
Synthesis and Antiproliferative Insights of Lipophilic Ru(II)-Hydroxy Stearic Acid Hybrid Species
 ,
,  ,
,  , ,
, ,  , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
2.2. Spectroscopic Studies
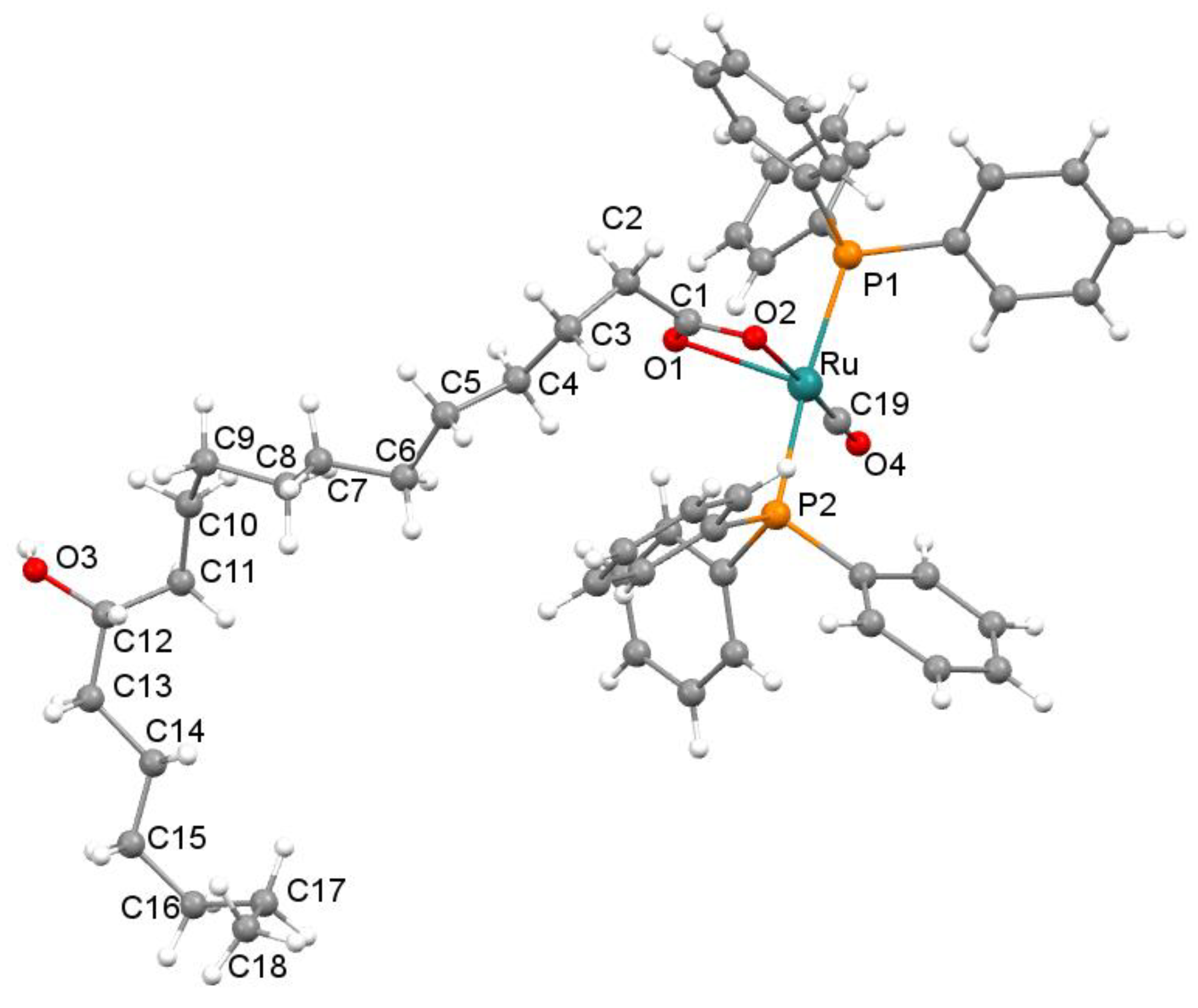
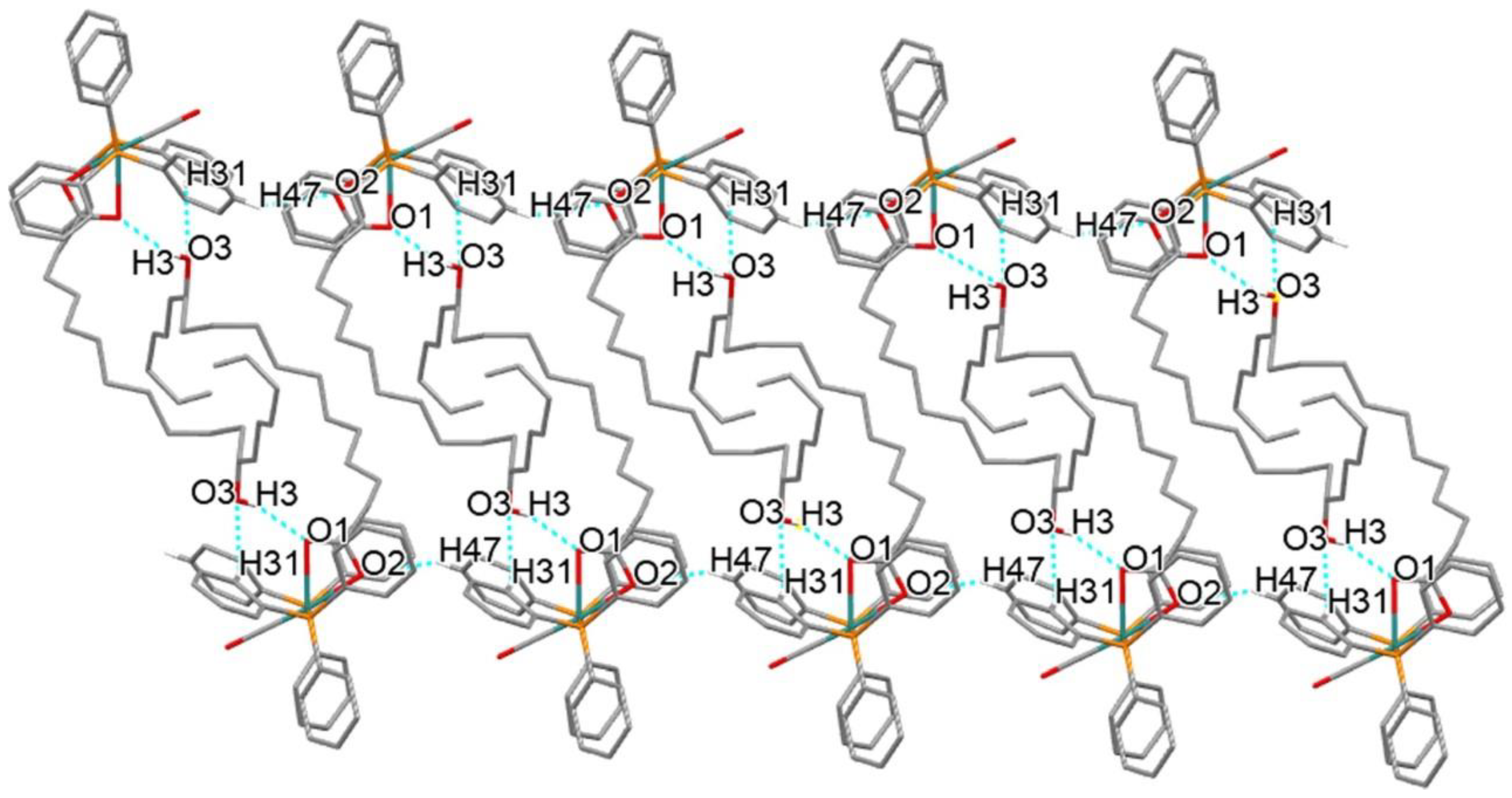
2.3. Crystal Structure Description of 4
2.4. Stability Studies in Solution
2.5. Lipophilicity Evaluation of 2–4
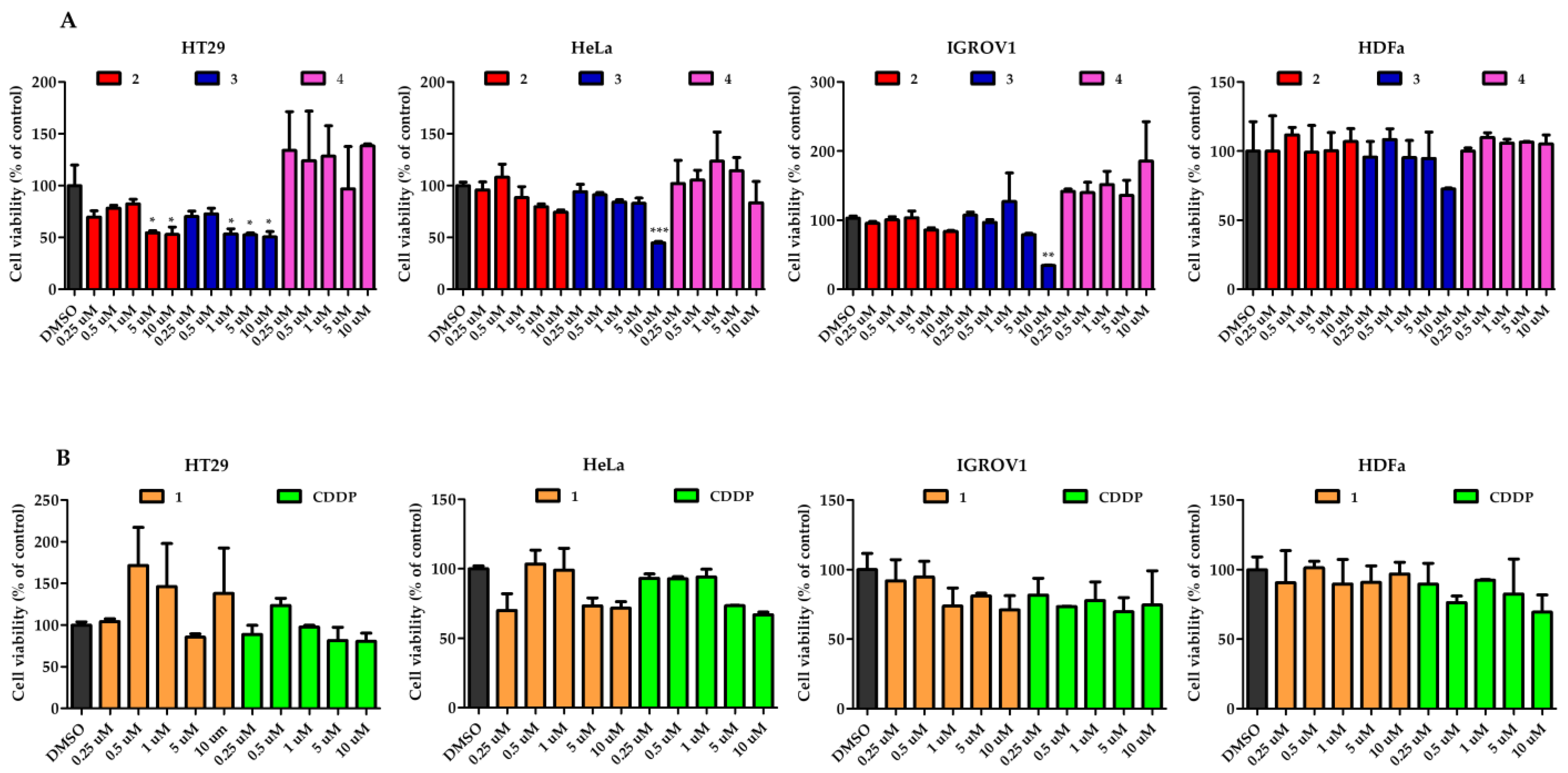
2.6. Antiproliferative Activity
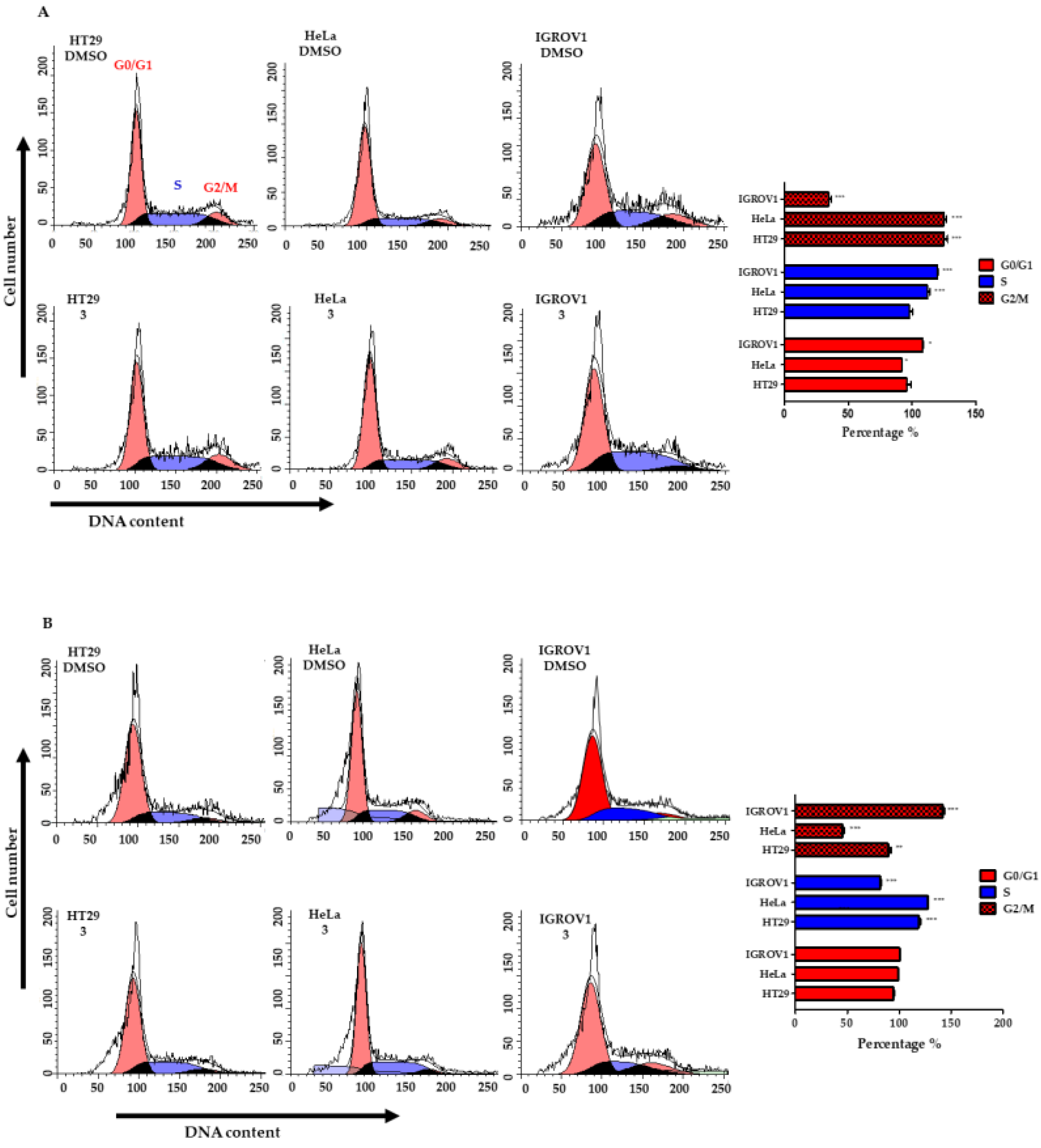
2.7. Cell Cycle Analysis by Quantification of DNA Content
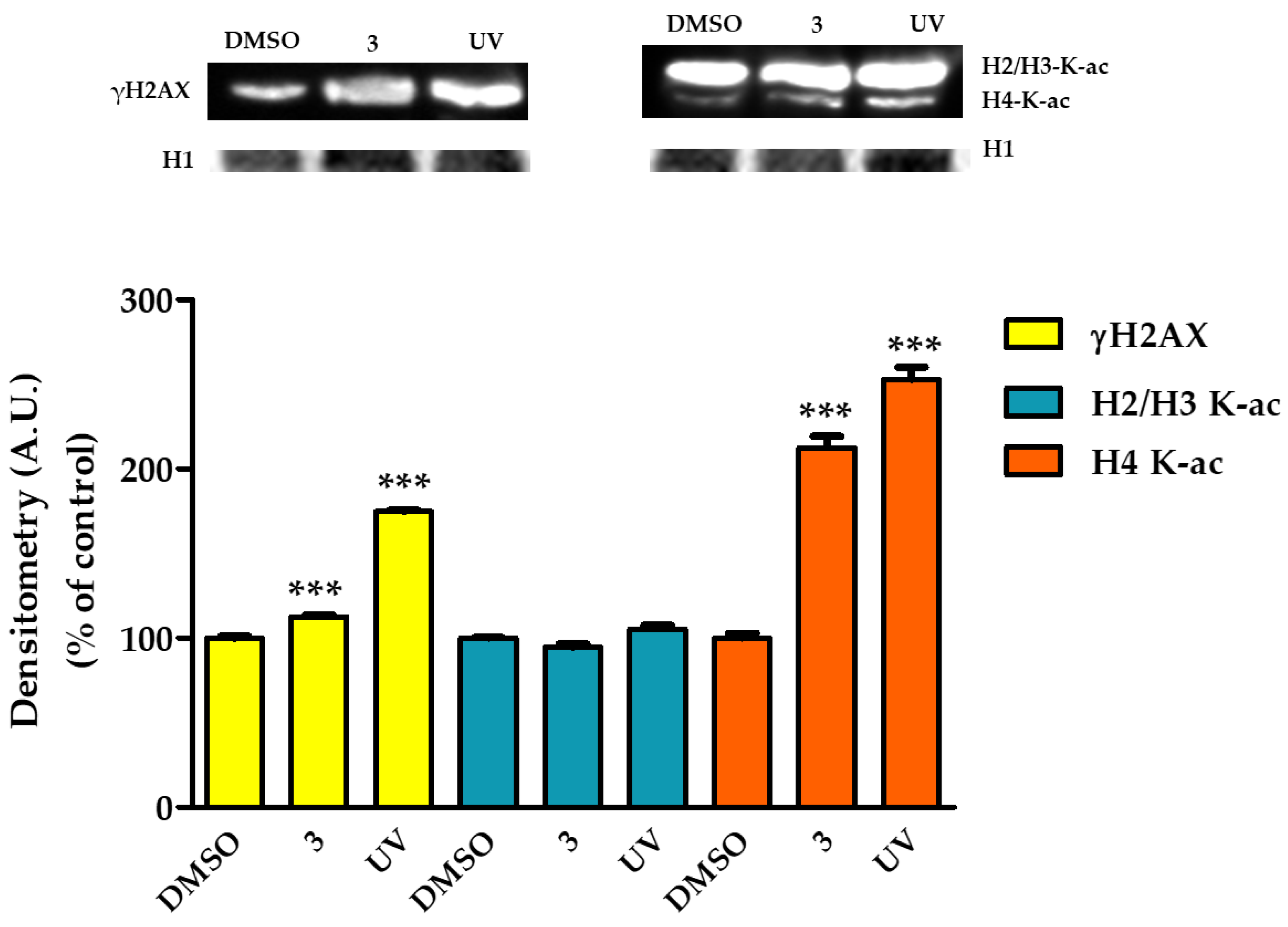
2.8. Complex 3 Induced DNA Damage in HT29 Cells
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. General
3.1.2. Synthesis of [RuH(CO)12-HSA(PPh3)2]
3.1.3. Synthesis of [RuH(CO)9-HSA(PPh3)2]
3.1.4. Synthesis of [RuH(CO)7-HSA(PPh3)2]
3.2. Lipophilicity Evaluation
3.3. X-ray Crystallography
3.4. Biology
3.4.1. Malignant and Non-Malignant Cells Culture
3.4.2. MTT Assay
3.4.3. Cell Cycle Analysis
3.4.4. Histone Post-Translational Modification
3.4.5. Statistical Analysis for Biological Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ohn, J.; Ailar, C.B.; Ornik, L.G. Cancer Undefeated. N. Engl. J. Med. 1997, 336, 1569–1574. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Yabroff, K.R.; Wu, X.C.; Negoita, S.; Stevens, J.; Coyle, L.; Zhao, J.; Mumphrey, B.J.; Jemal, A.; Ward, K.C. Association of the COVID-19 Pandemic With Patterns of Statewide Cancer Services. J. Natl. Cancer Inst. 2022, 114, 907–909. [Google Scholar] [CrossRef]
- Ghoshal, S.; Rigney, G.; Cheng, D.; Brumit, R.; Gee, M.S.; Hodin, R.A.; Lillemoe, K.D.; Levine, W.C.; Succi, M.D. Institutional Surgical Response and Associated Volume Trends Throughout the COVID-19 Pandemic and Postvaccination Recovery Period. JAMA Netw. Open. 2022, 5, E2227443. [Google Scholar] [CrossRef]
- Giorgi, E.; Binacchi, F.; Marotta, C.; Cirri, D.; Gabbiani, C.; Pratesi, A. Highlights of New Strategies to Increase the Efficacy of Transition Metal Complexes for Cancer Treatments. Molecules 2023, 2, 273. [Google Scholar] [CrossRef]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic Anticancer Compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef]
- Mbugua, S.N.; Sibuyi, N.R.S.; Njenga, L.W.; Odhiambo, R.A.; Wandiga, S.O.; Meyer, M.; Lalancette, R.A.; Onani, M.O. New Palladium(II) and Platinum(II) Complexes Based on Pyrrole Schiff Bases: Synthesis, Characterization, X-ray Structure, and Anticancer Activity. ACS Omega 2020, 5, 14942–14954. [Google Scholar] [CrossRef]
- Murray, B.S.; Dyson, P.J. Recent Progress in the Development of Organometallics for the Treatment of Cancer. Curr. Opin. Chem. Biol. 2020, 56, 28–34. [Google Scholar] [CrossRef]
- Yang, Z.; Jiang, G.; Xu, Z.; Zhao, S.; Liu, W. Advances in Alkynyl Gold Complexes for Use as Potential Anticancer Agents. Coord. Chem. Rev. 2020, 423, 213492. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin Reveals Therapeutic Anticancer Potential by Triggering Distinct Molecular Cell Death Mechanisms and Innate Immunity in Mutant P53 Non-Small Cell Lung Cancer. Redox Biol. 2021, 42, 101949. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Kasparkova, J. Ruthenium Coordination Compounds of Biological and Biomedical Significance. DNA Binding Agents. Coord. Chem. Rev. 2018, 376, 75–94. [Google Scholar] [CrossRef]
- Wee, H.A.; Dyson, P.J. Classical and Non-Classical Ruthenium-Based Anticancer Drugs: Towards Targeted Chemotherapy. Eur. J. Inorg. Chem. 2006, 2006, 4003–4018. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dorcier, A.; Scolaro, C.; Dyson, P.J. Development of Organometallic (Organo-Transition Metal) Pharmaceuticals. Appl. Organomet. Chem. 2005, 19, 1–10. [Google Scholar] [CrossRef]
- Muhammad, N.; Guo, Z. Metal-Based Anticancer Chemotherapeutic Agents. Curr. Opin. Chem. Biol. 2014, 19, 144–153. [Google Scholar] [CrossRef]
- Li, A.; Turro, C.; Kodanko, J.J. Ru(II) Polypyridyl Complexes Derived from Tetradentate Ancillary Ligands for Effective Photocaging. Acc. Chem. Res. 2018, 51, 1415–1421. [Google Scholar] [CrossRef]
- Gandioso, A.; Purkait, K.; Gasser, G. Recent Approaches towards the Development of Ru(Ii) Polypyridyl Complexes for Anticancer Photodynamic Therapy. Chimia 2021, 75, 845–855. [Google Scholar] [CrossRef]
- Conti, L.; Macedi, E.; Giorgi, C.; Valtancoli, B.; Fusi, V. Combination of Light and Ru(II) Polypyridyl Complexes: Recent Advances in the Development of New Anticancer Drugs. Coord. Chem. Rev. 2022, 469, 214656. [Google Scholar] [CrossRef]
- Ankathatti Munegowda, M.; Manalac, A.; Weersink, M.; McFarland, S.A.; Lilge, L. Ru(II) Containing Photosensitizers for Photodynamic Therapy: A Critique on Reporting and an Attempt to Compare Efficacy. Coord. Chem. Rev. 2022, 470, 214712. [Google Scholar] [CrossRef]
- Shumi, G.; Desalegn, T.; Demissie, T.B.; Ramachandran, V.P.; Eswaramoorthy, R. Metal Complexes in Target-Specific Anticancer Therapy: Recent Trends and Challenges. J. Chem. 2022, 2022, 9261683. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal Complexes in Cancer Therapy—An Update from Drug Design Perspective. Drug. Des. Devel Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Turel, I.; Kljun, J. Interactions of Metal Ions with DNA, Its Constituents and Derivatives, Which May Be Relevant for Anticancer Research. Curr. Top. Med. Chem. 2011, 11, 2661–2687. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Nováková, O. DNA Binding Mode of Ruthenium Complexes and Relationship to Tumor Cell Toxicity. Drug. Resist. Updates 2006, 9, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Bratsos, I.; Jedner, S.; Gianferrara, T.; Alessio, E. Ruthenium Anticancer Compounds: Challenges and Expectations. Chimia 2007, 61, 692–697. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the First Ruthenium-Based Anticancer Drug on the Edge to Clinical Application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, a New Redox-Active Anticancer Agent—Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; Van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H.M. Phase I/II Study with Ruthenium Compound NAMI-A and Gemcitabine in Patients with Non-Small Cell Lung Cancer after First Line Therapy. Invest. New. Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef]
- Zeng, L.; Gupta, P.; Chen, Y.; Wang, E.; Ji, L.; Chao, H.; Chen, Z.S. The Development of Anticancer Ruthenium(II) Complexes: From Single Molecule Compounds to Nanomaterials. Chem. Soc. Rev. 2017, 46, 5771–5804. [Google Scholar] [CrossRef]
- Wang, F.; Habtemariam, A.; Van Der Geer, E.P.L.; Ferná Ndez, R.; Melchart, M.; Deeth, R.J.; Aird, R.; Guichard, S.; Fabbiani, F.P.A.; Lozano-Casal, P.; et al. Controlling Ligand Substitution Reactions of Organometallic Complexes: Tuning Cancer Cell Cytotoxicity. Proc. Natl. Acad. Sci. USA 2005, 20, 18269–18274. [Google Scholar] [CrossRef]
- Riddell, I.A.; Lippard, S.J. Cisplatin and Oxaliplatin: Our Current Understanding of Their Metallo-Drugs: Development and Action of Anticancer Agents Actions. Met. Ions Life Sci. 2018, 18, 1–42. [Google Scholar] [CrossRef]
- Bordoni, S.; Cerini, S.; Tarroni, R.; Monari, M.; Micheletti, G.; Boga, C. Ruthenium-Thymine Acetate Binding Modes: Experimental and Theoretical Studies. Appl. Sci. 2021, 11, 3113. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Deng, Y.; Wang, T.; Miao, T.; Li, C.; Cai, X.; Liu, Y.; Henri, J.; Chen, L. Ru(II) Complexes Bearing O, O-Chelated Ligands Induced Apoptosis in A549 Cells through the Mitochondrial Apoptotic Pathway. Bioinorg. Chem. Appl. 2020, 2020, 8890950. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, A.; Melchart, M.; Fernández, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure-Activity Relationships for Cytotoxic Ruthenium(II) Arene Complexes Containing N,N-, N,O-, and O,O-Chelating Ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef]
- Puckett, C.A.; Barton, J.K. Methods to Explore Cellular Uptake of Ruthenium Complexes. J. Am. Chem. Soc. 2007, 129, 46–47. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, P.; Chen, H.; Ji, L.; Chao, H. Comparison between Polypyridyl and Cyclometalated Ruthenium(II) Complexes: Anticancer Activities against 2d and 3d Cancer Models. Chem. Eur. J. 2015, 21, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Sáez, R.; Lorenzo, J.; Prieto, M.J.; Font-Bardia, M.; Calvet, T.; Omeñaca, N.; Vilaseca, M.; Moreno, V. Influence of PPh3 Moiety in the Anticancer Activity of New Organometallic Ruthenium Complexes. J. Inorg. Biochem. 2014, 136, 1–12. [Google Scholar] [CrossRef]
- Calonghi, N.; Boga, C.; Nitti, P.; Telese, D.; Bordoni, S.; Farruggia, G.; Asaro, F.; Grandi, M.; Zalambani, C.; Micheletti, G. Effects of Regioisomerism on the Antiproliferative Activity of Hydroxystearic Acids on Human Cancer Cell Lines. Molecules 2022, 27, 2396. [Google Scholar] [CrossRef]
- Babu, T.; Sarkar, A.; Karmakar, S.; Schmidt, C.; Gibson, D. Multiaction Pt(IV) Carbamate Complexes Can Codeliver Pt(II) Drugs and Amine Containing Bioactive Molecules. Inorg. Chem. 2020, 59, 5182–5193. [Google Scholar] [CrossRef]
- Emam, S.M.; El Sayed, I.E.T.; Nassar, N. Transition Metal Complexes of Neocryptolepine Analogues. Part I: Synthesis, Spectroscopic Characterization, and in Vitro Anticancer Activity of Copper(II) Complexes. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 138, 942–953. [Google Scholar] [CrossRef]
- Atilla-Gokcumen, G.E.; Williams, D.S.; Bregman, H.; Pagano, N.; Meggers, E. Organometallic Compounds with Biological Activity: A Very Selective and Highly Potent Cellular Inhibitor for Glycogen Synthase Kinase 3. ChemBioChem 2006, 7, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, K.M.; Corrêa, R.S.; Barbosa, M.I.F.; Ellena, J.; Cominetti, M.R.; Batista, A.A. Ruthenium(II)/Triphenylphosphine Complexes: An Effective Way to Improve the Cytotoxicity of Lapachol. Polyhedron 2017, 130, 108–114. [Google Scholar] [CrossRef]
- Graminha, A.E.; Honorato, J.; Dulcey, L.L.; Godoy, L.R.; Barbosa, M.F.; Cominetti, M.R.; Menezes, A.C.; Batista, A.A. Evaluation of the Biological Potential of Ruthenium(II) Complexes with Cinnamic Acid. J. Inorg. Biochem. 2020, 206, 111021. [Google Scholar] [CrossRef] [PubMed]
- Nayeem, N.; Contel, M. Exploring the Potential of Metallodrugs as Chemotherapeutics for Triple Negative Breast Cancer. Chem. Eur. J. 2021, 27, 8891–8917. [Google Scholar] [CrossRef]
- Sun, R.W.Y.; Zhang, M.; Li, D.; Li, M.; Wong, A.S.T. Enhanced Anti-Cancer Activities of a Gold(III) Pyrrolidinedithiocarbamato Complex Incorporated in a Biodegradable Metal-Organic Framework. J. Inorg. Biochem. 2016, 163, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. A View on Multi-Action Pt(IV) Antitumor Prodrugs. Inorg. Chim. Acta 2019, 492, 32–47. [Google Scholar] [CrossRef]
- Riccardi, C.; Piccolo, M.; Ferraro, M.G.; Graziano, R.; Musumeci, D.; Trifuoggi, M.; Irace, C.; Montesarchio, D. Bioengineered Lipophilic Ru(III) Complexes as Potential Anticancer Agents. Biomater. Adv. 2022, 139, 213016. [Google Scholar] [CrossRef]
- Calonghi, N.; Boga, C.; Telese, D.; Bordoni, S.; Sartor, G.; Torsello, C.; Micheletti, G. Synthesis of 9-Hydroxystearic Acid Derivatives and Their Antiproliferative Activity on HT 29 Cancer Cells. Molecules 2019, 24, 3714. [Google Scholar] [CrossRef]
- Samouei, H.; Grushin, V.V. New, Highly Efficient, Simple, Safe, and Scalable Synthesis of [(Ph3P)3Ru(CO)(H)2]. Organometallics 2013, 32, 4440–4443. [Google Scholar] [CrossRef]
- Oliveira, K.M.; Honorato, J.; Gonçalves, G.R.; Cominetti, M.R.; Batista, A.A.; Correa, R.S. Ru(Ii)/Diclofenac-Based Complexes: DNA, BSA Interaction and Their Anticancer Evaluation against Lung and Breast Tumor Cells. Dalton Trans. 2020, 49, 12643–12652. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part. A: Theory and Applications in Inorganic Chemistry, 6th ed.; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach; John Wiley & Sons Ltd.: Chichester, UK, 2000. [Google Scholar]
- Sahi, A.K.; Anjali; Varshney, N.; Poddar, S.; Vajanthri, K.Y.; Mahto, S.K. Optimizing a Detection Method for Estimating Polyunsaturated Fatty Acid in Human Milk Based on Colorimetric Sensors. Mater. Sci. Energy Technol. 2019, 2, 624–628. [Google Scholar] [CrossRef]
- Asaro, F.; Boga, C.; Zorzi, D.L.; Geremia, S.; Gigli, L.; Nitti, P.; Semeraro, S. (R)-10-Hydroxystearic Acid: Crystals vs. Orgartogel. Int. J. Mol. Sci. 2020, 21, 8124. [Google Scholar] [CrossRef] [PubMed]
- Asaro, F.; Boga, C.; Demitri, N.; De Zorzi, R.; Drioli, S.; Gigli, L.; Micheletti, G.; Nitti, P.; Zangrando, E. X-Ray Crystal Structures and Organogelator Properties of (R)-9-Hydroxystearic Acid. Molecules 2019, 24, 2854. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Nagase, H.; Endo, T.; Ueda, H.; Nakagaki, M. Crystal Structure of DL-12-Hydroxystearic Acid. Chem. Lett. 1996, 25, 435–436. [Google Scholar] [CrossRef]
- Kamtjo, M.; Nagase, H.; Endo, T.; Veda, H.; Nakagakt, M. Polymorphic Structure of Dl-12Hydroxystearic Acid. Anal. Sci. 1999, 15, 1291–1292. [Google Scholar]
- Redrado, M.; Benedi, A.; Marzo, I.; García-Otín, A.L.; Fernández-Moreira, V.; Concepción Gimeno, M. Multifunctional Heterometallic IrIII−AuI Probes as Promising Anticancer and Antiangiogenic Agents. Chem. Eur. J. 2021, 27, 9885–9897. [Google Scholar] [CrossRef]
- Ali, I.; Wani, W.A.; Saleem, K.; Hseih, M.F. Design and Synthesis of Thalidomide Based Dithiocarbamate Cu(II), Ni(II) and Ru(III) Complexes as Anticancer Agents. Polyhedron 2013, 56, 134–143. [Google Scholar] [CrossRef]
- Łakomska, I.; Fandzloch, M.; Muzioł, T.; Lis, T.; Jezierska, J. Synthesis, Characterization and Antitumor Properties of Two Highly Cytotoxic Ruthenium(Iii) Complexes with Bulky Triazolopyrimidine Ligands. Dalton Trans. 2013, 42, 6219–6226. [Google Scholar] [CrossRef]
- Riccardi, C.; Musumeci, D.; Capuozzo, A.; Irace, C.; King, S.; Russo Krauss, I.; Paduano, L.; Montesarchio, D. “dressing up” an Old Drug: An Aminoacyl Lipid for the Functionalization of Ru(III)-Based Anticancer Agents. ACS Biomater. Sci. Eng. 2018, 4, 163–174. [Google Scholar] [CrossRef]
- Calonghi, N.; Cappadone, C.; Pagnotta, E.; Farruggia, G.; Buontempo, F.; Boga, C.; Brusa, G.L.; Santucci, M.A.; Masotti, L. 9-Hydroxystearic Acid Upregulates P21WAF1 in HT29 Cancer Cells. Biochem. Biophys. Res. Commun. 2004, 314, 138–142. [Google Scholar] [CrossRef]
- Calonghi, N.; Cappadone, C.; Pagnotta, E.; Boga, C.; Bertucci, C.; Fiori, J.; Tasco, G.; Casadio, R.; Masotti, L. Histone Deacetylase 1: A Target of 9-Hydroxystearic Acid in the Inhibition of Cell Growth in Human Colon Cancer. J. Lipid Res. 2005, 46, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Kokotou, M.G.; Kokotos, A.C.; Gkikas, D.; Mountanea, O.G.; Mantzourani, C.; Almutairi, A.; Lei, X.; Ramanadham, S.; Politis, P.K.; Kokotos, G. Saturated Hydroxy Fatty Acids Exhibit a Cell Growth Inhibitory Activity and Suppress the Cytokine-Induced β-Cell Apoptosis. J. Med. Chem. 2020, 63, 12666–12681. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; O’driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK Function Redundantly to Phosphorylate H2AX after Exposure to Ionizing Radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-Stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Dhuppar, S.; Mazumder, A. Measuring Cell Cycle-Dependent DNA Damage Responses and P53 Regulation on a Cell-by-Cell Basis from Image Analysis. Cell Cycle 2018, 17, 1358–1371. [Google Scholar] [CrossRef]
- Prendergast, Á.M.; Cruet-Hennequart, S.; Shaw, G.; Barry, F.P.; Carty, M.P. Activation of DNA Damage Response Pathways in Human Mesenchymal Stem Cells Exposed to Cisplatin or γ-Irradiation. Cell Cycle 2011, 10, 3768–3777. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Traganos, F.; Darzynkiewicz, Z. Kinetics of the UV-Induced DNA Damage Response in Relation to Cell Cycle Phase. Correlation with DNA Replication. Cytometry A 2010, 77, 285–293. [Google Scholar] [CrossRef]
- APEX3, SAINT and SADABS, v2015.5-2. Software for Chemical Crystallography Bruker AXS. Bruker AXS Inc.: Madison, WI, USA, 2015.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Calonghi, N.; Farruggia, G.; Boga, C.; Micheletti, G.; Fini, E.; Romani, L.; Telese, D.; Faraci, E.; Bergamini, C.; Cerini, S.; et al. Root Extracts of Two Cultivars of Paeonia Species: Lipid Composition and Biological Effects on Different Cell Lines: Preliminary Results. Molecules 2021, 26, 655. [Google Scholar] [CrossRef]
- Åmellem, Ø.; Stokke, T.; Sandvik, J.A.; Pettersen, E.O. The Retinoblastoma Gene Product Is Reversibly Dephosphorylated and Bound in the Nucleus in S and G2Phases during Hypoxic Stress. Exp. Cell Res. 1996, 227, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Micheletti, G.; Calonghi, N.; Farruggia, G.; Strocchi, E.; Palmacci, V.; Telese, D.; Bordoni, S.; Frisco, G.; Boga, C. Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells. Molecules 2020, 25, 404. [Google Scholar] [CrossRef] [PubMed]
- Calonghi, N.; Pagnotta, E.; Parolin, C.; Tognoli, C.; Boga, C.; Masotti, L. 9-Hydroxystearic Acid Interferes with EGF Signalling in a Human Colon Adenocarcinoma. Biochem. Biophys. Res. Commun. 2006, 342, 585–588. [Google Scholar] [CrossRef] [PubMed]








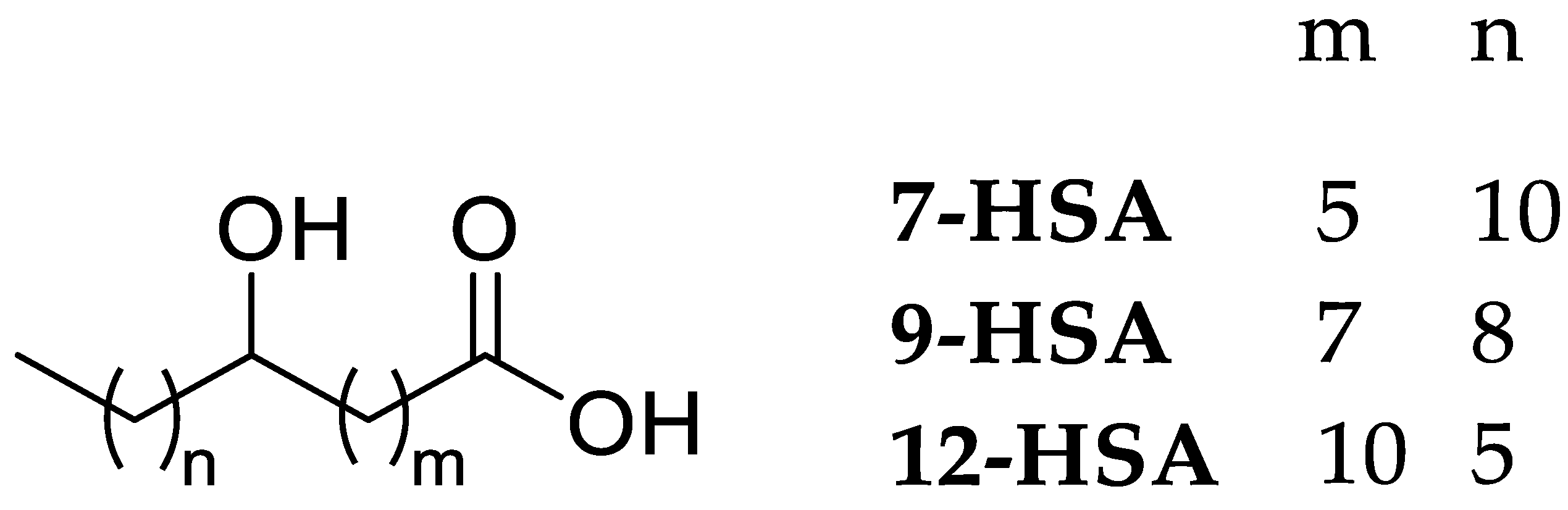{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
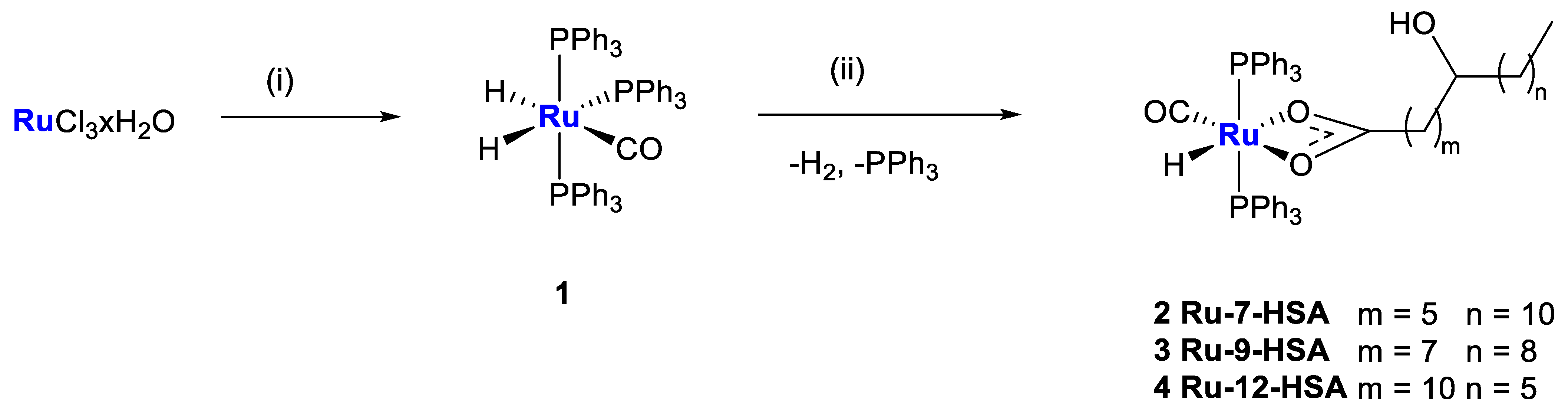{kind=link}
| Complex | HT29 | HeLa | IGROV1 | HDFa |
|---|---|---|---|---|
| 2 | 2.43 ± 0.01 | >10 | >10 | >10 |
| 3 | 0.17 ± 0.02 | 10.43 ± 0.05 | 8.55 ± 0.01 | >10 |
| 4 | >10 | >10 | >10 | >10 |
| 1 | >10 | >10 | >10 | >10 |
| CDDP | >10 | >10 | >10 | >10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drius, G.; Bordoni, S.; Boga, C.; Monari, M.; Fiori, J.; Esposito, E.; Zalambani, C.; Pincigher, L.; Farruggia, G.; Calonghi, N.; et al. Synthesis and Antiproliferative Insights of Lipophilic Ru(II)-Hydroxy Stearic Acid Hybrid Species. Molecules 2023, 28, 4051. https://doi.org/10.3390/molecules28104051
Drius G, Bordoni S, Boga C, Monari M, Fiori J, Esposito E, Zalambani C, Pincigher L, Farruggia G, Calonghi N, et al. Synthesis and Antiproliferative Insights of Lipophilic Ru(II)-Hydroxy Stearic Acid Hybrid Species. Molecules. 2023; 28(10):4051. https://doi.org/10.3390/molecules28104051
Chicago/Turabian StyleDrius, Giacomo, Silvia Bordoni, Carla Boga, Magda Monari, Jessica Fiori, Erika Esposito, Chiara Zalambani, Luca Pincigher, Giovanna Farruggia, Natalia Calonghi, and et al. 2023. "Synthesis and Antiproliferative Insights of Lipophilic Ru(II)-Hydroxy Stearic Acid Hybrid Species" Molecules 28, no. 10: 4051. https://doi.org/10.3390/molecules28104051
APA StyleDrius, G., Bordoni, S., Boga, C., Monari, M., Fiori, J., Esposito, E., Zalambani, C., Pincigher, L., Farruggia, G., Calonghi, N., & Micheletti, G. (2023). Synthesis and Antiproliferative Insights of Lipophilic Ru(II)-Hydroxy Stearic Acid Hybrid Species. Molecules, 28(10), 4051. https://doi.org/10.3390/molecules28104051