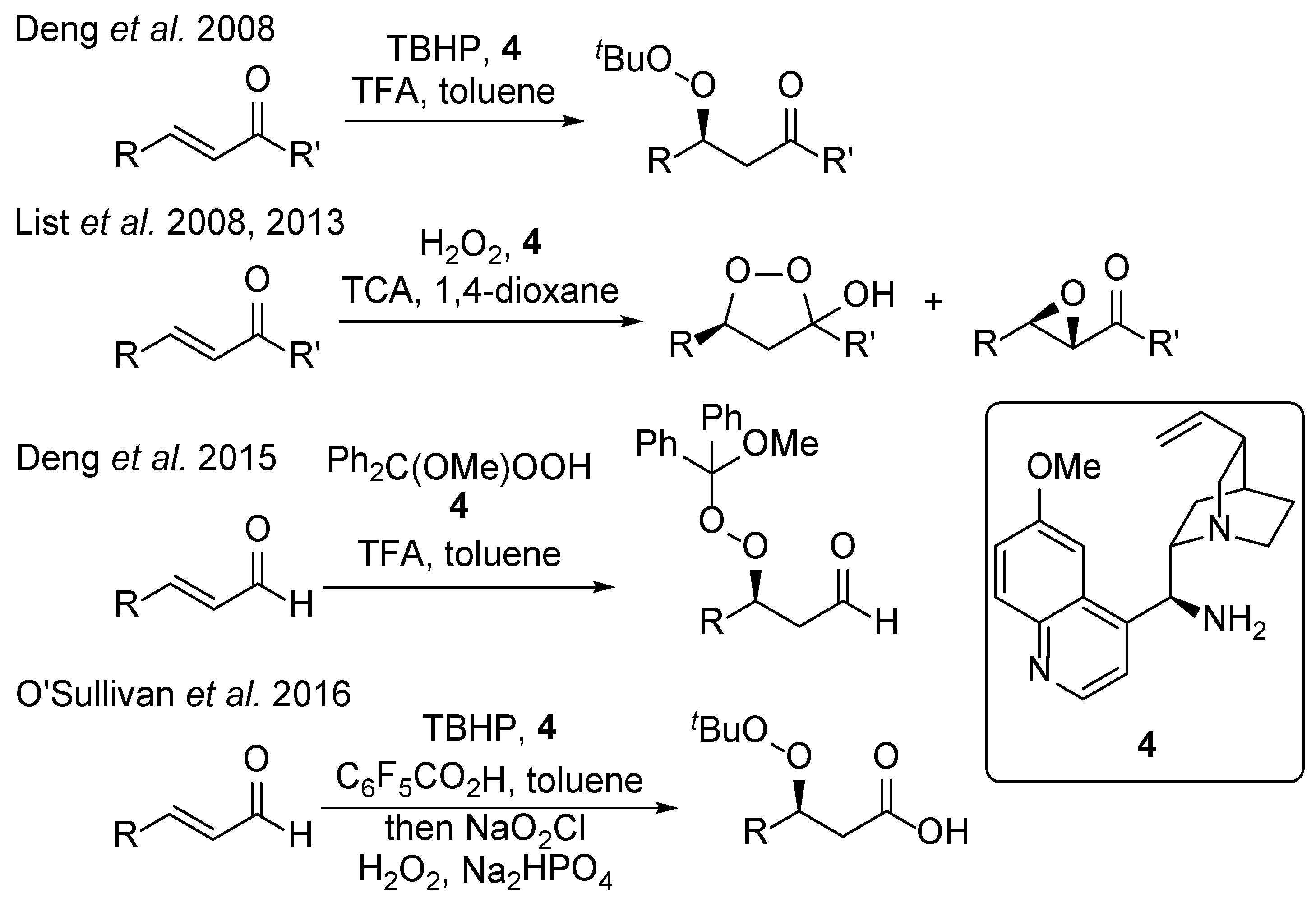
1. Introduction
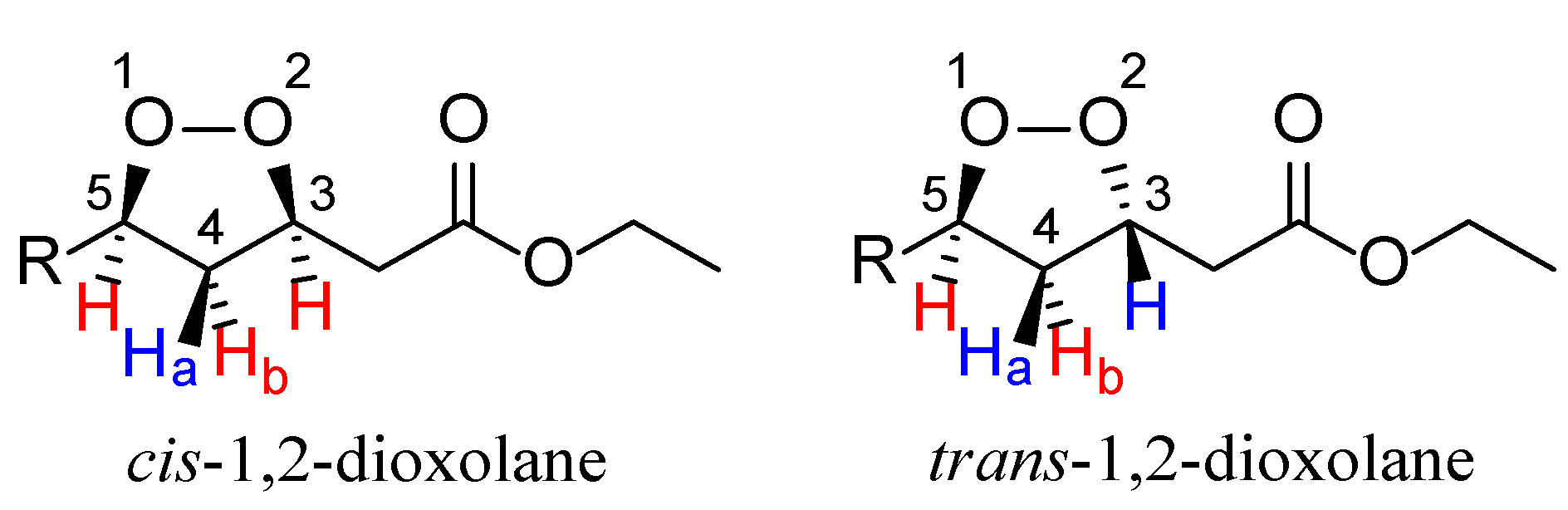
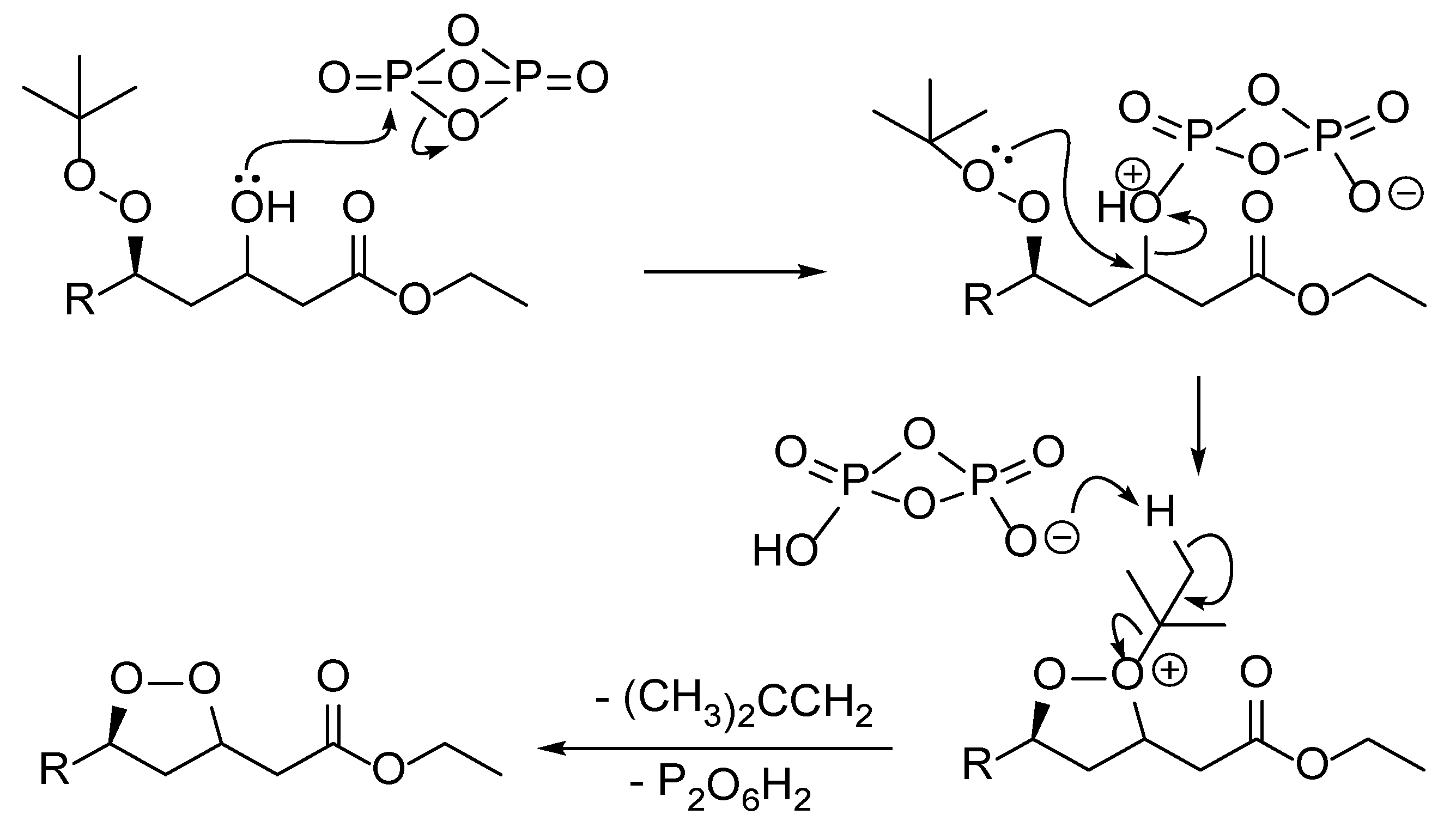
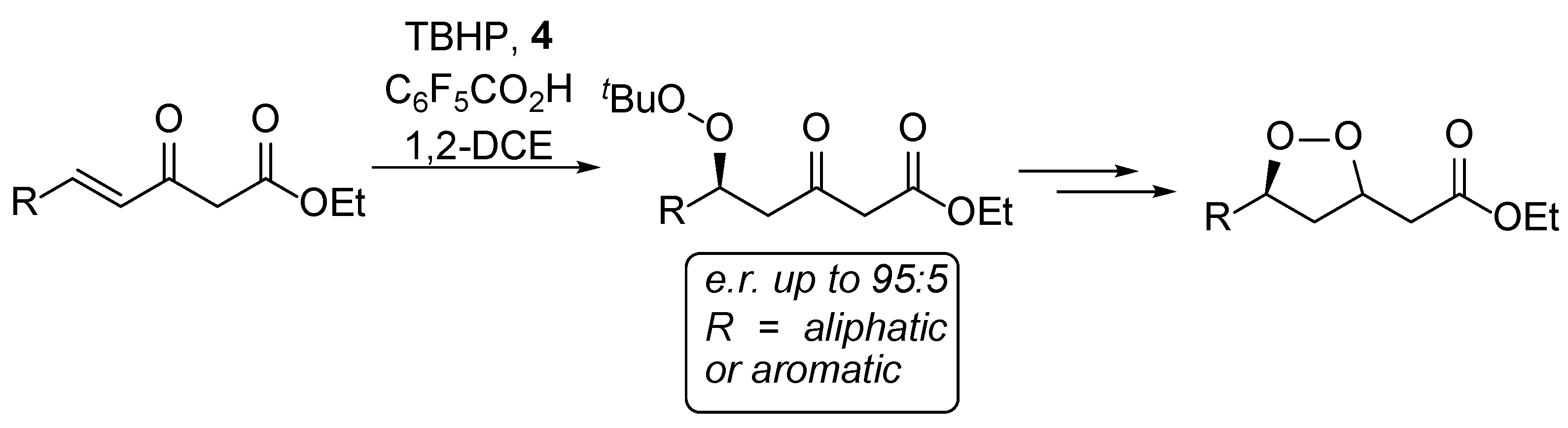
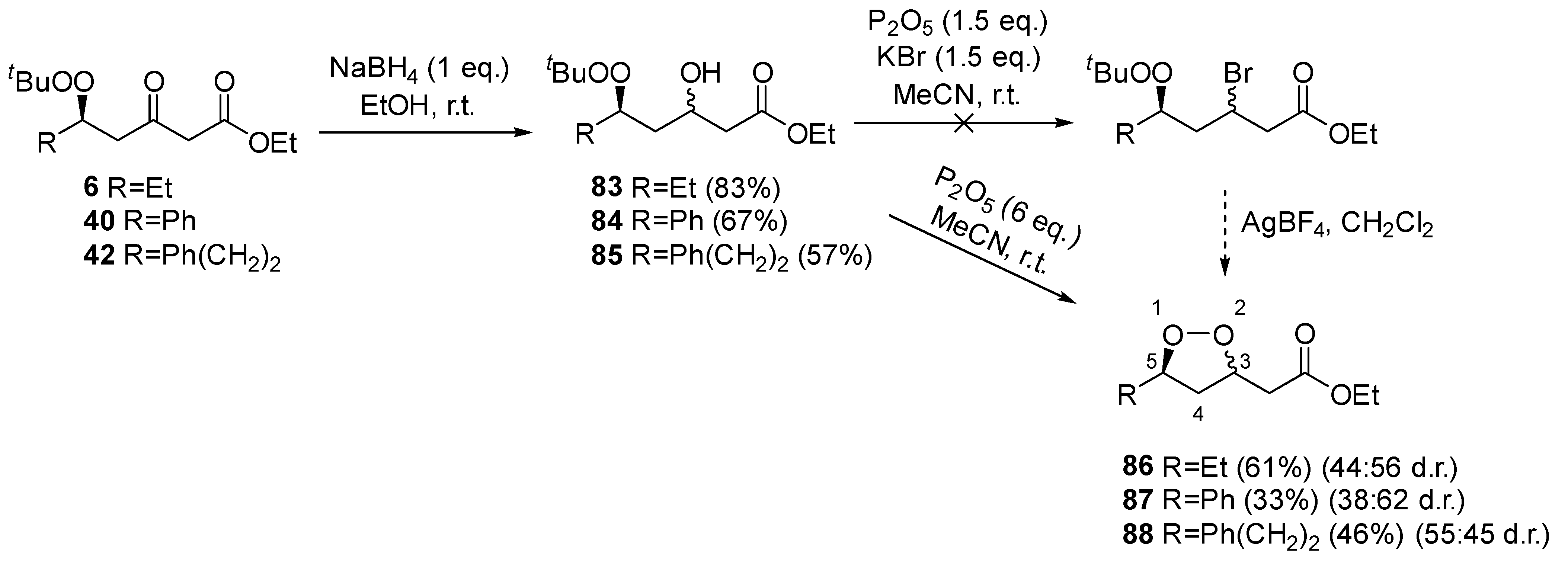
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Casteel, D.A. Peroxy natural products. Nat. Prod. Rep. 1992, 9, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Casteel, D.A. Peroxy natural products. Nat. Prod. Rep. 1999, 16, 55–73. [Google Scholar] [CrossRef]
- Liu, D.-Z.; Liu, J.-K. Peroxy natural products. Nat. Prod. Bioprospecting 2013, 3, 161–206. [Google Scholar] [CrossRef]
- Norris, M.D.; Perkins, M.V. Structural diversity and chemical synthesis of peroxide and peroxide-derived polyketide metabolites from marine sponges. Nat. Prod. Rep. 2016, 33, 861–880. [Google Scholar] [CrossRef]
- Kossuga, M.H.; Nascimento, A.M.; Reimão, J.Q.; Tempone, A.G.; Taniwaki, N.N.; Veloso, K.; Ferreira, A.G.; Cavalcanti, B.C.; Pessoa, C.; Moraes, M.O.; et al. Antiparasitic, Antineuroinflammatory, and Cytotoxic Polyketides from the Marine Sponge Plakortis angulospiculatus Collected in Brazil. J. Nat. Prod. 2008, 71, 334–339. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Bioactive peroxides as potential therapeutic agents. Eur. J. Med. Chem. 2008, 43, 223–251. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Astonishing Diversity of Natural Peroxides as Potential Therapeutic Agents. J. Mol. Genet. Med. 2015, 9, 1000163. [Google Scholar]
- Su, X.-Z.; Miller, L.H. The discovery of artemisinin and the Nobel Prize in Physiology or Medicine. Sci. China Life Sci. 2015, 58, 1175–1179. [Google Scholar] [CrossRef]
- WHO. WHO Guidelines for Malaria; World Health Organisation: Geneva, Switzerland, 2022; Available online: https://www.who.int/publications/i/item/guidelines-for-malaria (accessed on 10 April 2023).
- Chen, Y.; Killday, K.B.; McCarthy, P.J.; Schimoler, R.; Chilson, K.; Selitrennikoff, C.; Pomponi, S.A.; Wright, A.E. Three New Peroxides from the Sponge Plakinastrella Species. J. Nat. Prod. 2001, 64, 262–264. [Google Scholar] [CrossRef]
- Jiménez-Romero, C.; Ortiz, I.; Vicente, J.; Vera, B.; Rodríguez, A.D.; Nam, S.; Jove, R. Bioactive Cycloperoxides Isolated from the Puerto Rican Sponge Plakortis halichondrioides. J. Nat. Prod. 2010, 73, 1694–1700. [Google Scholar] [CrossRef]
- Terent’ev, A.O.; Borisov, D.A.; Vil’, V.A.; Dembitsky, V.M. Synthesis of five- and six-membered cyclic organic peroxides: Key transformations into peroxide ring-retaining products. Beilstein J. Org. Chem. 2014, 10, 34–114. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, H.; O’Reilly, K.; Gupta, M.K.; Horgan, C.; O’Leary, E.M.; O’Sullivan, T.P. Advances in the synthesis of acyclic peroxides. RSC Adv. 2017, 7, 19506–19556. [Google Scholar] [CrossRef]
- Lassaletta, J.M. Spotting trends in organocatalysis for the next decade. Nat. Commun. 2020, 11, 3787. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef]
- Hughes, D.L. Asymmetric Organocatalysis in Drug Development—Highlights of Recent Patent Literature. Org. Process Res. Dev. 2018, 22, 574–584. [Google Scholar] [CrossRef]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef]
- Marqués-López, E.; Herrera, R.P.; Christmann, M. Asymmetric organocatalysis in total synthesis—A trial by fire. Nat. Prod. Rep. 2010, 27, 1138–1167. [Google Scholar] [CrossRef]
- Abbasov, M.E.; Romo, D. The ever-expanding role of asymmetric covalent organocatalysis in scalable, natural product synthesis. Nat. Prod. Rep. 2014, 31, 1318–1327. [Google Scholar] [CrossRef]
- Xiang, S.-H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786. [Google Scholar] [CrossRef]
- García Mancheño, O.; Waser, M. Recent Developments and Trends in Asymmetric Organocatalysis. Eur. J. Org. Chem. 2023, 26, e202200950. [Google Scholar] [CrossRef]
- Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]
- List, B. Introduction: Organocatalysis. Chem. Rev. 2007, 107, 5413–5415. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Marcelli, T. Organocatalysis: Cinchona catalysts. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 142–152. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, Y.-C. Recent advances in asymmetric catalysis with cinchona alkaloid-based primary amines. Catal. Sci. Technol. 2011, 1, 354–365. [Google Scholar] [CrossRef]
- Marcelli, T.; Hiemstra, H. Cinchona Alkaloids in Asymmetric Organocatalysis. Synthesis 2010, 2010, 1229–1279. [Google Scholar] [CrossRef]
- Lu, X.; Liu, Y.; Sun, B.; Cindric, B.; Deng, L. Catalytic Enantioselective Peroxidation of α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2008, 130, 8134–8135. [Google Scholar] [CrossRef]
- Reisinger, C.M.; Wang, X.; List, B. Catalytic Asymmetric Hydroperoxidation of α,β-Unsaturated Ketones: An Approach to Enantiopure Peroxyhemiketals, Epoxides, and Aldols. Angew. Chem. Int. Ed. 2008, 47, 8112–8115. [Google Scholar] [CrossRef]
- Lifchits, O.; Mahlau, M.; Reisinger, C.M.; Lee, A.; Farès, C.; Polyak, I.; Gopakumar, G.; Thiel, W.; List, B. The Cinchona Primary Amine-Catalyzed Asymmetric Epoxidation and Hydroperoxidation of α,β-Unsaturated Carbonyl Compounds with Hydrogen Peroxide. J. Am. Chem. Soc. 2013, 135, 6677–6693. [Google Scholar] [CrossRef]
- Hu, L.; Lu, X.; Deng, L. Catalytic Enantioselective Peroxidation of α,β-Unsaturated Aldehydes for the Asymmetric Synthesis of Biologically Important Chiral Endoperoxides. J. Am. Chem. Soc. 2015, 137, 8400–8403. [Google Scholar] [CrossRef]
- O’Reilly, K.; Gupta, M.K.; Gandhi, H.K.; Kumar, V.P.; Eccles, K.S.; Lawrence, S.E.; O’Sullivan, T.P. Cinchona-catalysed, Enantioselective Synthesis of β-Peroxycarboxylic Acids, β-Peroxyesters and β-Peroxyalcohols. Curr. Org. Chem. 2016, 20, 2633–2638. [Google Scholar] [CrossRef]
- O’Reilly, K.; Gupta, M.K.; Gandhi, H.; Kumar, V.P.; O’Sullivan, T.P. Asymmetric Peroxidation of α,β-Unsaturated Aldehydes under Diarylprolinol Ether Catalysis. Curr. Org. Chem. 2017, 21, 2013–2016. [Google Scholar] [CrossRef]
- Russo, A.; Lattanzi, A. Catalytic Asymmetric β-Peroxidation of Nitroalkenes. Adv. Synth. Catal. 2008, 350, 1991–1995. [Google Scholar] [CrossRef]
- Lu, X.; Deng, L. Catalytic Asymmetric Peroxidation of α,β-Unsaturated Nitroalkenes by a Bifunctional Organic Catalyst. Org. Lett. 2014, 16, 2358–2361. [Google Scholar] [CrossRef]
- Benetti, S.; Romagnoli, R.; De Risi, C.; Spalluto, G.; Zanirato, V. Mastering beta-Keto Esters. Chem. Rev. 1995, 95, 1065–1114. [Google Scholar] [CrossRef]
- Everaere, K.; Mortreux, A.; Carpentier, J.-F. Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Carbonyl Compounds with 2-Propanol and Ephedrine-Type Ligands. Adv. Synth. Catal. 2003, 345, 67–77. [Google Scholar] [CrossRef]
- Ratovelomanana-Vidal, V.; Girard, C.; Touati, R.; Tranchier, J.P.; Hassine, B.B.; Genêt, J.P. Enantioselective Hydrogenation of β-Keto Esters using Chiral Diphosphine-Ruthenium Complexes: Optimization for Academic and Industrial Purposes and Synthetic Applications. Adv. Synth. Catal. 2003, 345, 261–274. [Google Scholar] [CrossRef]
- Bariotaki, A.; Kalaitzakis, D.; Smonou, I. Enzymatic Reductions for the Regio- and Stereoselective Synthesis of Hydroxy-keto Esters and Dihydroxy Esters. Org. Lett. 2012, 14, 1792–1795. [Google Scholar] [CrossRef]
- Heravi, M.M.; Khaghaninejad, S.; Mostofi, M. Chapter one—Pechmann reaction in the synthesis of coumarin derivatives. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2014; Volume 112, pp. 1–50. [Google Scholar] [CrossRef]
- Hennessy, M.C.; O’Sullivan, T.P. Recent advances in the transesterification of β-keto esters. RSC Adv. 2021, 11, 22859–22920. [Google Scholar] [CrossRef]
- Khademi, Z.; Heravi, M.M. Applications of Claisen condensations in total synthesis of natural products. An old reaction, a new perspective. Tetrahedron 2022, 103, 132573. [Google Scholar] [CrossRef]
- Govender, T.; Arvidsson, P.I.; Maguire, G.E.M.; Kruger, H.G.; Naicker, T. Enantioselective Organocatalyzed Transformations of β-Ketoesters. Chem. Rev. 2016, 116, 9375–9437. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, S.; Schietroma, D.M.S.; Tulli, L.G.; Monaco, M.R.; Bella, M. Unsaturated β-ketoesters as versatile electrophiles in organocatalysis. Chem. Commun. 2010, 46, 5160–5162. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Tsutsumi, T.; Nakata, K.; Nakatsuji, H.; Tanabe, Y. Asymmetric Total Syntheses of Two 3-Acyl-5,6-dihydro-2H-pyrones: (R)-Podoblastin-S and (R)-Lachnelluloic Acid with Verification of the Absolute Configuration of (−)-Lachnelluloic Acid. Molecules 2017, 22, 69. [Google Scholar] [CrossRef] [PubMed]
- Song, C.E. An overview of cinchona alkaloids in chemistry. In Cinchona Alkaloids in Synthesis and Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 1–10. [Google Scholar] [CrossRef]
- Boratyński, P.J.; Zielińska-Błajet, M.; Skarżewski, J. Chapter Two—Cinchona Alkaloids—Derivatives and Applications. Alkaloids Chem. Bio. 2019, 82, 29–145. [Google Scholar] [CrossRef]
- Dijkstra, G.D.H.; Kellogg, R.M.; Wynberg, H. Conformational analysis of some chiral catalysts of the cinchona and ephedra family. The alkaloid catalyzed addition of aromatic thiols to cyclic α,β-unsaturated ketones. Red. Trav. Chim. Pays-Bas 1989, 108, 195–204. [Google Scholar] [CrossRef]
- Dijkstra, G.D.H.; Kellogg, R.M.; Wynberg, H.; Svendsen, J.S.; Marko, I.; Sharpless, K.B. Conformational study of cinchona alkaloids. A combined NMR, molecular mechanics and X-ray approach. J. Am. Chem. Soc. 1989, 111, 8069–8076. [Google Scholar] [CrossRef]
- Bürgi, T.; Baiker, A. Conformational Behavior of Cinchonidine in Different Solvents: A Combined NMR and ab Initio Investigation. J. Am. Chem. Soc. 1998, 120, 12920–12926. [Google Scholar] [CrossRef]
- Melchiorre, P. Cinchona-based Primary Amine Catalysis in the Asymmetric Functionalization of Carbonyl Compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770. [Google Scholar] [CrossRef]
- Evans, G.J.S.; White, K.; Platts, J.A.; Tomkinson, N.C.O. Computational study of iminium ion formation: Effects of amine structure. Org. Biomol. Chem. 2006, 4, 2616–2627. [Google Scholar] [CrossRef]
- Tian, X.; Cassani, C.; Liu, Y.; Moran, A.; Urakawa, A.; Galzerano, P.; Arceo, E.; Melchiorre, P. Diastereodivergent Asymmetric Sulfa-Michael Additions of α-Branched Enones using a Single Chiral Organic Catalyst. J. Am. Chem. Soc. 2011, 133, 17934–17941. [Google Scholar] [CrossRef]
- Marigo, M.; Franzén, J.; Poulsen, T.B.; Zhuang, W.; Jørgensen, K.A. Asymmetric Organocatalytic Epoxidation of α,β-Unsaturated Aldehydes with Hydrogen Peroxide. J. Am. Chem. Soc. 2005, 127, 6964–6965. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, H.; O’Sullivan, T.P. Preparation of γ,δ-unsaturated-β-ketoesters: Lewis acid-catalysed CH insertion of ethyl diazoacetate into α,β-unsaturated aldehydes. Tetrahedron Lett. 2017, 58, 3533–3535. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M.; Monkiewicz, J. Anionic activation of stabilized ylides. A highly Z-stereoselective wittig reaction of (3-ethoxycarbonyl-2-oxopropylidene)triphenyl-phosphorane with aliphatic aldehydes. Tetrahedron Lett. 1986, 27, 739–742. [Google Scholar] [CrossRef]
- Porter, N.A.; Mitchell, J.C. Intramolecular alkylation of peroxides and hydroperoxides; peroxide transfer via peroxonium intermediates. Tetrahedron Lett. 1983, 24, 543–546. [Google Scholar] [CrossRef]
- Khazdooz, L.; Zarei, A.; Aghaei, H.; Azizi, G.; Gheisari, M.M. An efficient and selective method for the iodination and bromination of alcohols under mild conditions. Tetrahedron Lett. 2016, 57, 168–171. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Ferrié, L.; Figadère, B. Synthesis of 3,5-disubstituted-1,2-dioxolanes: Access to analogues of mycangimycin and some rearrangement products. Tetrahedron Lett. 2016, 57, 5286–5289. [Google Scholar] [CrossRef]
- Pinet, A.; Nguyen, L.T.; Figadère, B.; Ferrié, L. Synthesis of 3,5-Disubstituted 1,2-Dioxolanes. Eur. J. Org. Chem. 2020, 2020, 7407–7416. [Google Scholar] [CrossRef]
- Pinet, A.; Figadère, B.; Ferrié, L. Access to Functionalized 3,5-Disubstituted 1,2-Dioxolanes under Mild Conditions through Indium(III) Chloride/Trimethylsilyl Chloride or Scandium(III) Triflate Catalysis. Adv. Synth. Catal. 2020, 362, 1190–1194. [Google Scholar] [CrossRef]
- Pinet, A.; Cojean, S.; Nguyen, L.T.; Vásquez-Ocmín, P.; Maciuk, A.; Loiseau, P.M.; Le Pape, P.; Figadère, B.; Ferrié, L. Anti-protozoal and anti-fungal evaluation of 3,5-disubstituted 1,2-dioxolanes. Bioorganic Med. Chem. Lett. 2021, 47, 128196. [Google Scholar] [CrossRef]
- Khazaei, A.; Rad, M.N.S.; Borazjani, M.K.; Saednia, S.; Borazjani, M.K.; Golbaghi, M.; Behrouz, S. Highly Efficient Etherification and Oxidation of Aromatic Alcohols Using Supported and Unsupported Phosphorus Pentoxide as a Heterogeneous Reagent. Synth. Commun. 2011, 41, 1544–1553. [Google Scholar] [CrossRef]
- Xu, W.; Wang, X.-B.; Wang, Z.-M.; Wu, J.-J.; Li, F.; Wang, J.; Kong, L.-Y. Synthesis and evaluation of donepezil–ferulic acid hybrids as multi-target-directed ligands against Alzheimer’s disease. MedChemComm 2016, 7, 990–998. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly Enantioselective Conjugate Addition of Nitromethane to Chalcones Using Bifunctional Cinchona Organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef] [PubMed]
- Cassani, C.; Martín-Rapún, R.; Arceo, E.; Bravo, F.; Melchiorre, P. Synthesis of 9-amino(9-deoxy)epi cinchona alkaloids, general chiral organocatalysts for the stereoselective functionalization of carbonyl compounds. Nat. Protoc. 2013, 8, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Han, T.-J.; Gao, X.; Yang, Y.-F.; Mei, G.-J. Further developments of β,γ-unsaturated α-ketoesters as versatile synthons in asymmetric catalysis. iScience 2022, 25, 103913. [Google Scholar] [CrossRef] [PubMed]
- Nagy, S.; Fehér, Z.; Dargó, G.; Barabás, J.; Garádi, Z.; Mátravölgyi, B.; Kisszékelyi, P.; Dargó, G.; Huszthy, P.; Höltzl, T.; et al. Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring. Materials 2019, 12, 3034. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Conversion [a] | Isolated Yield | e.r. |
| 1 | MeCN | 40% | 26% | 69:31 |
| 2 | THF | 50% | 31% | 92:8 |
| 3 | EtOAc | 64% | 33% | 92:8 |
| 4 | Tol | 73% | 42% | 95:5 |
| 5 | EtOH | n.d. [b] | 13% (19%) [c] | 79:21 |
| 6 | H2O | 54% | 45% | 88:12 |
| 7 | CH2Cl2 | 75% | 52% | 93:7 |
| 8 | 1,2-DCE | 87% | 69% | 93:7 |
| 9 | CHCl3 | 73% | 49% | 96:4 |
| 10 | CCl4 | 61% | 42% | 94.5:5.5 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Co-Catalyst | pKa | Temp. | Conversion [a] | Isolated Yield | e.r. |
| 1 | Triflic acid | −14.7 | r.t. | 60% | 41% | 57:43 |
| 2 | p-Toluenesulfonic acid | −2.8 | r.t. | 15% | 10% | 94:6 |
| 3 | Methanesulfonic acid | −1.9 | r.t. | 23% | 18% | 95.5:4.5 |
| 4 | Heptafluorobutyric acid | 0.4 | r.t. | 84% | 63% | 96:4 |
| 5 | Trifluoroacetic acid | 0.5 | r.t. | 77% | 58% | 96:4 |
| 6 | Pentafluorobenzoic acid | 1.5 | r.t. | 87% | 69% | 93:7 |
| 7 | Pentafluorobenzoic acid | 1.5 | 4 °C | 76% | 54% | 89:11 |
| 8 | Pentafluorobenzoic acid | 1.5 | 50 °C | 32% | 21% | 94:6 |
| 9 | Chloroacetic acid | 2.9 | r.t. | 25% | 17% | 86.5:13.5 |
| 10 | Tartaric acid | 2.9 | r.t. | 4% | n.d. | n.d. |
| 11 | 2,4-Bis(trifluoromethyl)benzoic acid | 3.3 | r.t. | 42% | 29% | 86:14 |
| 12 | 4-(Trifluoromethyl)benzoic acid | 3.6 | r.t. | 22% | 18% | 81:19 |
| 13 | Boc-L-phenylglycine | 3.9 | r.t. | 22% | 18% | 88:12 |
| 14 | Boc-D-phenylglycine | 3.9 | r.t. | 25% | 20% | 87:13 |
| 15 | Benzoic acid | 4.2 | r.t. | 8% | n.d. | n.d. |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | β-Keto Ester | Conversion [a] | Peroxide | Isolated Yield | e.r. | MW (g/mol) | Alcohol | Isolated Yield | MW (g/mol) |
| 1 | CH3 | 7 | 86% | 33 | 79% | 91:9 | 246.30 | 58 | 87% | 174.20 |
| 2 | CH3CH2 | 5 | 87% | 6 | 69% | 93:7 | 260.33 | 59 | 71% | 188.22 |
| 3 | CH3(CH2)2 | 8 | 74% | 34 | 52% | 93:7 | 274.36 | 60 | 77% | 202.25 |
| 4 | CH3(CH2)3 | 9 | 46% | 35 | 34% | 95:5 | 288.38 | 61 | 93% | 216.28 |
| 5 | CH3(CH2)4 | 10 | 50% | 36 | 43% | 95:5 | 302.41 | 62 | 93% | 230.30 |
| 6 | (CH3)2CH | 11 | n.d. | 37 | 27% | 90:10 | 274.36 | 63 | 93% | 202.25 |
| 7 | (CH3)2CHCH2 | 12 | 33% | 38 | 30% | 92:8 | 288.39 | 64 | 97% | 216.28 |
| 8 | Cy | 13 | 42% | 39 | 35% | 92:8 | 314.42 | 65 | 91% | 242.32 |
| 9 | C6H5 | 14 | 51% | 40 | 49% | 74:26 | 308.37 | 66 | 83% | 236.27 |
| 10 | 2-Naphthyl | 15 | 24% | 41 | 17% | 67:33 | 358.43 | 67 | 57% | 286.33 |
| 11 | C6H5CH2CH2 | 16 | 54% | 42 | 47% | 92:8 | 336.43 | 68 | 74% | 264.32 |
| 12 | 2-FC6H4 | 17 | 25% | 43 | 22% | 83:17 | 326.36 | 69 | 80% | 254.26 |
| 13 | 2-ClC6H4 | 18 | 29% | 44 | 25% | 91:9 | 342.82 | 70 | 84% | 270.71 |
| 14 | 2-BrC6H4 | 19 | 32% | 45 | 29% | 92:8 | 387.27 | 71 | 97% | 315.16 |
| 15 | 2-IC6H4 | 20 | 29% | 46 | 20% | 93:7 | 434.27 | 72 | 34% | 362.16 |
| 16 | 3-FC6H4 | 21 | 37% | 47 | 26% | 78:22 | 326.36 | 73 | 89% | 254.26 |
| 17 | 3-ClC6H4 | 22 | 42% | 48 | 17% | 78:22 | 342.82 | 74 | 86% | 270.71 |
| 18 | 3-BrC6H4 | 23 | 35% | 49 | 28% | 80:20 | 387.27 | 75 | 83% | 315.16 |
| 19 | 4-FC6H4 | 24 | 29% | 50 | 28% | 73:27 | 326.36 | 76 | 83% | 254.26 |
| 20 | 4-ClC6H4 | 25 | 26% | 51 | 24% | 76:24 | 342.82 | 77 | 80% | 270.71 |
| 21 | 4-BrC6H4 | 26 | 34% | 52 | 18% | 78:22 | 387.27 | 78 | 90% | 315.16 |
| 22 | 4-CF3C6H4 | 27 | 38% | 53 | 31% | 67:33 | 376.37 | 79 | 94% | 304.27 |
| 23 | 4-MeOC6H4 | 28 | 43% | 54 | 38% | 57:43 | 338.40 | 80 | 74% | 266.30 |
| 24 | BnOCH2 | 29 | 27% | 55 | 26% | 82:18 | 352.43 | 81 | 61% | 280.32 |
| 25 |  | 30 | 25% | 56 | 22% | n.d [b] | 352.38 | 82 | 94% | 280.28 |
| 26 | 2-Furyl | 31 | 7% | 57 | 6% | n.d [b] | 298.34 | - | - | - |
| 27 |  | 32 | 0% | - | 0% | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hennessy, M.C.; Gandhi, H.; O’Sullivan, T.P. Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides. Molecules 2023, 28, 4317. https://doi.org/10.3390/molecules28114317
Hennessy MC, Gandhi H, O’Sullivan TP. Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides. Molecules. 2023; 28(11):4317. https://doi.org/10.3390/molecules28114317
Chicago/Turabian StyleHennessy, Mary C., Hirenkumar Gandhi, and Timothy P. O’Sullivan. 2023. "Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides" Molecules 28, no. 11: 4317. https://doi.org/10.3390/molecules28114317
APA StyleHennessy, M. C., Gandhi, H., & O’Sullivan, T. P. (2023). Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides. Molecules, 28(11), 4317. https://doi.org/10.3390/molecules28114317