
Modulation Effect on Tubulin Polymerization, Cytotoxicity and Antioxidant Activity of 1H-Benzimidazole-2-Yl Hydrazones


 , ,
, ,  ,
,  , ,
, , 
Abstract
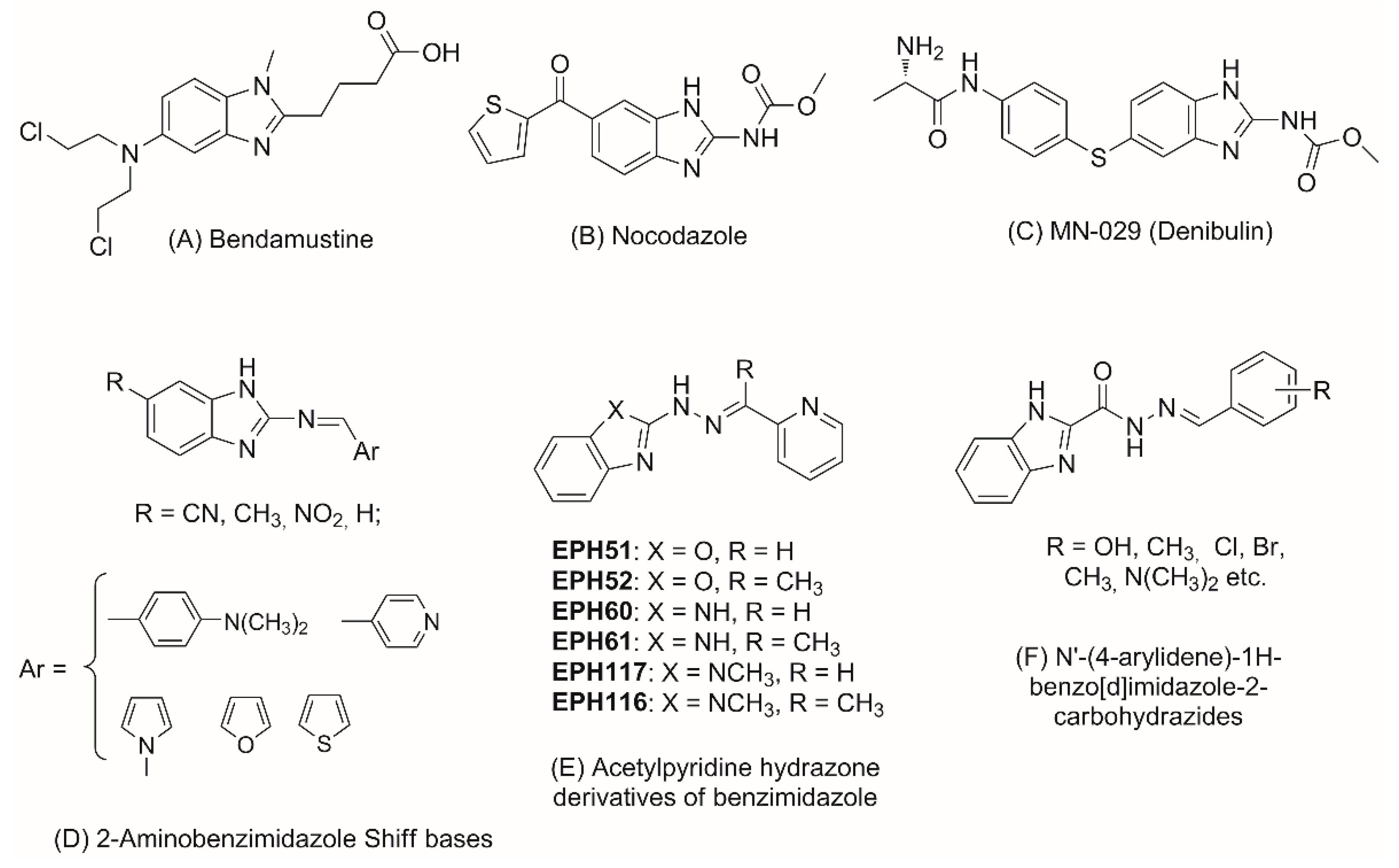
1. Introduction
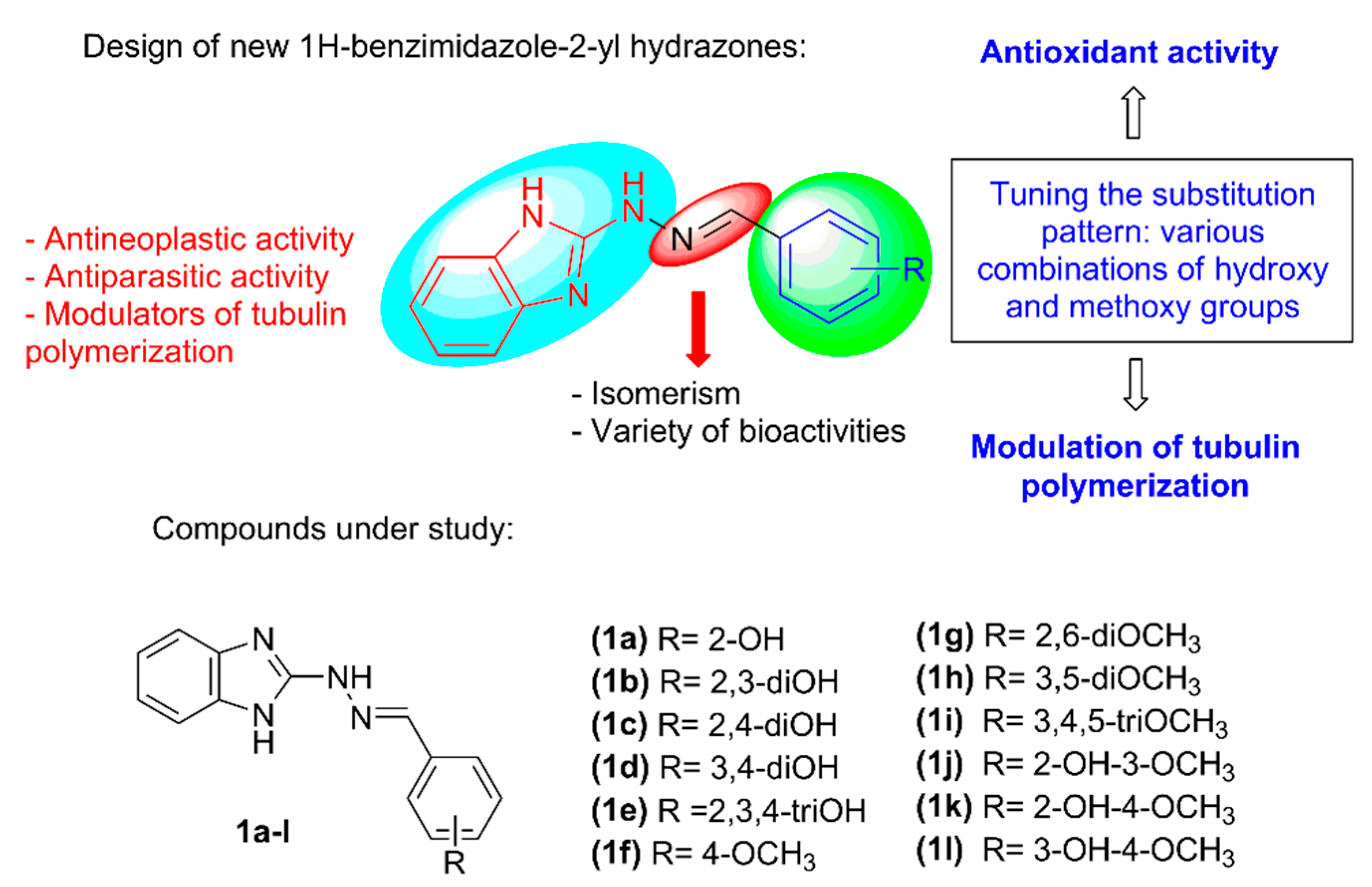
2. Results and Discussion
2.1. Synthesis
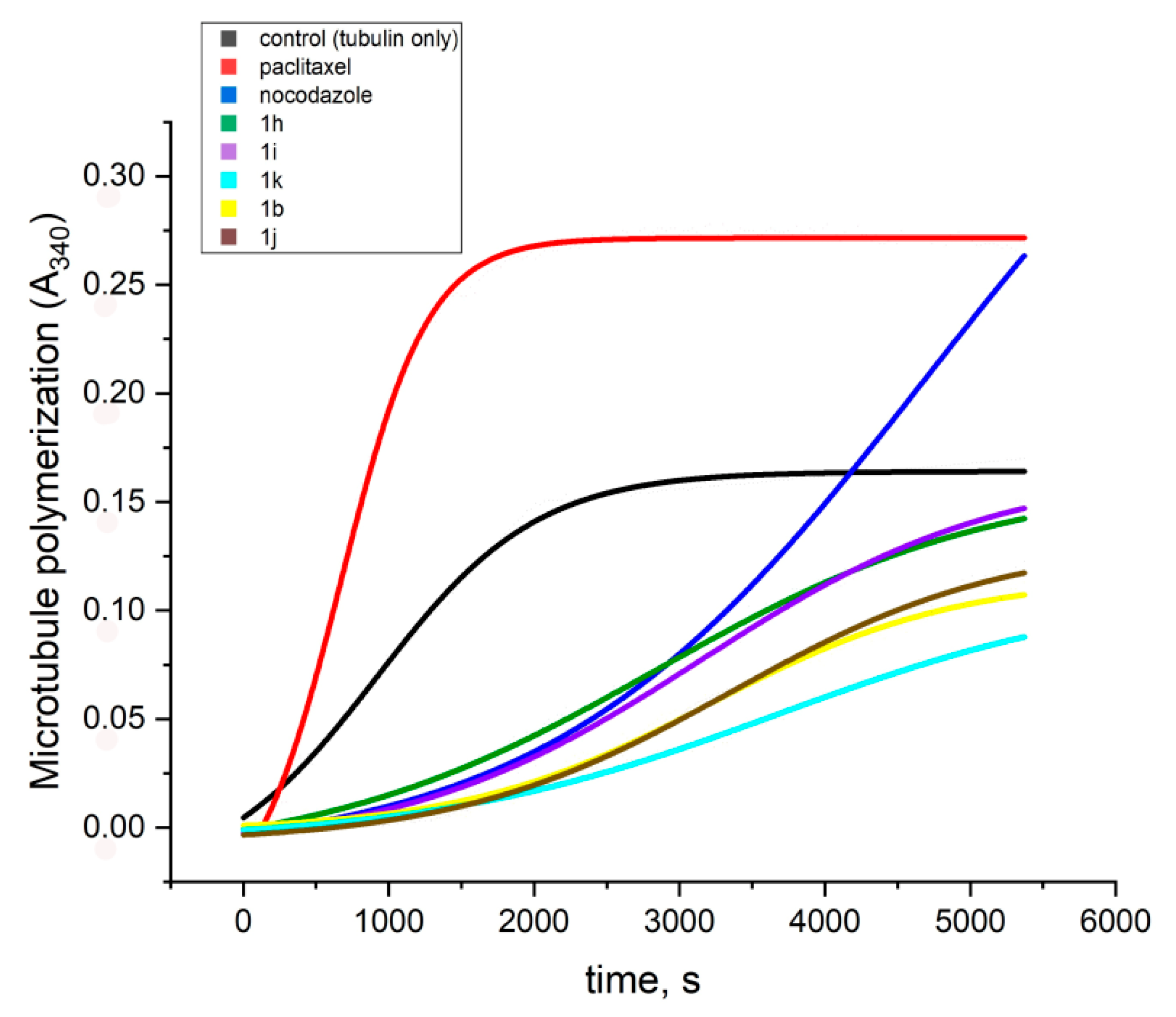
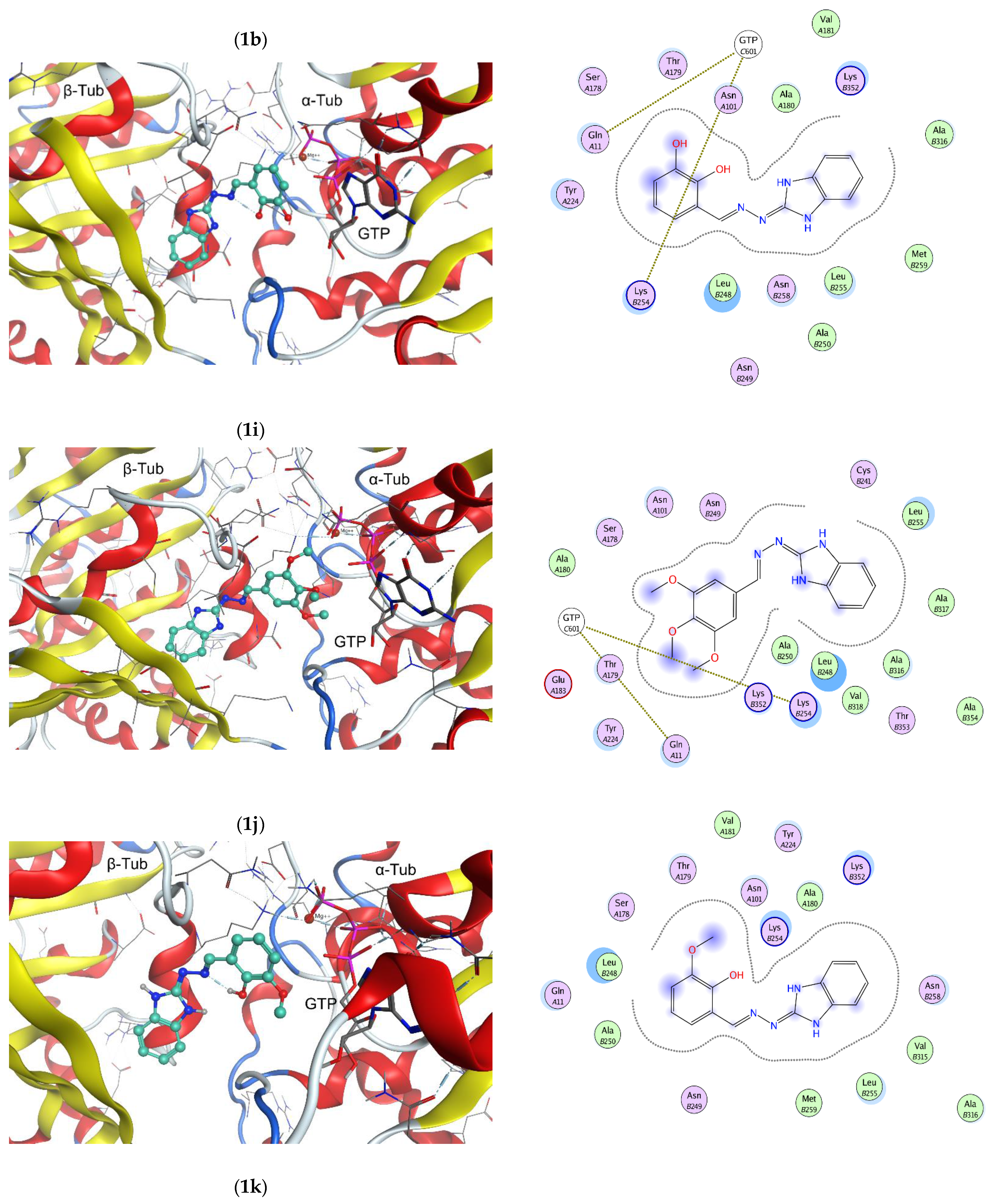
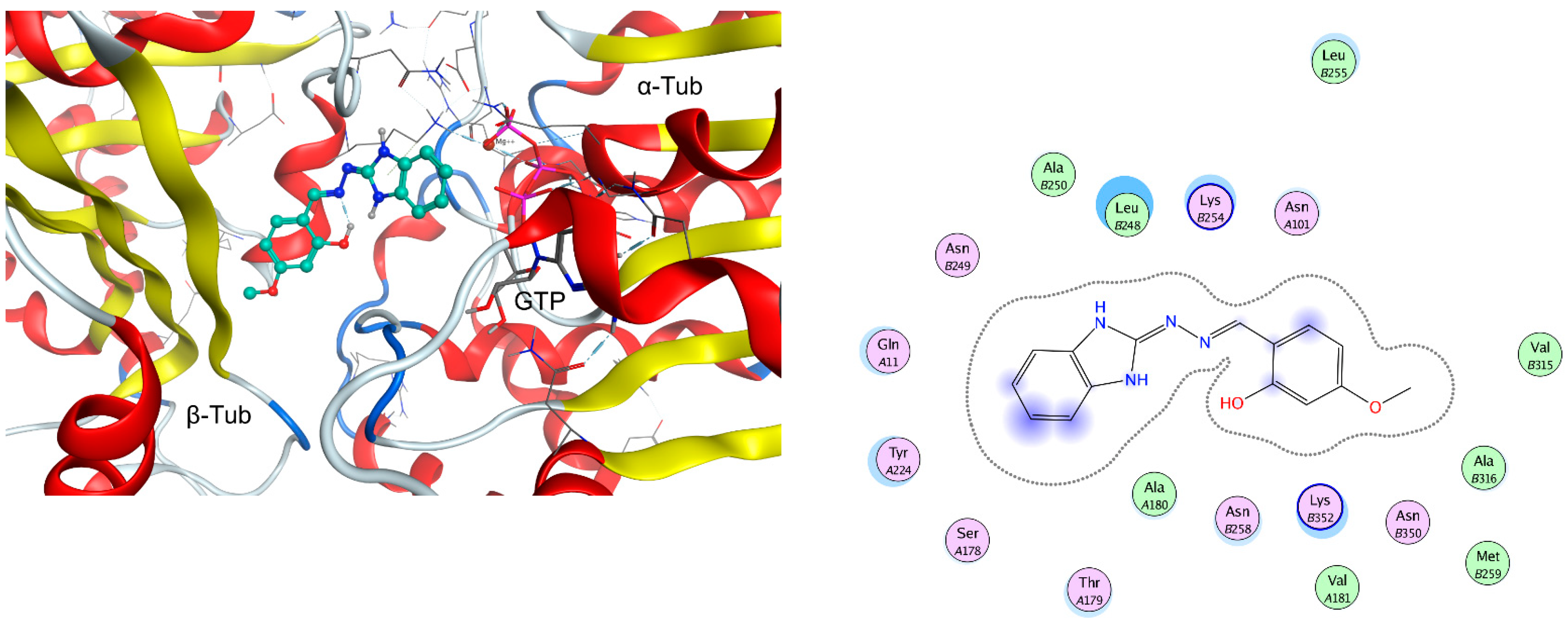
2.2. In Vitro Effect on Tubulin Polymerization and Docking Study on the Tubulin-Ligands Interactions
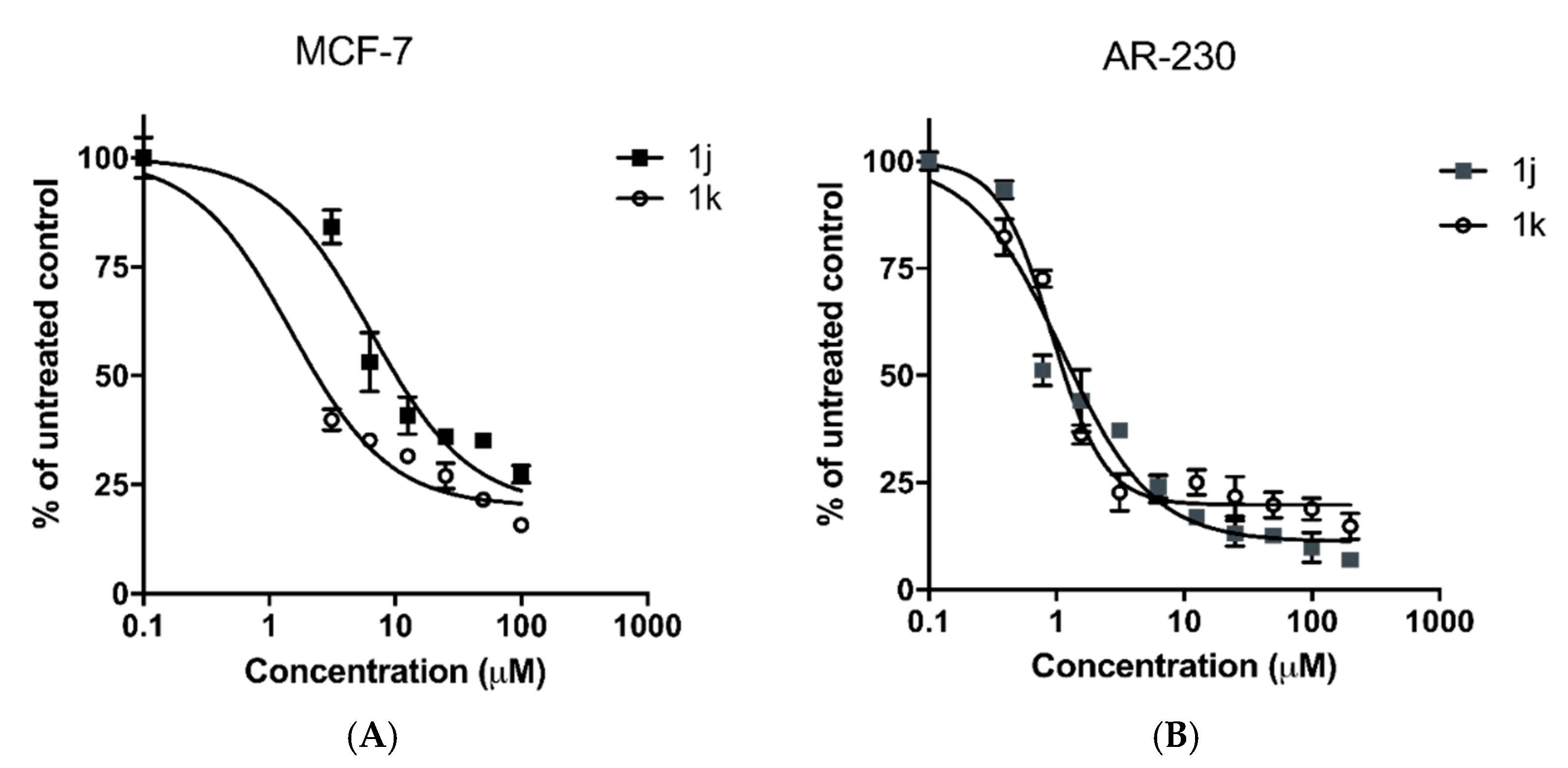
2.3. In Vitro Cytotoxicity
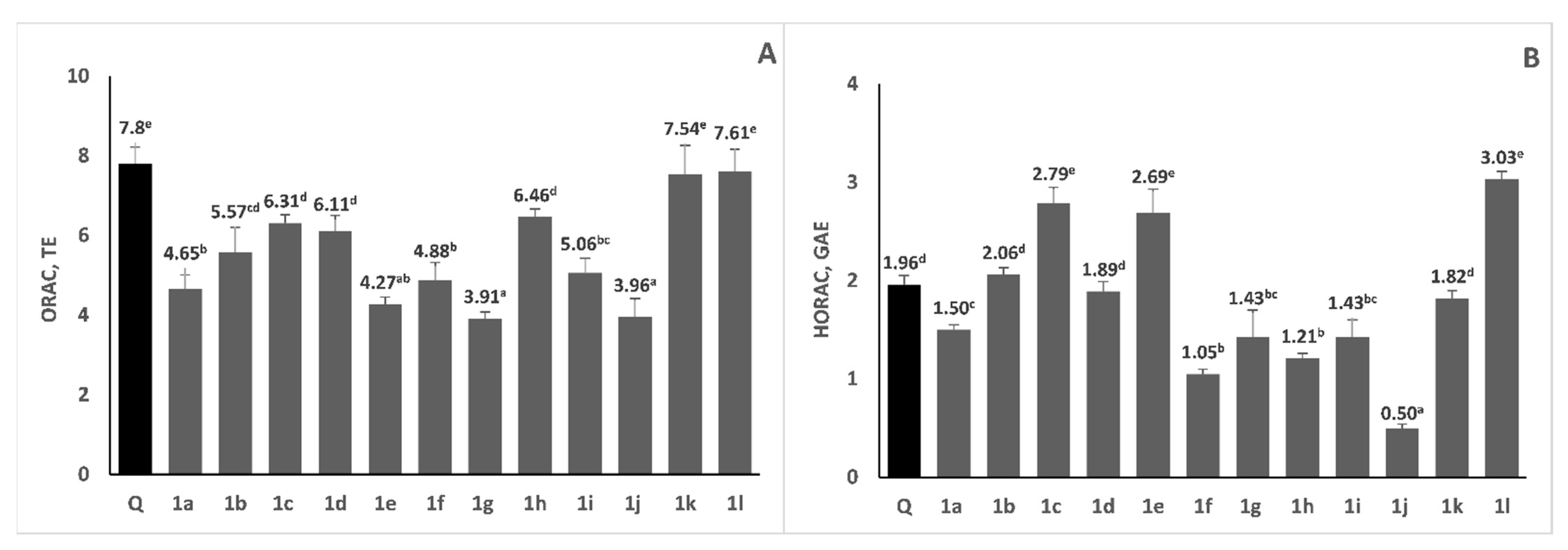
2.4. Radical Scavenging Activity
2.5. Prediction of the Physico-Chemical Properties and Drug-Likeness
3. Materials and Methods
3.1. In Vitro Tubulin Polymerization Assay
3.2. Molecular Docking
3.3. Cytotoxicity Study
3.4. Antioxidant Study
3.4.1. Chemicals
3.4.2. Oxygen Radical Absorbance Capacity (ORAC)
3.4.3. Hydroxyl Radical Averting Capacity (HORAC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hranjec, M.; Starčević, K.; Pavelić, S.K.; Lučin, P.; Pavelić, K.; Zamola, G.K. Synthesis, spectroscopic characterization and antiproliferative evaluation in vitro of novel Schiff bases related to benzimidazoles. Eur. J. Med. Chem. 2011, 46, 2274–2279. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Ojha, H.; Tiwari, A.K.; Kumar, N.; Singh, B.; Mishra, A.K. Design, Synthesis, and In Vitro Antiproliferative Activity of Benzimidazole Analogues for Radiopharmaceutical Efficacy. Cancer Biother. Radiopharm. 2010, 25, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Nawrocka, W.; Sztuba, B.; Kowalska, M.W.; Liszkiewicz, H.; Wietrzyk, J.; Nasulewicz, A.; Peczyska, M.; Opolski, A. Synthesis and antiproliferative activity in vitro of 2-aminobenzimidazole derivatives. II Farmaco. 2004, 59, 83–91. [Google Scholar] [CrossRef] [PubMed]
- de Dios, C.; Shih, B.; Lopez de Uralde, C.; Sanchez, M.; del Prado, L.M.; Cabrejas, M.; Pleite, S.; Blanco-Urgoiti, J.; Lorite, M.J.; Nevill, C., Jr.; et al. Design of Potent and Selective 2-Aminobenzimidazole-Based p38r MAP Kinase Inhibitors with Excellent in Vivo Efficacy. J. Med. Chem. 2005, 48, 2270–2273. [Google Scholar] [CrossRef]
- Hall, I.H.; Peaty, N.J.; Henry, J.R.; Easmon, J.; Heinisch, G.; Pürstinger, G. Investigations on the Mechanism of Action of the Novel Antitumor Agents 2-Benzothiazolyl, 2-Benzoxazolyl, and 2-Benzimidazolyl Hydrazones Derived from 2-Acetylpyridine. Arch. Pharm. 1999, 332, 115–123. [Google Scholar] [CrossRef]
- Easmon, J.; Pürstinger, G.; Roth, T.; Fiebig, H.-H.; Marcel, J.; Jaeger, W.; Heinisch, G.; Hofmann, J. 2-benzoxazolyl and 2-benzimidazolyl hydrazones derived from 2-acetylpyridine: A novel class of antitumor agents. Int. J. Cancer 2001, 9, 89–96. [Google Scholar] [CrossRef]
- Onnis, V.; Demurtas, M.; Deplano, A.; Balboni, G.; Baldisserotto, A.; Manfredini, S.; Pacifico, S.; Liekens, S.; Balzarini, J. Design, Synthesis and Evaluation of Antiproliferative Activity of New Benzimidazolehydrazones. Molecules 2016, 21, 579–588. [Google Scholar] [CrossRef]
- Boiteux, S.; Radicella, J.P. Base excision repair of 8-hydroxyguanine protects DNA from endogenous oxidative stress. Biochim 1999, 81, 59–67. [Google Scholar] [CrossRef]
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302. [Google Scholar] [CrossRef]
- Khandrika, L.; Kumar, B.; Koul, S.; Maroni, P.; Koul, H.K. Oxidative stress in prostate cancer. Cancer Lett. 2009, 282, 125–136. [Google Scholar] [CrossRef]
- Visconti, R.; Grieco, D. New insights on oxidative stress in cancer. Curr. Opin. Drug Discov. Devel. 2009, 12, 240–245. [Google Scholar] [PubMed]
- Bella, G.D.; Mascia, F.; Gualano, L.; Di Bella, L. Melatonin Anticancer Effects: Review. Int. J. Mol. Sci. 2013, 14, 2410–2430. [Google Scholar] [CrossRef] [PubMed]
- Karbownik, M. Potential anticarcinogenic action of melatonin and other antioxidants mediated by antioxidative mechanisms. Neuro Endocrinol. Lett. 2002, 23, 39–44. [Google Scholar] [PubMed]
- Reiter, R.J.; Tan, D.X.; Herman, T.S.; Thomas, C.R., Jr. Melatonin as a radioprotective agent: A review. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 639–653. [Google Scholar]
- Kadoma, Y.; Fujisawa, S. Radical-scavenging activity of melatonin, either alone or in combination with vitamin E, ascorbate or 2-mercaptoethanol as co-antioxidants, using the induction period method. In Vivo 2011, 25, 49–53. [Google Scholar]
- Galano, A. On the direct scavenging activity of melatonin towards hydroxyl and a series of peroxyl radicals. Phys. Chem. Chem. Phys. 2011, 13, 7178–7188. [Google Scholar] [CrossRef]
- Lissoni, P.; Bastone, A.; Sala, R.; Mauri, R.; Rovelli, F.; Viviani, S.; Bajetta, E.; Esposti, D.; Esposti, G.; Di Bella, L. The clinical significance of melatonin serum determination in oncological patients and its correlations with GH and PRL blood levels. Eur. J. Cancer Clin. Oncol. 1987, 23, 949–957. [Google Scholar] [CrossRef]
- Di Bella, L. Melatonin: An Essential Factor for the Treatment and Recovery from Leukemia and Cancer. In Proceedings of the International Symposium on Melatonin, Bremen, Germany, 28–30 September 1980; pp. 161–162. [Google Scholar]
- Lissoni, P.; Barni, S.; Cattaneo, G.; Tancini, G.; Esposti, G.; Esposti, D.; Fraschini, F. Clinical result with the pineal hormone melatonin in advanced cancer resistant to standard antitumor therapies. Oncology 1991, 48, 48–50. [Google Scholar] [CrossRef]
- Bubenik, G.A.; Blask, D.E.; Brown, G.M.; Maestroni, G.J.; Pang, S.F.; Reiter, R.J.; Viswanathan, M.; Zisapel, N. Prospects of the clinical utilization of melatonin. Biol. Signals Recept. 1998, 7, 195–219. [Google Scholar] [CrossRef]
- Di Bella, G. The Di Bella Method (DBM). Neuro. Endocrinol. Lett. 2010, 31, 1–42. [Google Scholar]
- Anwar, M.M.; Mahfouz, H.A.; Sayed, A.S. Potential protective effects of melatonin on bone marrow of rats exposed to cytotoxic drugs. Comp. Biochem. Physiol. A 1998, 119, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Rapozzi, V.; Zorzet, S.; Comelli, M.; Mavelli, I.; Perissin, L.; Giraldi, T. Melatonin decreases bone marrow and lymphatic toxicity of adriamycin in mice bearing TLX5 lymphoma. Life Sci. 1998, 63, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Anichina, K.; Argirova, M.; Tzoneva, R.; Uzunova, V.; Mavrova, A.; Vuchev, D.; Popova-Daskalova, G.; Fratev, F.; Guncheva, M.; Yancheva, D. 1H-benzimidazole-2-yl hydrazones as tubulin-targeting agents: Synthesis, structural characterization, anthelmintic activity and antiproliferative activity against MCF-7 breast carcinoma cells and molecular docking studies. Chem. Biol. Interact. 2021, 345, 109540. [Google Scholar] [CrossRef] [PubMed]
- Argirova, M.A.; Georgieva, M.K.; Hristova-Avakumova, N.G.; Vuchev, D.I.; Popova-Daskalova, G.V.; Anichina, K.K.; Yancheva, D.Y. New 1H-benzimidazole-2-yl hydrazones with combined antiparasitic and antioxidant activity. RSC Adv. 2021, 11, 39848–39868. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); H3A 2R7; Chemical Computing Group Inc.: Montreal, QC, Canada, 2020.
- Knossow, M.; Campanacci, V.; Khodja, L.A.; Gigant, B. The Mechanism of Tubulin Assembly into Microtubules: Insights from Structural Studies. iScience 2020, 23, 101511. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Gigant, B.; Yu, Y.; Wu, Y.; Chen, X.; Lai, Q.; Yang, Z.; Chen, Q.; Yang, J. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283, 102–111. [Google Scholar] [CrossRef]
- de la Roche, N.M.; Mühlethaler, T.; Di Martino, R.M.C.; Ortega, J.A.; Gioia, D.; Royd, B.; Prota, A.E.; Steinmetz, M.O.; Cavalli, A. Novel fragment-derived colchicine-site binders as microtubule-destabilizing agents. Eur. J. Med. Chem. 2022, 241, 114614. [Google Scholar] [CrossRef]
- Dettmann, S.; Szymanowitz, K.; Wellner, A.; Schiedel, A.; Müller, C.E.; Gust, R. 2-Phenyl-1-[4-(2-piperidine-1-yl-ethoxy) benzyl]-1H-benzimidazoles as ligands for the estrogen receptor: Synthesis and pharmacological evaluation. Bioorg. Med. Chem. 2010, 18, 4905–4916. [Google Scholar] [CrossRef]
- Singla, R.; Gupta, K.B.; Upadhyay, S.; Dhiman, M.; Jaitak, V. Design, synthesis and biological evaluation of novel indole-benzimidazole hybrids targeting estrogen receptor alpha (ER-α). Eur. J. Med. Chem. 2018, 146, 206–219. [Google Scholar] [CrossRef]
- Karadayi, F.Z.; Yaman, M.; Kisla, M.M. Keskus, A.G., Konu, O.; Ates-Alagoz, Z. Design, synthesis and anticancer/antiestrogenic activities of novel indole-benzimidazoles. Bioorg. Chem. 2020, 100, 103929. [Google Scholar] [CrossRef] [PubMed]
- Payton-Stewart, F.; Tilghman, S.L.; Williams, L.G.; Winfield, L.L. Benzimidazoles diminish ERE transcriptional activity and cell growth in breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 450, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Çevik, U.A.; Celik, I.; Mella, J.; Mellado, M.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, and Molecular Modeling Studies of a Novel Benzimidazole as an Aromatase Inhibitor. ACS Omega 2022, 7, 16152–16163. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Kim, J.; Yun, S.-M.; Lee, H.; Park, Y.; Hong, S.-S.; Hong, S. Discovery of New Benzothiazole-Based Inhibitors of Breakpoint Cluster Region-Abelson Kinase Including the T315I Mutant. J. Med. Chem. 2013, 56, 3531–3545. [Google Scholar] [CrossRef]
- Ou, B.X.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescence probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Žuvela, P.; David, J.; Yang, X.; Huang, D.; Wong, M.W. Non-linear quantitative structure-activity relationships modelling, mechanistic study and in-silico design of flavonoids as potent antioxidants. Int. J. Mol. Sci. 2019, 20, 2328. [Google Scholar] [CrossRef]
- Dávalos, A.; Gómez-Cordovés, C.; Bartolomé, B. Extending applicability of the oxygen radical absorbance capacity (ORAC-fluorescein) assay. J. Agric. Food. Chem. 2004, 52, 48–54. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Flanagan, J.; Deemer, E.K.; Prior, R.L.; Huang, D. Novel fluorometric assay for hydroxyl radical prevention capacity using fluorescein as the probe. J. Agric. Food Chem. 2002, 50, 2772–2777. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Ai, H.; Hu, H.; Li, S.; Zhao, J.; Liu, H. Applications of machine learning methods in drug toxicity prediction. Curr. Top. Med. Chem. 2018, 18, 987–997. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics, Molinspiration Property Engine v2013.09. Available online: https://www.molinspiration.com (accessed on 21 November 2022).
- Daina, A.; Zoete, V. A BOILED-egg to predict gastrointestinal Absorption and brain Penetration of small molecules. Chem. Med. Chem. 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Druglikeness and Medicinal Chemistry Friendliness of Small Molecules. Available online: http://www.swissadme.ch (accessed on 22 November 2022).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, U.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, S.R.; Doerksen, J. Topological polar surface area: A useful descriptor in 2D-QSAR, Curr. Med. Chem. 2009, 16, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harbor Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Ottaviani, G.; Gosling, D.J.; Patissier, C.; Rodde, S.; Zhou, L.; Faller, B. What is modulating solubility in simulated intestinal fluids? Eur. J. Pharm. Sci. 2010, 41, 452–457. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Testa, B.; Kraemer, S.D. The biochemistry of drug metabolism—An introduction: Part 4. reactions of conjugation and their enzymes. Chem. Biodivers. 2008, 5, 2171–2336. [Google Scholar] [CrossRef]
- Hollenberg, P.F. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab. Rev. 2002, 34, 17–35. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef]
- Osiris, Property Explorer. Available online: http://www.Organic-chemistry.org/prog/peo (accessed on 25 November 2022).
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | Lag Time, s | Initial Rate of the Exponential Phase, Signal Response a.u.s−1 × 10−6 |
|---|---|---|
| tubulin (spontaneous polymerization) | - | 75.3 |
| paclitaxel | 151 | 167 |
| nocodazole | 935 | 52 |
| 1a | 1390 | 8.77 |
| 1b | 1461 | 13.4 |
| 1c | 1190 | 4.9 |
| 1d | 1407 | 13.3 |
| 1f | 807 | 17.1 |
| 1g | 1000 | 10.7 |
| 1h | 1090 | 23.1 |
| 1i | 988 | 20.6 |
| 1j | 1200 | 16.9 |
| 1k | 1295 | 9.7 |
| 1l | no lag phase | 16 |
| Cell Line/Compound | IC50 (μM ± SD) | CSIMCF-7 | CSIAR230 | ||
|---|---|---|---|---|---|
| MCF-7 1 | AR-230 2 | 3T3 3/ CCL-1 4 | |||
| Podophyllotoxin | 0.3 ± 0.06 | 0.47 ± 0.09 | 2.7 4 | ≈9 | ≈5.7 |
| 1a | 18.7 ± 1.1 | 21.2 ± 2.3 | 204 ± 5.1 3 | ≈11 | ≈9.6 |
| 1b | 64.5 ± 6.2 | 3.3 ± 0.3 | 61.1 ± 1.8 3 | ≈1 | ≈18.4 |
| 1d | - | 61.8 ± 5.7 | 112.8 ± 9.1 4 | - | ≈1.8 |
| 1f | 11.2 ± 1.3 | 19.4 ± 2.2 | 355 ± 5.3 3 | ≈31.6 | ≈18.2 |
| 1g | 7.9 ± 2.1 | 11.5 ± 1.0 | 261 ± 4.7 3 | ≈33 | ≈22.6 |
| 1h | 4.6 ± 0.7 | 5.9 ±0.9 | 335 ± 7.0 3 | ≈72.8 | ≈56.7 |
| 1i | 1.2 ± 0.2 | 1.7 ± 0.3 | 71 ± 2.4 3 | ≈59.1 | ≈41.7 |
| 1j | 6.4 ± 1.2 | 1.1 ± 0.2 | >400 4 | ≈62.5 | ≈363.6 |
| 1k | 1.5 ± 0.3 | 0.9 ± 0.1 | 101.8 ± 7.3 4 | ≈67.8 | ≈113 |
| 1l | 27.6 ± 3.5 | 16.8 ± 3.4 | 134.7 ± 1.33 | ≈4.8 | ≈8 |
| Compound | logP a | TPSA b | Natoms c | MW d | NON e | NOHNH f | Nviol g | Nrotb h | Vol i |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 2.98 | 73.30 | 19 | 252.28 | 5 | 3 | 0 | 3 | 223.95 |
| 1b | 2.28 | 93.53 | 20 | 268.28 | 6 | 4 | 0 | 3 | 231.97 |
| 1c | 2.48 | 93.53 | 20 | 268.28 | 6 | 4 | 0 | 3 | 231.97 |
| 1d | 2.07 | 93.53 | 20 | 268.28 | 6 | 4 | 0 | 3 | 231.97 |
| 1e | 2.01 | 113.76 | 21 | 284.27 | 7 | 5 | 0 | 3 | 239.99 |
| 1f | 3.10 | 62.31 | 20 | 266.30 | 5 | 2 | 0 | 4 | 241.48 |
| 1g | 3.06 | 71.54 | 22 | 296.33 | 6 | 2 | 0 | 5 | 267.03 |
| 1h | 3.08 | 71.54 | 22 | 296.33 | 6 | 2 | 0 | 5 | 267.03 |
| 1i | 2.67 | 80.78 | 24 | 326.36 | 7 | 2 | 0 | 6 | 292.57 |
| 1j | 2.59 | 82.54 | 21 | 282.30 | 6 | 3 | 0 | 4 | 249.50 |
| 1k | 3.02 | 82.54 | 21 | 282.30 | 6 | 3 | 0 | 4 | 249.50 |
| 1l | 2.38 | 82.54 | 21 | 282.30 | 6 | 3 | 0 | 4 | 249.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Argirova, M.; Guncheva, M.; Momekov, G.; Cherneva, E.; Mihaylova, R.; Rangelov, M.; Todorova, N.; Denev, P.; Anichina, K.; Mavrova, A.; et al. Modulation Effect on Tubulin Polymerization, Cytotoxicity and Antioxidant Activity of 1H-Benzimidazole-2-Yl Hydrazones. Molecules 2023, 28, 291. https://doi.org/10.3390/molecules28010291
Argirova M, Guncheva M, Momekov G, Cherneva E, Mihaylova R, Rangelov M, Todorova N, Denev P, Anichina K, Mavrova A, et al. Modulation Effect on Tubulin Polymerization, Cytotoxicity and Antioxidant Activity of 1H-Benzimidazole-2-Yl Hydrazones. Molecules. 2023; 28(1):291. https://doi.org/10.3390/molecules28010291
Chicago/Turabian StyleArgirova, Maria, Maya Guncheva, Georgi Momekov, Emiliya Cherneva, Rositsa Mihaylova, Miroslav Rangelov, Nadezhda Todorova, Petko Denev, Kameliya Anichina, Anelia Mavrova, and et al. 2023. "Modulation Effect on Tubulin Polymerization, Cytotoxicity and Antioxidant Activity of 1H-Benzimidazole-2-Yl Hydrazones" Molecules 28, no. 1: 291. https://doi.org/10.3390/molecules28010291
APA StyleArgirova, M., Guncheva, M., Momekov, G., Cherneva, E., Mihaylova, R., Rangelov, M., Todorova, N., Denev, P., Anichina, K., Mavrova, A., & Yancheva, D. (2023). Modulation Effect on Tubulin Polymerization, Cytotoxicity and Antioxidant Activity of 1H-Benzimidazole-2-Yl Hydrazones. Molecules, 28(1), 291. https://doi.org/10.3390/molecules28010291