

Co-Crystallization between Aliphatic Polyesters through Co-Inclusion Complexation with Small Molecule

Abstract
1. Introduction
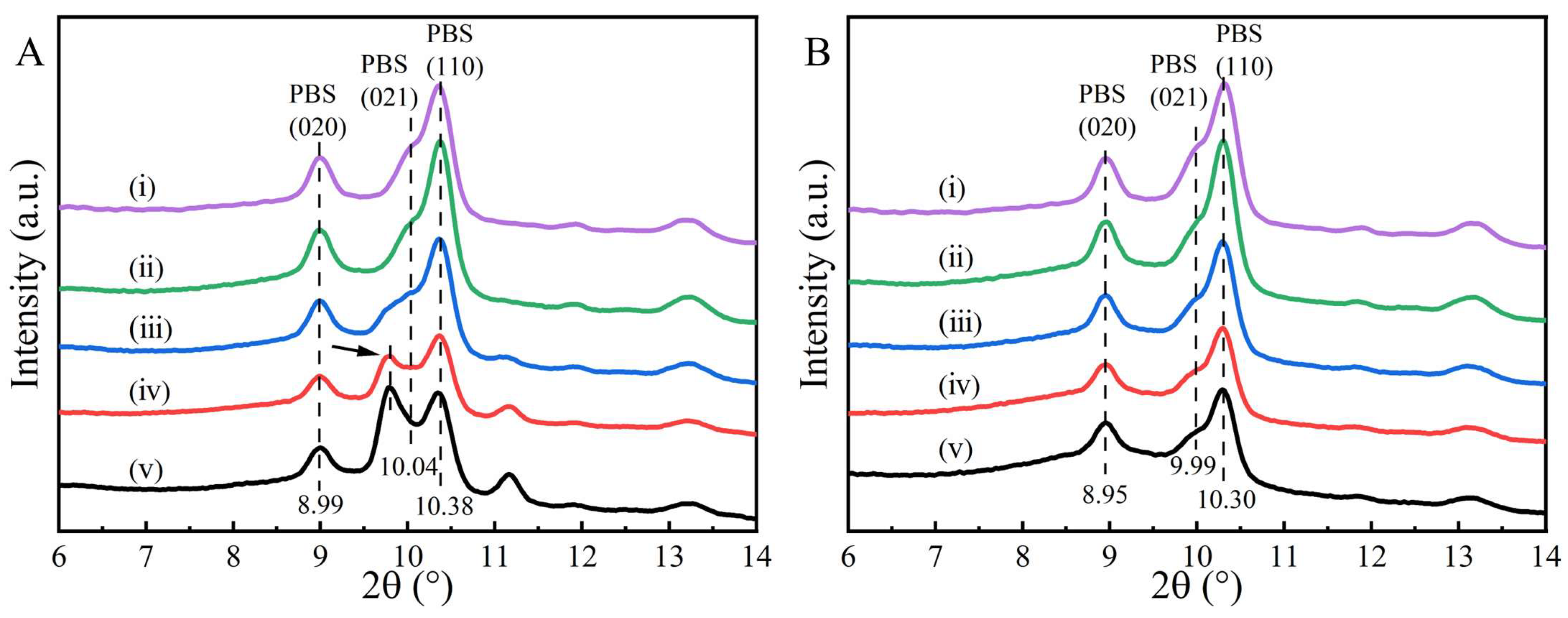
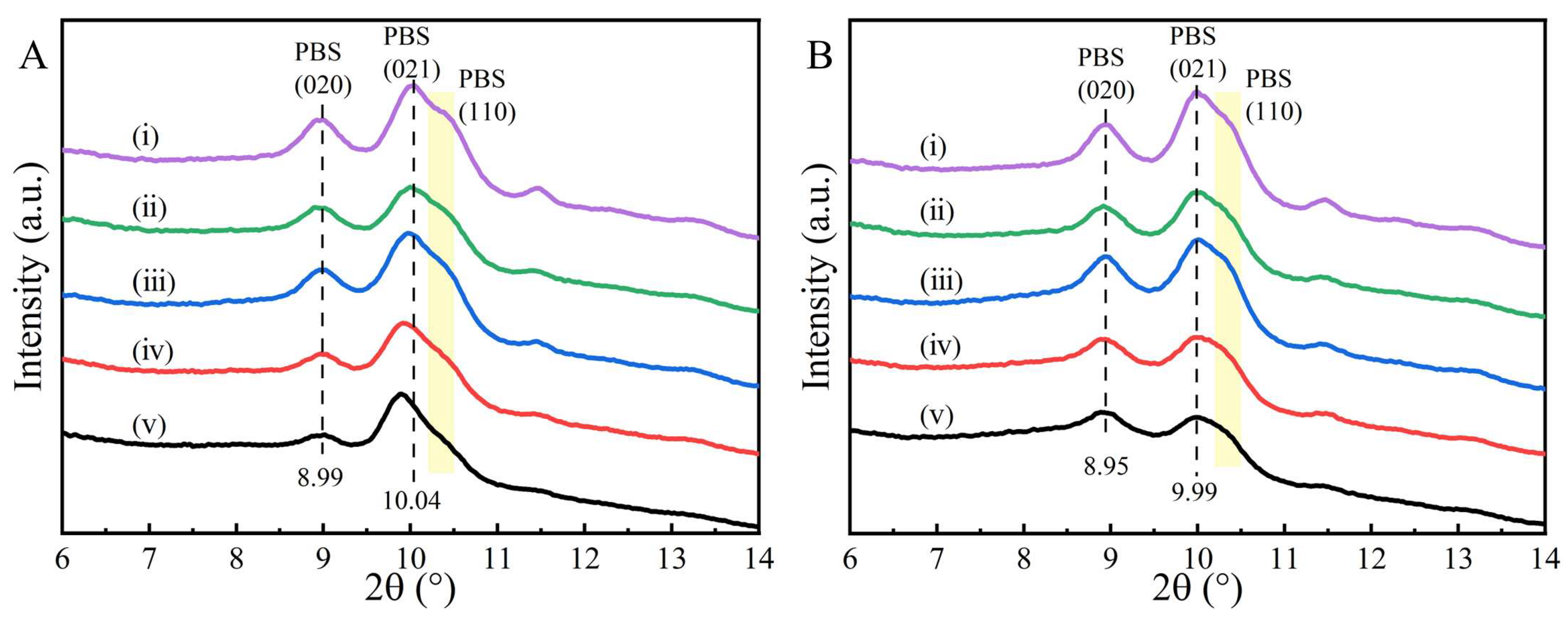
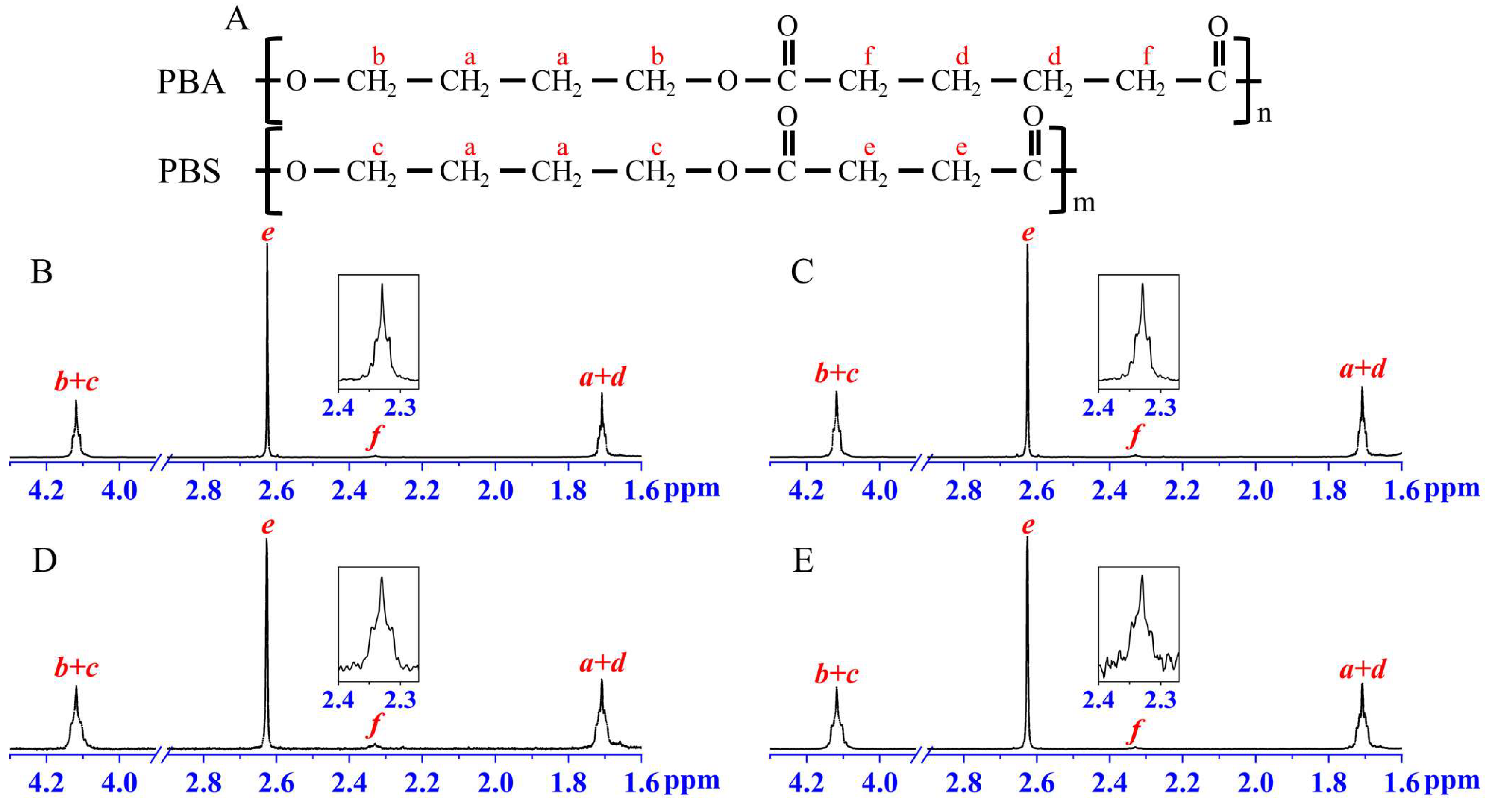
2. Results and Discussion
3. Experimental
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wang, M.T.; Qiu, Z.B. Unusual Fractional Crystallization Behavior of Novel Crystalline/Crystalline Polymer Blends of Poly(ethylene suberate) and Poly(ethylene oxide) with Similar Melting Points. Macromolecules 2014, 47, 8351–8358. [Google Scholar] [CrossRef]
- Yang, J.J.; Pan, P.J.; Hua, L.; Xie, Y.H.; Dong, T.; Zhu, B.; Inoue, Y.; Feng, X. Fractionated crystallization, polymorphic crystalline structure, and spherulite morphology of poly(butylene adipate) in its miscible blend with poly(butylene succinate). Polymer 2011, 52, 3460–3468. [Google Scholar] [CrossRef]
- Jiang, N.; Abe, H. Morphological changes in poly(L-lactide)/poly(3-hydroxybutyrate-co-3-hydroxyvalerate) blends induced by different miscibility. Polymer 2015, 66, 259–267. [Google Scholar] [CrossRef]
- Yoshie, N.; Asaka, A.; Inoue, Y. Cocrystallization and Phase Segregation in Crystalline/Crystalline Polymer Blends of Bacterial Copolyesters. Macromolecules 2004, 37, 3770–3779. [Google Scholar] [CrossRef]
- Pucciariello, R.; Villani, V.; Ballesteros, O.R.D. Cocrystallization in Blends of Random Tetrafluoroethylene Fluorinated Copolymers: The Effect of the Chain Structure and Crystallization Conditions. J. Polym. Sci. B Polym. Phys. 2002, 40, 1477–1489. [Google Scholar] [CrossRef]
- Tsuji, K.; Tashiro, K.; Bouapao, L.; Hanesaka, M. Separate Crystallization and Cocrystallization of Poly(L-lactide) in the Presence of L-Lactide Based Copolymers with Low Crystallizability, Poly(L-lactide-co-glycolide) and Poly(L-lactide-co-D-lactide). Macromol. Chem. Phys. 2012, 213, 2099–2112. [Google Scholar] [CrossRef]
- Chen, H.Y.; Cheung, Y.W.; Hiltner, A.; Baer, E. Miscibility of ethylene-styrene copolymer blends. Polymer 2001, 42, 7819–7830. [Google Scholar] [CrossRef]
- Yu, P.Q.; Yan, L.T.; Chen, N.; Xie, X.M. Confined crystallization behaviors and phase morphologies of PVCH-PE-PVCH/PE homopolymer blends. Polymer 2012, 53, 4727–4736. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Nandan, B.; Chen, H.L.; Liao, C.S.; Jeng, U.S. Cocrystallization Behavior in Binary Blend of Crystalline-Amorphous Diblock Copolymers. Macromolecules 2004, 37, 8175–8179. [Google Scholar] [CrossRef]
- Reddy, K.R.; Tashiro, K.; Sakurai, T.; Yamaguchi, N. Cocrystallization Phenomenon between the H and D Species of Isotactic Polypropylene Blends As Revealed by Thermal and Infrared Spectroscopic Analyses for a Series of D/H Blend Samples. Macromolecules 2008, 41, 9807–9813. [Google Scholar] [CrossRef]
- Reddy, K.R.; Tashiro, K.; Sakurai, T.; Yamaguchi, N.; Sasaki, S.; Masunaga, H.; Takata, M. Isothermal Crystallization Behavior of Isotactic Polypropylene H/D Blends as Viewed from Time-Resolved FTIR and Synchrotron SAXS/WAXD Measurements. Macromolecules 2009, 42, 4191–4199. [Google Scholar] [CrossRef]
- Shin, T.J.; Lee, B.; Seong, B.S.; Han, Y.S.; Lee, C.H.; Song, H.H.; Stein, R.S.; Ree, M. Composition-dependent phase segregation and cocrystallization behaviors of blends of metallocene-catalyzed octene-LLDPE(D) and LDPE(H). Polymer 2010, 51, 5799–5806. [Google Scholar] [CrossRef]
- Kongkhlang, T.; Reddy, K.R.; Kitano, T.; Nishu, T.; Tashiro, K. Cocrystallization phenomenon of polyoxymethylene blend samples between the deuterated and hydrogenated species. Polym. J. 2011, 43, 66–73. [Google Scholar] [CrossRef]
- Tashiro, K.; Gose, N. Diffusion and aggregation of hydrogeneous and deuterated polyethylene chains at their interfacial boundary as studied by tine- and space- resolved FTIR microscopic measurements. Polymer 2001, 42, 8987–8998. [Google Scholar] [CrossRef]
- Reddy, K.R.; Tashiro, K.; Sakurai, T.; Yamaguchi, N. Isotope Effect on the Isothermal Crystallization Behavior of Isotactic Polypropylene Blends between the Deuterated and Hydrogenated Species. Macromolecules 2004, 42, 1672–1678. [Google Scholar] [CrossRef]
- Safari, M.; Otaegi, I.; Aramburu, N.; Wang, Y.; Liu, G.; Dong, X.; Wang, D.; Guerrica-Echevarria, G.; Müller, A.J. Composition Dependent Miscibility in the Crystalline State of Polyamide 6/Polyamide 4,10 Blends: From Single to Double Crystalline Blends. Polymer 2021, 219, 123570. [Google Scholar] [CrossRef]
- Wei, X.W.; Yang, L.L.; Li, Y.; Meng, X.Y.; Cai, L.H.; Zhou, Q.; Ye, H.M. Asymmetrical formation of isomorphism in the crystalline/crystalline blend of poly(butylene succinate) and poly(butylene fumarate). Polymer 2021, 235, 124282. [Google Scholar] [CrossRef]
- Vasanthan, N.; Shin, I.D.; Tonelli, A.E. Structure, Conformation, and Motions of Poly(ethylene oxide) and Poly(ethylene glycol) in Their Urea Inclusion Compounds. Macromolecules 1996, 29, 263–267. [Google Scholar] [CrossRef]
- Huh, K.M.; Ooya, T.; Sasaki, S.; Yui, N. Polymer Inclusion Complex Consisting of Poly(ε-lysine) and α-Cyclodextrin. Macromolecules 2001, 34, 2402–2404. [Google Scholar] [CrossRef]
- Tonelli, A.E. Restructuring polymers via nanoconfinement and subsequent release. Beilstein J. Org. Chem. 2012, 8, 1318–1332. [Google Scholar] [CrossRef]
- Rusa, C.C.; Tonelli, A.E. Polymer/Polymer Inclusion Compounds as a Novel Approach To Obtaining a PLLA/PCL Intimately Compatible Blend. Macromolecules 2000, 33, 5321–5324. [Google Scholar] [CrossRef]
- Wei, M.; Tonelli, A.E. Compatiblization of Polymers via Coalescence from Their Common Cyclodextrin Inclusion Compounds. Macromolecules 2001, 34, 4061–4065. [Google Scholar] [CrossRef]
- Shuai, X.T.; Porbeni, F.E.; Wei, M.; Bullions, T.; Tonelli, A.E. Formation of Inclusion Complexes of Poly(3-hydroxybutyrate)s with Cyclodextrins. 1. Immobilization of Atactic Poly(R,S-3-hydroxybutyrate) and Miscibility Enhancement between Poly(R,S-3-hydroxybutyrate) and Poly(ε-caprolactone). Macromolecules 2002, 35, 3126–3132. [Google Scholar] [CrossRef]
- Bullions, T.A.; Edeki, E.M.; Porbeni, F.E.; Wei, M.; Shuai, X.; Rusa, C.C.; Tonelli, A.E. Intimate Blend of Poly(ethylene terephthalate) and Poly(ethylene 2,6-naphthalate) via Formation with and Coalescence from Their Common Inclusion Compound with γ-Cyclodextrin. J. Polym. Sci. Part B Polym. Phys. 2002, 41, 139–148. [Google Scholar] [CrossRef]
- Rusa, C.C.; Wei, M.; Shuai, X.T.; Bullions, T.A.; Wang, X.; Rusa, M.; Uyar, T.; Tonelli, A.E. Molecular Mixing of In Miscib Polymers through Formation of and Coalescence from Their Common Crystalline Cyclodextrin Inclusion Compounds. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4207–4224. [Google Scholar] [CrossRef]
- Rusa, C.C.; Uyar, T.; Rusa, M.; Hunt, M.A.; Wang, X.W.; Tonelli, A.E. An Intimate Polycarbonate/Poly(methyl methacrylate)/Poly(vinyl acetate) Ternary Blend via Coalescence from Their Common Inclusion Compound with γ-Cyclodextrin. J. Polym. Sci. Part B Polym. Phys. 2004, 42, 4182–4194. [Google Scholar] [CrossRef]
- Carrasco, L.; Gargallo, L.; Deodato, R. Study of Miscibility in Polymer Blends Obtained from Binary Inclusion Complexes of γ-Cyclodextrin and Polycarbonate/Poly(Ethylene Terephthalate). J. Macromol. Sci. Part B Phys. 2012, 51, 1750–1765. [Google Scholar] [CrossRef]
- Liu, P.; Chen, X.T.; Ye, H.M. Enhancing Stereocomplexation Ability of Polylactide by Coalescing from Its Inclusion Complex with Urea. Polymers 2017, 9, 592. [Google Scholar] [CrossRef]
- Wang, H.J.; Feng, H.P.; Wang, W.X.; Guo, P.Y.; Zhao, T.S.; Ren, L.F.; Qiang, X.H. Effects of Crystallization Temperature and Blend Ratio on the Crystal Structure of Poly(butylene adipate) in the Poly(butylene adipate)/Poly(butylene succinate) Blends. Chin. J. Polym. Sci. 2014, 32, 488–496. [Google Scholar] [CrossRef]
- Ye, H.M.; Chen, X.T.; Liu, P.; Wu, S.Y.; Jiang, Z.Y.; Xiong, B.J.; Xu, J. Preparation of Poly(butylene succinate) Crystals with Exceptionally High Melting Point and Crystallinity from Its Inclusion Complex. Macromolecules 2017, 50, 5425–5433. [Google Scholar] [CrossRef]
- Perez-Camargo, R.A.; Liu, G.M.; Cavallo, D.; Wang, D.J.; Muller, A.J. EEffect of the Crystallization Conditions on the Exclusion/Inclusion Balance in Biodegradable Poly(butylene succinate-ran-butylene adipate) Copolymers. Biomacromolecules 2020, 21, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Arandia, I.; Mugica, A.; Zubitur, M.; Arbe, A.; Liu, G.; Wang, D.; Mincheva, R.; Dubois, P.; Mueller, A.J. How Composition Determines the Properties of Isodimorphic Poly(butylene succinate-ran-butylene azelate) Random Biobased Copolymers: From Single to Double Crystalline Random Copolymers. Macromolecules 2015, 48, 43–57. [Google Scholar] [CrossRef]
- Hallstein, J.; Gomoll, A.; Lieske, A.; Buesse, T.; Balko, J.; Bruell, R.; Malz, F.; Metzsch-Zilligen, E.; Pfaendner, R.; Zehm, D. Unraveling the cause for the unusual processing behavior of commercial partially bio-based poly(butylene succinates) and their stabilization. J. Appl. Polym. Sci. 2021, 138, e50669. [Google Scholar] [CrossRef]
- Soni, R.S.; Singh, S.; Dutt, K. Studies on Synthesis and Characterization of N-Alkyl Terephthalamides Using Different Amines from Polyethylene Terephthalate Waste. J. Appl. Polym. Sci. 2010, 115, 3074–3080. [Google Scholar] [CrossRef]
- Lugito, G.; Wang, L.Y.; Woo, E.M. Chemical and Morphological Alterations Effected by Methylamine Reactions on Polyesters. Macromol. Chem. Phys. 2014, 215, 1297–1305. [Google Scholar] [CrossRef]
- Tian, Y.P.; Wu, T.; Meng, X.; Ye, H.M. Thermodynamic Features of Extended-Chain Crystals of Poly (butylene succinate) and Its Random Copolymers with Adipic Acid. Macromolecules 2022, 55, 5669–5674. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feeding (before) | 1H-NMR (after) |
|---|---|
| 10/90 | 3.4/96.6 |
| 20/80 | 3.7/96.3 |
| 30/70 | 4.0/96.0 |
| 40/60 | 6.1/93.9 |
| PBS/PBA | PBS Lamellar Crystal Proportion (%) | Tm of Co-Crystal (°C) | ||
|---|---|---|---|---|
| before | after | before | after | |
| 90/10 | 10.9 | 12.3 | 132.8 | 132.9 |
| 80/20 | 18.0 | 21.9 | 131.9 | 132.2 |
| 70/30 | 21.8 | 23.1 | 126.1 | 129.4 |
| 60/40 | 23.8 | 31.5 | 124.0 | 128.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-Y.; Zhang, X.-W.; Wu, T.-Y.; Ye, H.-M. Co-Crystallization between Aliphatic Polyesters through Co-Inclusion Complexation with Small Molecule. Molecules 2023, 28, 4091. https://doi.org/10.3390/molecules28104091
Chen J-Y, Zhang X-W, Wu T-Y, Ye H-M. Co-Crystallization between Aliphatic Polyesters through Co-Inclusion Complexation with Small Molecule. Molecules. 2023; 28(10):4091. https://doi.org/10.3390/molecules28104091
Chicago/Turabian StyleChen, Jia-Yao, Xue-Wen Zhang, Tian-Yu Wu, and Hai-Mu Ye. 2023. "Co-Crystallization between Aliphatic Polyesters through Co-Inclusion Complexation with Small Molecule" Molecules 28, no. 10: 4091. https://doi.org/10.3390/molecules28104091
APA StyleChen, J.-Y., Zhang, X.-W., Wu, T.-Y., & Ye, H.-M. (2023). Co-Crystallization between Aliphatic Polyesters through Co-Inclusion Complexation with Small Molecule. Molecules, 28(10), 4091. https://doi.org/10.3390/molecules28104091