
Radical Decarboxylative Carbon–Nitrogen Bond Formation


Abstract
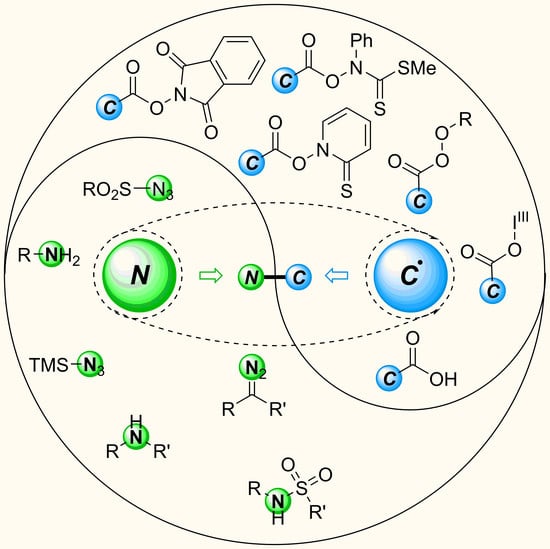
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Radical Decarboxylative Coupling with Barton Esters
3. Radical Decarboxylative Coupling with MPDOC Esters
4. Radical Decarboxylative Coupling with Carboxylic Acids
4.1. Transition–Metal–Catalyzed or Mediated Fashion
4.2. Photoredox or Dual Photoredox Transition–Metal–Catalyzed Fashion
4.3. Electrochemical Fashion
4.4. Metal–Free Fashion
5. Radical Decarboxylative Coupling with NHP Esters
6. Radical Decarboxylative Coupling with Iodocarboxylates
7. Radical Decarboxylative Coupling with Oxime Esters
8. Radical Decarboxylative Couplings with Diacyl Peroxides
9. Discussion and Conclusions
- (1)
- Radical decarboxylation often requires the pre–activation of carboxylic acids, which accompanies multiple reaction steps and more reaction waste. From the viewpoint of atom economy and environmental benefits, it is desirable to develop more efficient strategies through directly using carboxylic acids as the reactants.
- (2)
- Another challenging task is the development of enantioselective radical decarboxylation fashion. It would be a great strategy for the synthesis of valuable chiral amines. However, enantiocontrolled radical decarboxylation is still underdeveloped to date. Transition–metal catalysis might be a feasible solution for this task.
- (3)
- Furthermore, it is known that the better understood the mechanism is, the better the synthetic applications will be. Therefore, mechanistic appreciation of radical decarboxylation will be of great value, especially when merged with dual or combined catalytic systems.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ullmann, F. Ueber eine neue Bildungsweise von Diphenylaminderivaten. Ber. Dtsch. Chem. Ges. 1903, 36, 2382–2384. [Google Scholar] [CrossRef]
- Monnier, F.; Taillefer, M. Catalytic C–C, C–N, and C–O Ullmann–type coupling reactions. Angew. Chem. Int. Ed. 2009, 48, 6954–6971. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.M.T.; Monaco, K.L.; Wang, R.-P.; Winters, M.-P. New N– and O–arylations with phenylboronic acids and cupric acetate. Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar] [CrossRef]
- Lam, P.Y.S.; Clark, C.G.; Saubern, S.; Adams, J.; Winters, M.P.; Chan, D.M.T.; Combs, A. New aryl/heteroaryl C–N bond cross–coupling reactions via arylboronic acid/cupric acetate arylation. Tetrahedron Lett. 1998, 39, 2941–2944. [Google Scholar] [CrossRef]
- Guram, A.S.; Rennels, R.A.; Buchwald, S.L. A Simple Catalytic Method for the Conversion of Aryl Bromides to Arylamines. Angew. Chem. Int. Ed. 1995, 34, 1348–1350. [Google Scholar] [CrossRef]
- Louie, J.; Hartwig, J.F. Palladium–catalyzed synthesis of arylamines from aryl halides. Mechanistic studies lead to coupling in the absence of tin reagents. Tetrahedron Lett. 1995, 36, 3609–3612. [Google Scholar] [CrossRef]
- Kolbe, H. Zersetzung der Valeriansäure durch den elektrischen Strom. Justus Liebigs Ann. Chem. 1848, 64, 339–341. [Google Scholar]
- Kolbe, H. Untersuchungen über die Elektrolyse organischer Verbindungen. Justus Liebigs Ann. Chem. 1849, 69, 257–294. [Google Scholar] [CrossRef]
- Ugi, I.; Steinbrückner, C. Über ein neues Kondensations–Prinzip. Angew. Chem. 1960, 72, 267–268. [Google Scholar] [CrossRef]
- Curtius, T. Ueber Stickstoffwasserstoffsäure (Azoimid) N3H. Ber. Dtsch. Chem. Ges. 1890, 23, 3023–3033. [Google Scholar] [CrossRef]
- Curtius, T. 20. Hydrazide und Azide organischer Säuren I. Abhandlung. J. Prakt. Chem. 1894, 50, 275–294. [Google Scholar] [CrossRef]
- Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M. Nucleophilic character of alkyl radicals—VI. Tetrahedron 1971, 27, 3575–3579. [Google Scholar] [CrossRef]
- Corey, E.J.; Nicolaou, K.C. Efficient and mild lactonization method for the synthesis of macrolides. J. Am. Chem. Soc. 1974, 96, 5614–5616. [Google Scholar] [CrossRef]
- Cory, E.J.; Nicolaou, K.C.; Melvin, L.S., Jr. Letter: Synthesis of novel macrocyclic lactones in the prostaglandin and polyether antibiotic series. J. Am. Chem. Soc. 1975, 97, 653–654. [Google Scholar] [CrossRef]
- Xuan, J.; Zhang, Z.G.; Xiao, W.J. Visible–Light–Induced Decarboxylative Functionalization of Carboxylic Acids and Their Derivatives. Angew. Chem. Int. Ed. 2015, 54, 15632–15641. [Google Scholar] [CrossRef]
- Konev, M.O.; Jarvo, E.R. Decarboxylative Alkyl–Alkyl Cross–Coupling Reactions. Angew. Chem. Int. Ed. 2016, 55, 11340–11342. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, G.; Sun, P. Transition metal–free decarboxylative alkylation reactions. Org. Biomol. Chem. 2016, 14, 10763–10777. [Google Scholar] [CrossRef]
- Tóth, B.L.; Tischler, O.; Novák, Z. Recent advances in dual transition metal–visible light photoredox catalysis. Tetrahedron Lett. 2016, 57, 4505–4513. [Google Scholar] [CrossRef]
- Patra, T.; Maiti, D. Decarboxylation as the Key Step in C–C Bond–Forming Reactions. Chem. Eur. J. 2017, 23, 7382–7401. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Ozbalik, N.; Vacher, B. The invention of radical reactions part XVIII. A convenient solution to the 1–carbon problem (R–CO2H → R–13CO2H). Tetrahedron 1988, 44, 3501–3512. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Jaszberenyi, J.C.; Theodorakis, E.A.; Reibenspies, J.H. The invention of radical reactions. 30. Diazirines as carbon radical traps. Mechanistic aspects and synthetic applications of a novel and efficient amination process. J. Am. Chem. Soc. 1993, 115, 8050–8059. [Google Scholar] [CrossRef]
- Masterson, D.S.; Porter, N.A. Diastereoselective free radical halogenation, azidation, and rearrangement of beta–silyl Barton esters. Org. Lett. 2002, 4, 4253–4256. [Google Scholar] [CrossRef]
- Nyfeler, E.; Renaud, P. Decarboxylative radical azidation using MPDOC and MMDOC esters. Org. Lett. 2008, 10, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Mitsunobu, O.; Yamada, M.; Mukaiyama, T. Preparation of Esters of Phosphoric Acid by the Reaction of Trivalent Phosphorus Compounds with Diethyl Azodicarboxylate in the Presence of Alcohols. Bull. Chem. Soc. Jpn. 1967, 40, 935–939. [Google Scholar] [CrossRef]
- Arshadi, S.; Ebrahimiasl, S.; Hosseinian, A.; Monfared, A.; Vessally, E. Recent developments in decarboxylative cross–coupling reactions between carboxylic acids and N–H compounds. RSC Adv. 2019, 9, 8964–8976. [Google Scholar] [CrossRef]
- Singh, S.; Roy, V.J.; Dagar, N.; Sen, P.P.; Roy, S.R. Photocatalysis in Dual Catalysis Systems for Carbon–Nitrogen Bond Formation. Adv. Synth. Catal. 2020, 363, 937–979. [Google Scholar] [CrossRef]
- Yu, W.-Y.; Chan, C.-M.; Chow, Y.-C. Recent Advances in Photocatalytic C–N Bond Coupling Reactions. Synthesis 2020, 52, 2899–2921. [Google Scholar] [CrossRef]
- Zeng, Z.; Feceu, A.; Sivendran, N.; Gooßen, L.J. Decarboxylation–Initiated Intermolecular Carbon–Heteroatom Bond Formation. Adv. Synth. Catal. 2021, 363, 2678–2722. [Google Scholar] [CrossRef]
- Rivas, M.; Palchykov, V.; Jia, X.; Gevorgyan, V. Recent advances in visible light–induced C(sp3)–N bond formation. Nat. Rev. Chem. 2022, 6, 544–561. [Google Scholar] [CrossRef]
- Goossen, L.J.; Rodriguez, N.; Goossen, K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 2008, 47, 3100–3120. [Google Scholar] [CrossRef]
- Rodriguez, N.; Goossen, L.J. Decarboxylative coupling reactions: A modern strategy for C–C–bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [Google Scholar] [CrossRef]
- Shang, R.; Liu, L. Transition metal–catalyzed decarboxylative cross–coupling reactions. Sci. China Chem. 2011, 54, 1670–1687. [Google Scholar] [CrossRef]
- Weaver, J.D.; Recio, A., 3rd; Grenning, A.J.; Tunge, J.A. Transition metal–catalyzed decarboxylative allylation and benzylation reactions. Chem. Rev. 2011, 111, 1846–1913. [Google Scholar] [CrossRef]
- Dzik, W.I.; Lange, P.P.; Goossen, L.J. Carboxylates as sources of carbon nucleophiles and electrophiles: Comparison of decarboxylative and decarbonylative pathways. Chem. Sci. 2012, 3, 2671–2678. [Google Scholar] [CrossRef]
- Larrosa, I.; Cornella, J. Decarboxylative Carbon–Carbon Bond–Forming Transformations of (Hetero)aromatic Carboxylic Acids. Synthesis 2012, 44, 653–676. [Google Scholar] [CrossRef]
- Park, K.; Lee, S. Transition metal–catalyzed decarboxylative coupling reactions of alkynyl carboxylic acids. RSC Adv. 2013, 3, 14165–14182. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T.S.; Liu, X. Recent advances in C–S bond formation via C–H bond functionalization and decarboxylation. Chem. Soc. Rev. 2015, 44, 291–314. [Google Scholar] [CrossRef]
- Yin, X.; Li, W.; Zhao, B.; Cheng, K. Research Progress on Silver–Catalyzed Decarboxylative Coupling Reaction. Chin. J. Org. Chem. 2018, 38, 2879–2887. [Google Scholar] [CrossRef]
- Jamison, C.R.; Overman, L.E. Fragment Coupling with Tertiary Radicals Generated by Visible–Light Photocatalysis. Acc. Chem. Res. 2016, 49, 1578–1586. [Google Scholar] [CrossRef]
- Marzo, L.; Pagire, S.K.; Reiser, O.; Konig, B. Visible–Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef]
- Murarka, S. N–(Acyloxy)phthalimides as Redox–Active Esters in Cross–Coupling Reactions. Adv. Synth. Catal. 2018, 360, 1735–1753. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, L.; Zheng, Y.; Shao, X.; Ramadoss, V. Recent Developments in Photochemical and Electrochemical Decarboxylative C(sp3)–N Bond Formation. Synthesis 2020, 52, 1357–1368. [Google Scholar]
- Parida, S.K.; Mandal, T.; Das, S.; Hota, S.K.; De Sarkar, S.; Murarka, S. Single Electron Transfer–Induced Redox Processes Involving N–(Acyloxy)phthalimides. ACS Catal. 2021, 11, 1640–1683. [Google Scholar] [CrossRef]
- Kitcatt, D.M.; Nicolle, S.; Lee, A.L. Direct decarboxylative Giese reactions. Chem. Soc. Rev. 2022, 51, 1415–1453. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, X.; Li, Z.; Cui, L.; Li, C. Silver–Catalyzed Decarboxylative Radical Azidation of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc. 2015, 137, 9820–9823. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; Wang, X.; Huang, X.; Shen, T.; Zhang, Y.; Sun, X.; Zou, M.; Song, S.; Jiao, N. Silver–Catalyzed Decarboxylative Azidation of Aliphatic Carboxylic Acids. Org. Lett. 2015, 17, 4702–4705. [Google Scholar] [CrossRef]
- Agasti, S.; Maiti, S.; Maity, S.; Anniyappan, M.; Talawar, M.B.; Maiti, D. Bismuth nitrate as a source of nitro radical in ipso–nitration of carboxylic acids. Polyhedron 2019, 172, 120–124. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, C.; Wang, Q. Copper–Catalyzed Decarboxylative Functionalization of Conjugated beta, gamma–Unsaturated Carboxylic Acids. ACS Catal. 2020, 10, 13179–13185. [Google Scholar] [CrossRef]
- Fang, Z.; Feng, Y.; Dong, H.; Li, D.; Tang, T. Copper(i)–catalyzed radical decarboxylative imidation of carboxylic acids with N–fluoroarylsulfonimides. Chem. Commun. 2016, 52, 11120–11123. [Google Scholar] [CrossRef]
- Li, Q.Y.; Gockel, S.N.; Lutovsky, G.A.; DeGlopper, K.S.; Baldwin, N.J.; Bundesmann, M.W.; Tucker, J.W.; Bagley, S.W.; Yoon, T.P. Decarboxylative cross–nucleophile coupling via ligand–to–metal charge transfer photoexcitation of Cu(II) carboxylates. Nat. Chem. 2022, 14, 94–99. [Google Scholar] [CrossRef]
- Soorukram, D.; Phae–nok, S.; Kuhakarn, C.; Leowanawat, P.; Reutrakul, V. Decarboxylation of Paraconic Acids by a Silver(I) Nitrate/Persulfate Combination: An Entry to β–Nitro– and β–Hydroxy γ–Butyrolactones. Synlett 2022, 33, 1323–1328. [Google Scholar] [CrossRef]
- Kao, S.-C.; Bian, K.-J.; Chen, X.-W.; Chen, Y.; Martí, A.A.; West, J.G. Photochemical iron–catalyzed decarboxylative azidation via the merger of ligand–to–metal charge transfer and radical ligand transfer catalysis. Chem Catal. 2023, 3, 100603. [Google Scholar] [CrossRef]
- Marcote, D.C.; Street-Jeakings, R.; Dauncey, E.; Douglas, J.J.; Ruffoni, A.; Leonori, D. Photoinduced decarboxylative azidation of cyclic amino acids. Org. Biomol. Chem. 2019, 17, 1839–1842. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Q.; Yi, H.; Qin, C.; Bai, R.; Qi, X.; Lan, Y.; Lei, A. Visible–light–mediated decarboxylation/oxidative amidation of alpha–keto acids with amines under mild reaction conditions using O2. Angew. Chem. Int. Ed. 2014, 53, 502–506. [Google Scholar] [CrossRef]
- Xu, N.; Liu, J.; Li, D.; Wang, L. Synthesis of imides via palladium–catalyzed decarboxylative amidation of alpha–oxocarboxylic acids with secondary amides. Org. Biomol. Chem. 2016, 14, 4749–4757. [Google Scholar] [CrossRef]
- Pimpasri, C.; Sumunnee, L.; Yotphan, S. Copper–catalyzed oxidative decarboxylative coupling of alpha–keto acids and sulfoximines. Org. Biomol. Chem. 2017, 15, 4320–4327. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Nguyen, V.D.; Haug, G.C.; Vuong, N.T.H.; Dang, H.T.; Arman, H.D.; Larionov, O.V. Visible–Light–Enabled Direct Decarboxylative N–Alkylation. Angew. Chem. Int. Ed. 2020, 59, 7921–7927. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Nguyen, V.D.; Haug, G.C.; Dang, H.T.; Jin, S.; Li, Z.; Flores-Hansen, C.; Benavides, B.S.; Arman, H.D.; Larionov, O.V. Alkene Synthesis by Photocatalytic Chemoenzymatically Compatible Dehydrodecarboxylation of Carboxylic Acids and Biomass. ACS Catal. 2019, 9, 9485–9498. [Google Scholar] [CrossRef]
- Li, P.; Zbieg, J.R.; Terrett, J.A. The Direct Decarboxylative N–Alkylation of Azoles, Sulfonamides, Ureas, and Carbamates with Carboxylic Acids via Photoredox Catalysis. Org. Lett. 2021, 23, 9563–9568. [Google Scholar] [CrossRef]
- Wang, S.; Li, T.; Gu, C.; Han, J.; Zhao, C.G.; Zhu, C.; Tan, H.; Xie, J. Decarboxylative tandem C–N coupling with nitroarenes via SH2 mechanism. Nat. Commun. 2022, 13, 2432. [Google Scholar] [CrossRef]
- Xiong, N.; Li, Y.; Zeng, R. Merging Photoinduced Iron–Catalyzed Decarboxylation with Copper Catalysis for C–N and C–C Couplings. ACS Catal. 2023, 13, 1678–1685. [Google Scholar] [CrossRef]
- Shao, X.; Zheng, Y.; Tian, L.; Martin-Torres, I.; Echavarren, A.M.; Wang, Y. Decarboxylative Csp(3)–N Bond Formation by Electrochemical Oxidation of Amino Acids. Org. Lett. 2019, 21, 9262–9267. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Zhang, H.J.; Shang, M.; He, C.; Vantourout, J.C.; Baran, P.S. Electrochemical Decarboxylative N–Alkylation of Heterocycles. Org. Lett. 2020, 22, 7594–7598. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Fu, N. Electrophotochemical Decarboxylative Azidation of Aliphatic Carboxylic Acids. ACS Catal. 2022, 12, 10661–10667. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, Z. Metal–free intramolecular oxidative decarboxylative amination of primary alpha–amino acids with product selectivity. Chem. Commun. 2011, 47, 9513–9515. [Google Scholar] [CrossRef]
- Lang, S.B.; Cartwright, K.C.; Welter, R.S.; Locascio, T.M.; Tunge, J.A. Photocatalytic Aminodecarboxylation of Carboxylic Acids. Eur. J. Org. Chem. 2016, 2016, 3331–3334. [Google Scholar] [CrossRef]
- Shi, J.; Yuan, T.; Wang, R.; Zheng, M.; Wang, X. Boron carbonitride photocatalysts for direct decarboxylation: The construction of C(sp3)–N or C(sp3)–C(sp2) bonds with visible light. Green Chem. 2021, 23, 3945–3949. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Fu, H. Thiophenol–Catalyzed Visible–Light Photoredox Decarboxylative Couplings of N–(Acetoxy)phthalimides. Org. Lett. 2016, 18, 6400–6403. [Google Scholar] [CrossRef]
- Zhao, W.; Wurz, R.P.; Peters, J.C.; Fu, G.C. Photoinduced, Copper–Catalyzed Decarboxylative C–N Coupling to Generate Protected Amines: An Alternative to the Curtius Rearrangement. J. Am. Chem. Soc. 2017, 139, 12153–12156. [Google Scholar] [CrossRef]
- Mao, R.; Frey, A.; Balon, J.; Hu, X. Decarboxylative C(sp3)–N cross–coupling via synergetic photoredox and copper catalysis. Nat. Catal. 2018, 1, 120–126. [Google Scholar] [CrossRef]
- Mao, R.; Balon, J.; Hu, X. Cross–Coupling of Alkyl Redox–Active Esters with Benzophenone Imines: Tandem Photoredox and Copper Catalysis. Angew. Chem. Int. Ed. 2018, 57, 9501–9504. [Google Scholar] [CrossRef]
- Bosque, I.; Bach, T. 3–Acetoxyquinuclidine as Catalyst in Electron Donor–Acceptor Complex–Mediated Reactions Triggered by Visible Light. ACS Catal. 2019, 9, 9103–9109. [Google Scholar] [CrossRef]
- Chan, C.M.; Xing, Q.; Chow, Y.C.; Hung, S.F.; Yu, W.Y. Photoredox Decarboxylative C(sp(3))–N Coupling of alpha–Diazoacetates with Alkyl N–Hydroxyphthalimide Esters for Diversified Synthesis of Functionalized N–Alkyl Hydrazones. Org. Lett. 2019, 21, 8037–8043. [Google Scholar] [CrossRef]
- Barzano, G.; Mao, R.; Garreau, M.; Waser, J.; Hu, X. Tandem Photoredox and Copper–Catalyzed Decarboxylative C(sp(3))–N Coupling of Anilines and Imines Using an Organic Photocatalyst. Org. Lett. 2020, 22, 5412–5416. [Google Scholar] [CrossRef]
- Chandrachud, P.P.; Wojtas, L.; Lopchuk, J.M. Decarboxylative Amination: Diazirines as Single and Double Electrophilic Nitrogen Transfer Reagents. J. Am. Chem. Soc. 2020, 142, 21743–21750. [Google Scholar] [CrossRef]
- Chen, K.Q.; Wang, Z.X.; Chen, X.Y. Photochemical Decarboxylative C(sp(3))–X Coupling Facilitated by Weak Interaction of N–Heterocyclic Carbene. Org. Lett. 2020, 22, 8059–8064. [Google Scholar] [CrossRef]
- Kobayashi, R.; Shibutani, S.; Nagao, K.; Ikeda, Z.; Wang, J.; Ibanez, I.; Reynolds, M.; Sasaki, Y.; Ohmiya, H. Decarboxylative N–Alkylation of Azoles through Visible–Light–Mediated Organophotoredox Catalysis. Org. Lett. 2021, 23, 5415–5419. [Google Scholar] [CrossRef]
- Shu, X.; Xu, R.; Liao, S. Photocatalytic divergent decarboxylative amination: A metal–free access to aliphatic amines and hydrazines. Sci. China Chem. 2021, 64, 1756–1762. [Google Scholar] [CrossRef]
- Liu, Z.J.; Lu, X.; Wang, G.; Li, L.; Jiang, W.T.; Wang, Y.D.; Xiao, B.; Fu, Y. Directing Group in Decarboxylative Cross–Coupling: Copper–Catalyzed Site–Selective C–N Bond Formation from Nonactivated Aliphatic Carboxylic Acids. J. Am. Chem. Soc. 2016, 138, 9714–9719. [Google Scholar] [CrossRef]
- Yang, Y.-N.; Jiang, J.-L.; Shi, J. Mechanistic Study of Copper–Catalyzed Decarboxylative C–N Cross–Coupling with Hypervalent Iodine Oxidant. Organometallics 2017, 36, 2081–2087. [Google Scholar] [CrossRef]
- Kiyokawa, K.; Watanabe, T.; Fra, L.; Kojima, T.; Minakata, S. Hypervalent Iodine(III)–Mediated Decarboxylative Ritter–Type Amination Leading to the Production of alpha–Tertiary Amine Derivatives. J. Org. Chem. 2017, 82, 11711–11720. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, X.; MacMillan, D.W.C. Decarboxylative sp(3) C–N coupling via dual copper and photoredox catalysis. Nature 2018, 559, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Li, R.H.; Wang, S.; Zhao, Z.W.; Geng, Y.; Wang, X.L.; Su, Z.M.; Guan, W. Springboard Role for Iridium Photocatalyst: Theoretical Insight of C(sp3)–N Cross–Coupling by Photoredox–Mediated Iridium/Copper Dual Catalysis versus Single–Copper Catalysis. ChemCatChem 2022, 14, e202101737. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Ito, E.; Fukushima, T.; Murakami, K.; Itami, K. Late–Stage Functionalization of Arylacetic Acids by Photoredox–Catalyzed Decarboxylative Carbon–Heteroatom Bond Formation. Chem. Eur. J. 2018, 24, 9254–9258. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, M.; Shi, Z. Single–Electron–Transfer–Induced C(sp(3))–N Couplings via C–C Bond Cleavage of Cycloketoxime Esters. J. Org. Chem. 2019, 84, 10145–10159. [Google Scholar] [CrossRef]
- Min, Q.-Q.; Lin, N.; Chen, G.-L.; Liu, F. Copper–catalysed C(sp3)–N coupling initiated by selective C–C bond cleavage of cyclobutanone oxime esters. Org. Chem. Front. 2019, 6, 1200–1204. [Google Scholar] [CrossRef]
- Tian, L.; Gao, S.; Wang, R.; Li, Y.; Tang, C.; Shi, L.; Fu, J. Copper–catalyzed ring–opening C(sp(3))–N coupling of cycloketone oxime esters: Access to 1 degrees, 2 degrees and 3 degrees alkyl amines. Chem. Commun. 2019, 55, 5347–5350. [Google Scholar] [CrossRef]
- Soni, V.K.; Lee, S.; Kang, J.; Moon, Y.K.; Hwang, H.S.; You, Y.; Cho, E.J. Reactivity Tuning for Radical–Radical Cross–Coupling via Selective Photocatalytic Energy Transfer: Access to Amine Building Blocks. ACS Catal. 2019, 9, 10454–10463. [Google Scholar] [CrossRef]
- Tu, J.L.; Liu, J.L.; Tang, W.; Su, M.; Liu, F. Radical Aza–Cyclization of alpha–Imino–oxy Acids for Synthesis of Alkene–Containing N–Heterocycles via Dual Cobaloxime and Photoredox Catalysis. Org. Lett. 2020, 22, 1222–1226. [Google Scholar] [CrossRef]
- Tang, Z.L.; Ouyang, X.H.; Song, R.J.; Li, J.H. Decarboxylative C(sp(3))–N Cross–Coupling of Diacyl Peroxides with Nitrogen Nucleophiles. Org. Lett. 2021, 23, 1000–1004. [Google Scholar] [CrossRef]
- Wang, K.; Li, Y.; Li, X.; Li, D.; Bao, H. Iron–Catalyzed Asymmetric Decarboxylative Azidation. Org. Lett. 2021, 23, 8847–8851. [Google Scholar] [CrossRef]









































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Yuan, X.; Hu, J.; Li, Y.; Bao, H. Radical Decarboxylative Carbon–Nitrogen Bond Formation. Molecules 2023, 28, 4249. https://doi.org/10.3390/molecules28104249
Li X, Yuan X, Hu J, Li Y, Bao H. Radical Decarboxylative Carbon–Nitrogen Bond Formation. Molecules. 2023; 28(10):4249. https://doi.org/10.3390/molecules28104249
Chicago/Turabian StyleLi, Xiangting, Xiaobin Yuan, Jiahao Hu, Yajun Li, and Hongli Bao. 2023. "Radical Decarboxylative Carbon–Nitrogen Bond Formation" Molecules 28, no. 10: 4249. https://doi.org/10.3390/molecules28104249
APA StyleLi, X., Yuan, X., Hu, J., Li, Y., & Bao, H. (2023). Radical Decarboxylative Carbon–Nitrogen Bond Formation. Molecules, 28(10), 4249. https://doi.org/10.3390/molecules28104249