

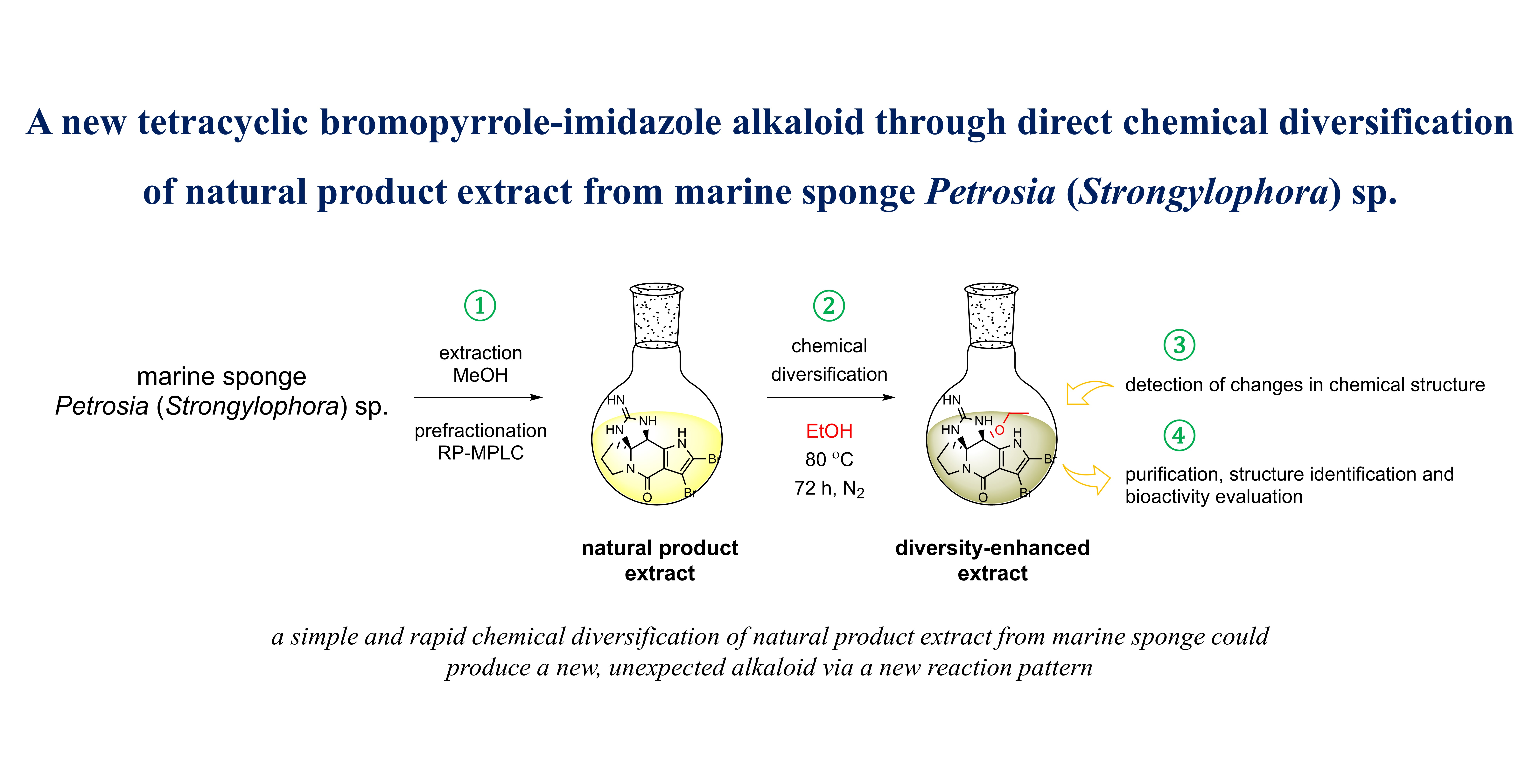
A New Tetracyclic Bromopyrrole-Imidazole Derivative through Direct Chemical Diversification of Substances Present in Natural Product Extract from Marine Sponge Petrosia (Strongylophora) sp.

 , ,
, ,  and
and
Abstract
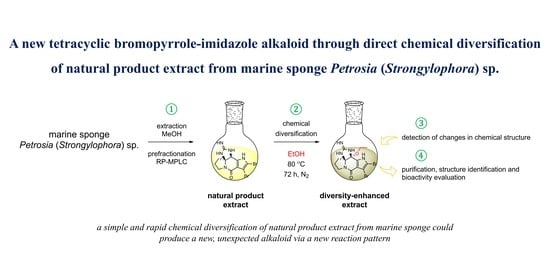
:
1. Introduction
2. Results and Discussion
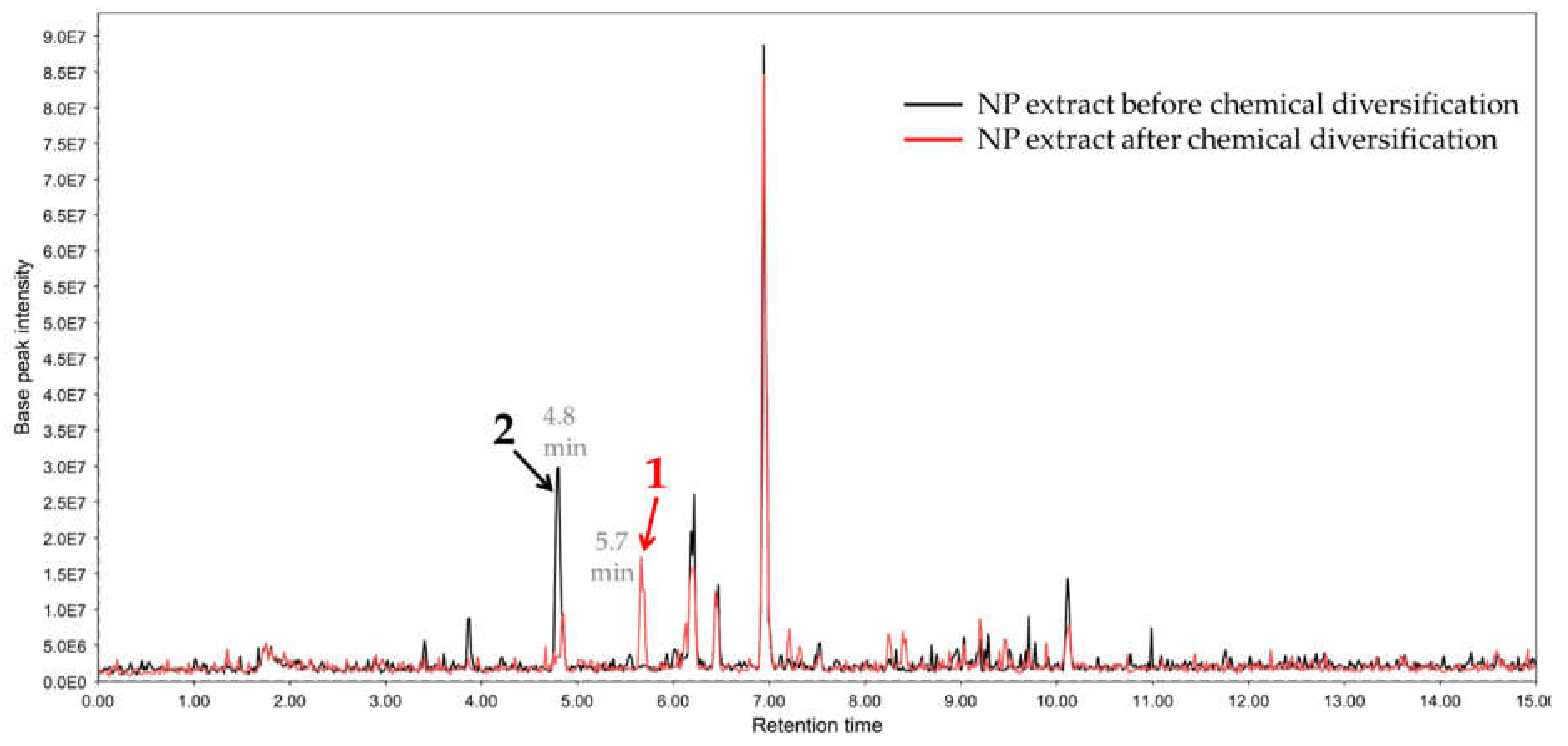
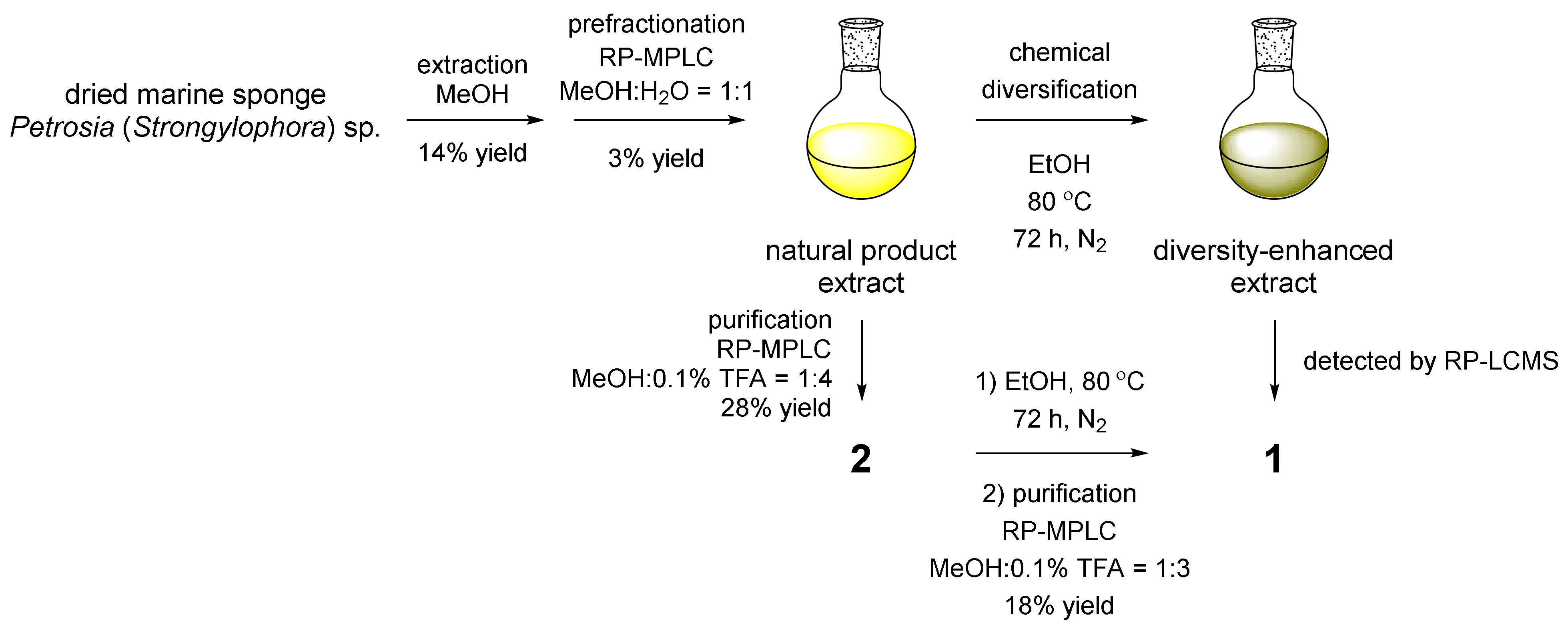
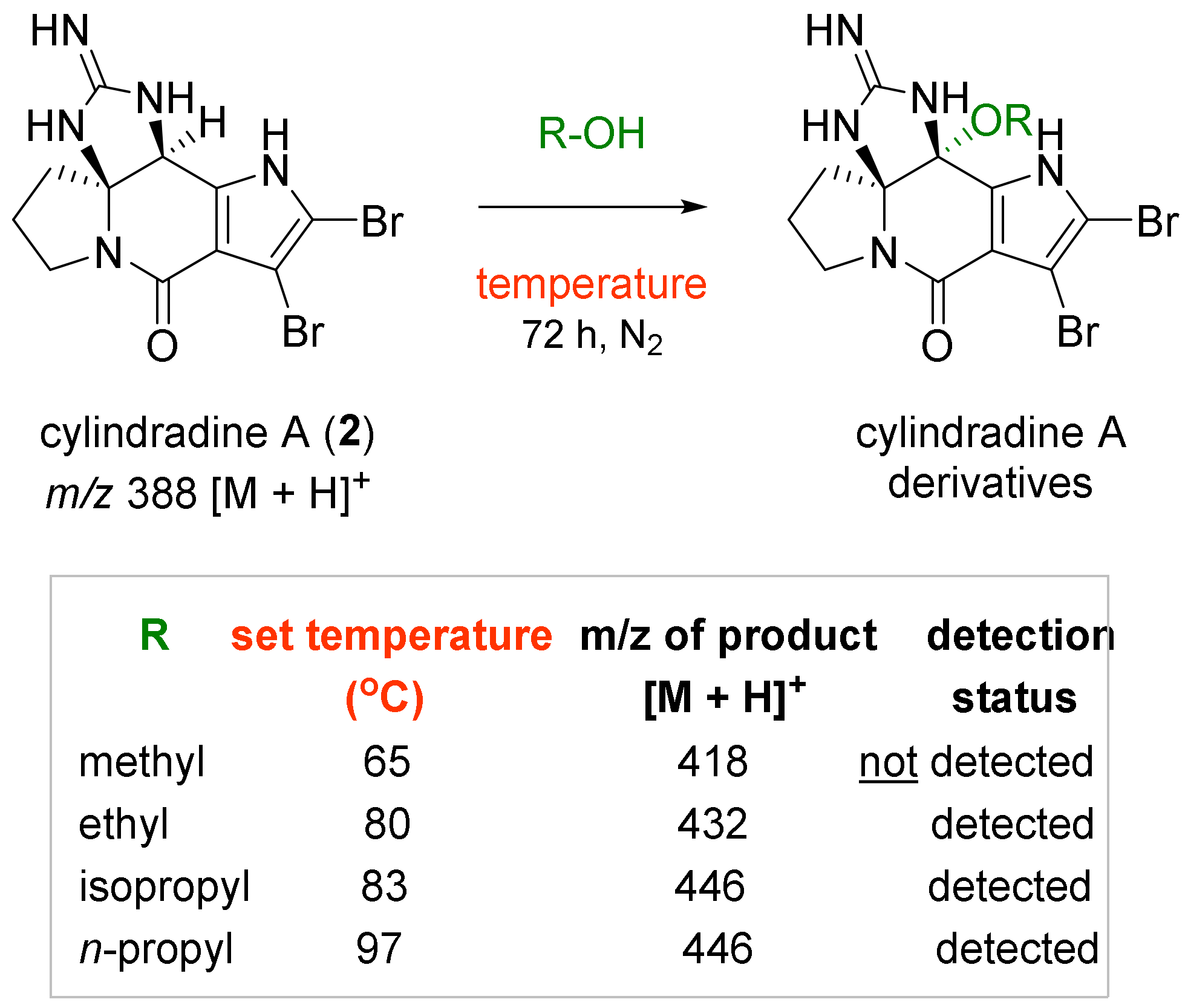
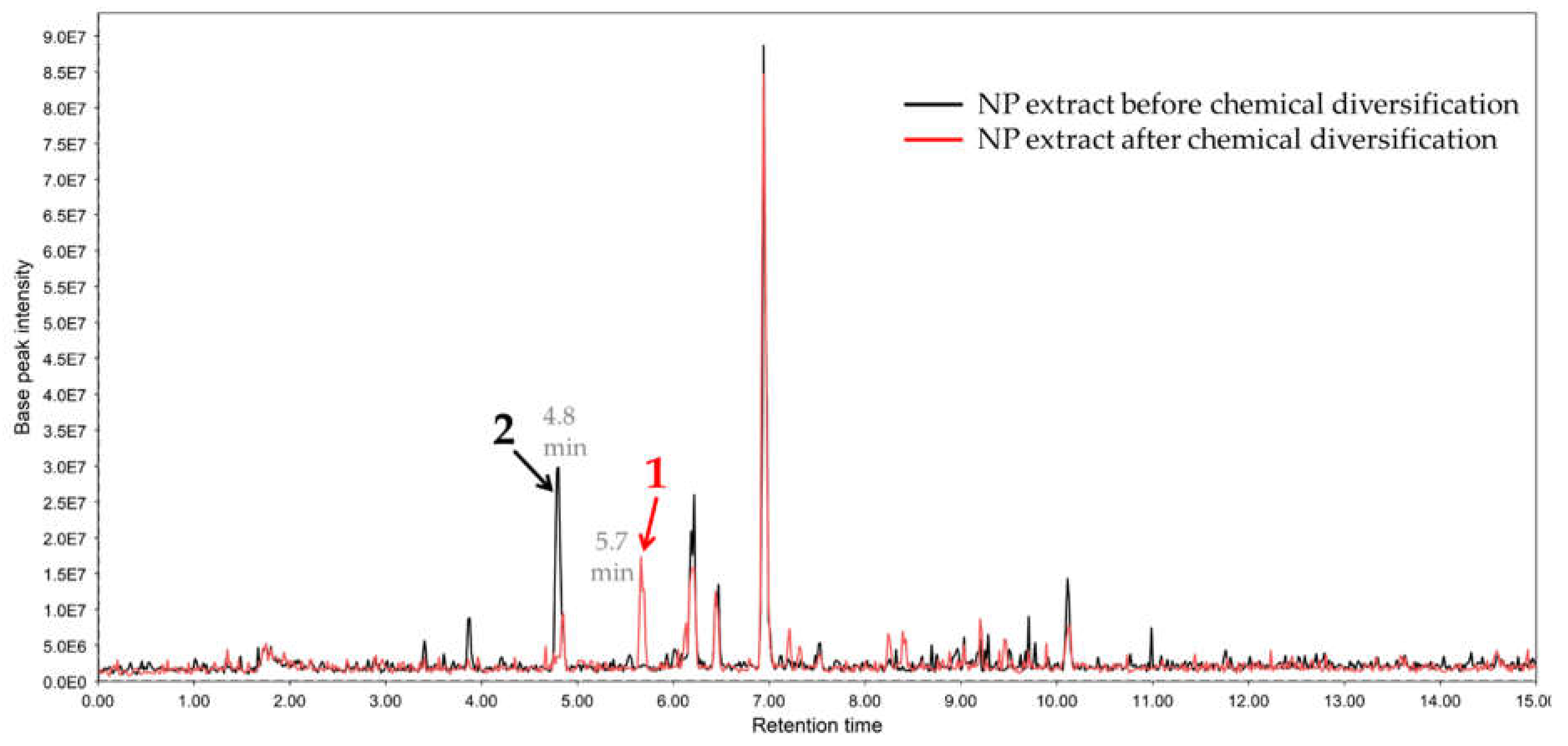
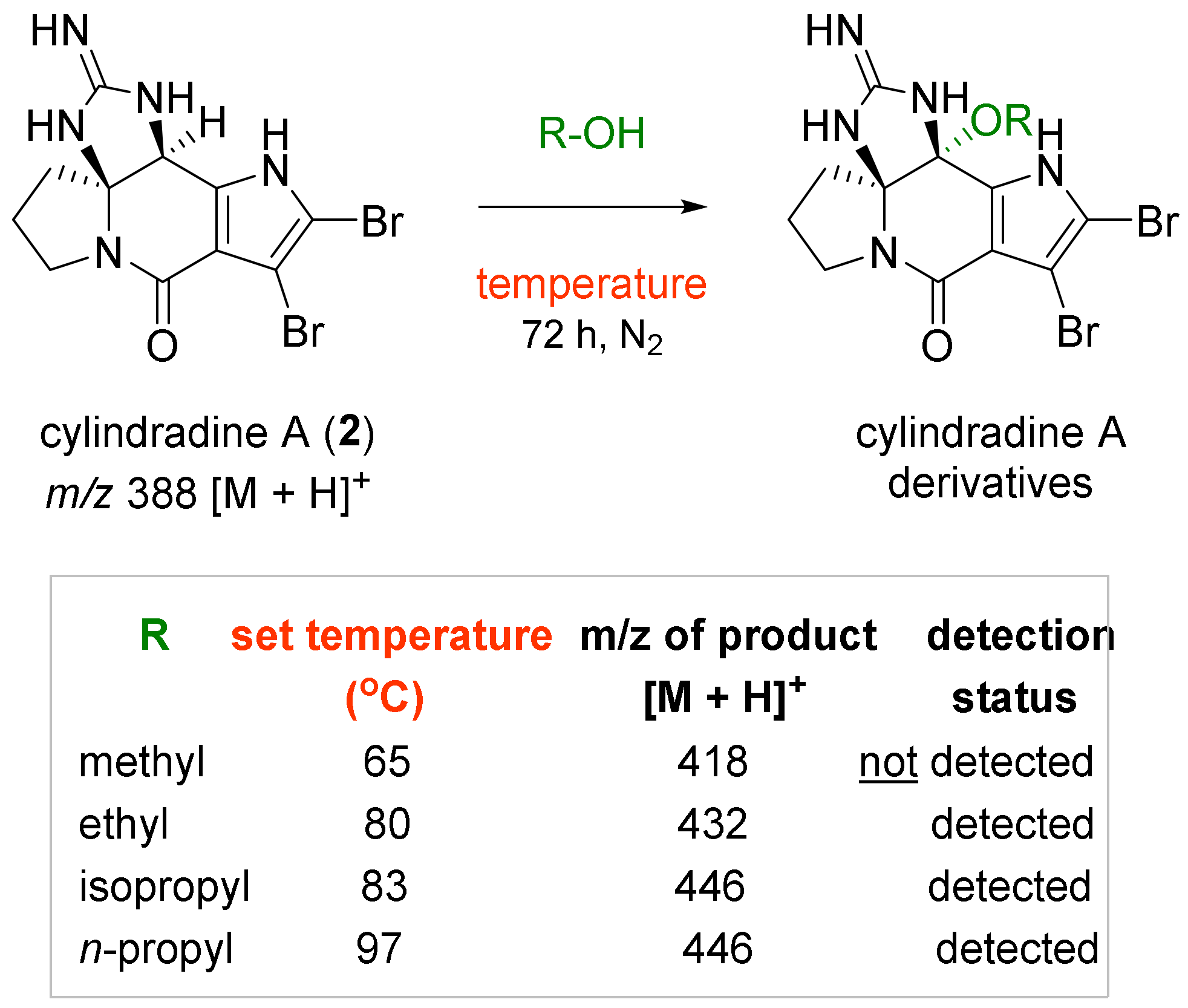
2.1. Chemical Diversification of Substances Present in NP Extract and Reversed-Phase Liquid Chromatography/Mass Spectrometry (RP-LCMS) Detection
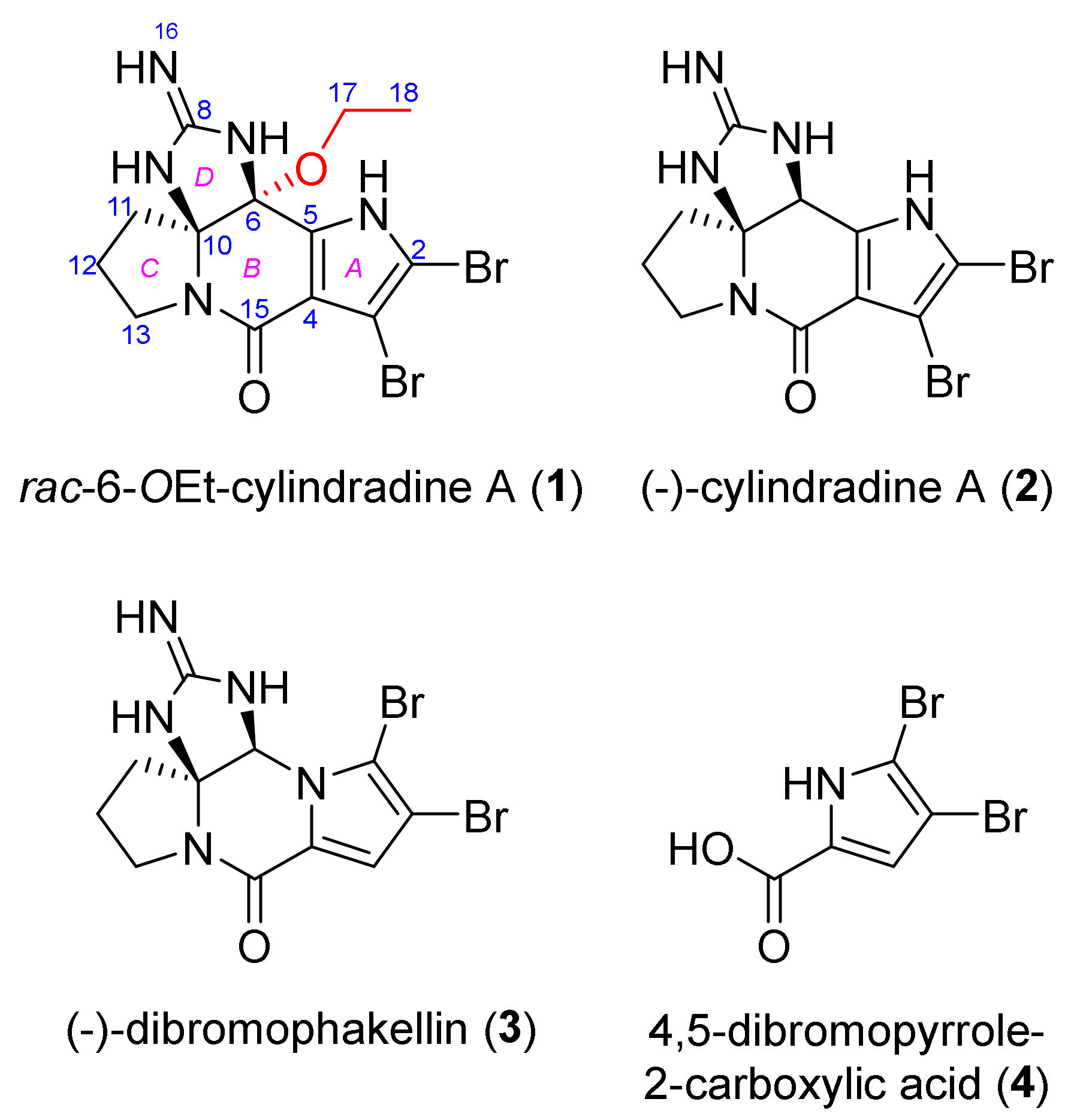
2.2. Structure Elucidation
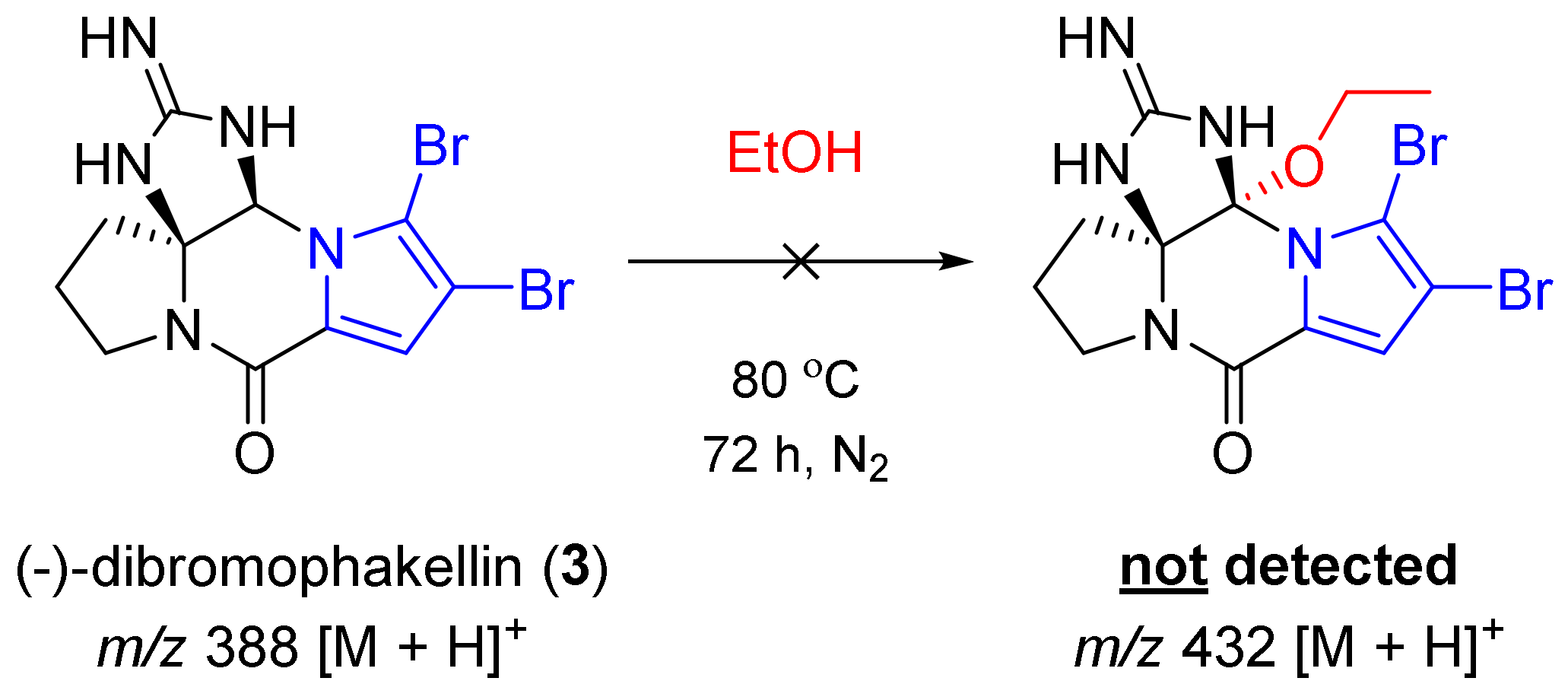
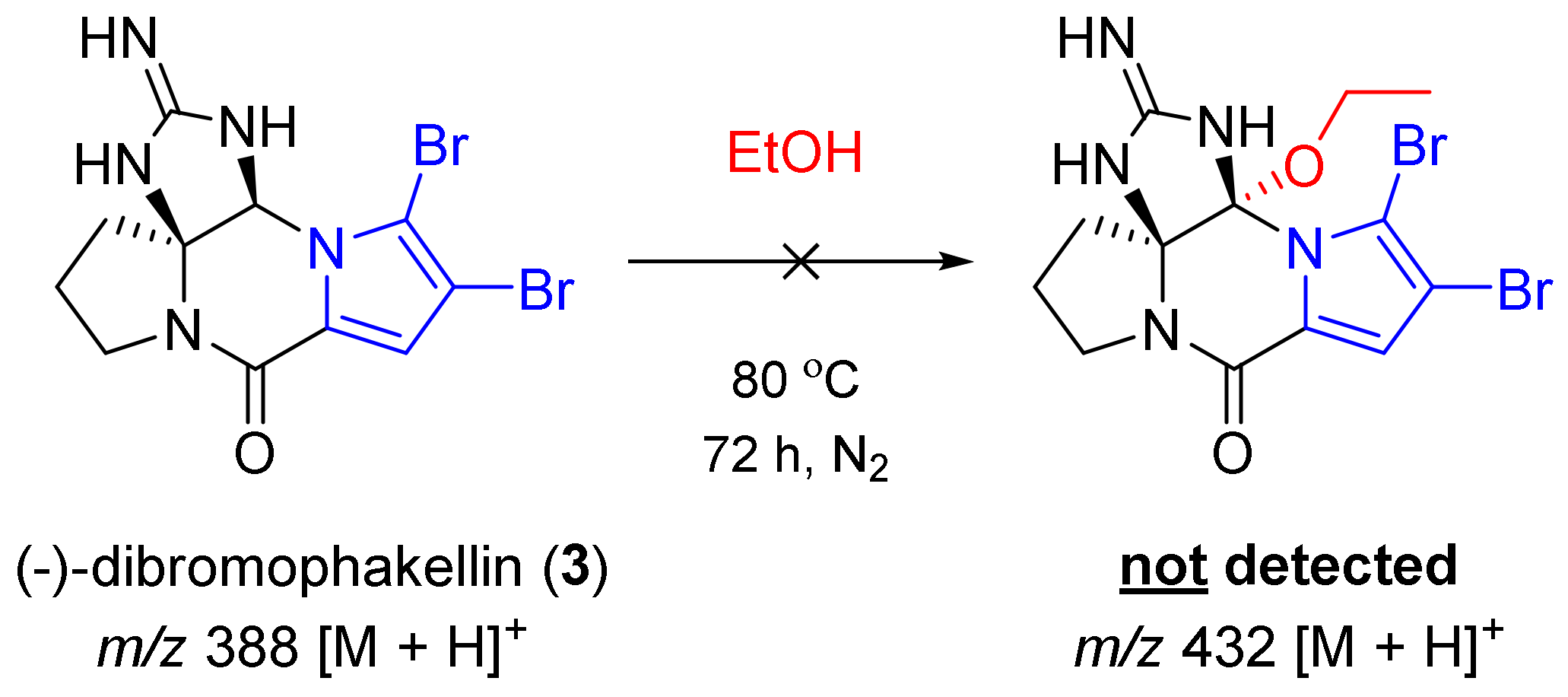
2.3. Experiments to Investigate the Possible Reaction Mechanisms
2.4. Biological Activities
2.5. Discussion: Further Consideration of Our Finding
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Sponge Material
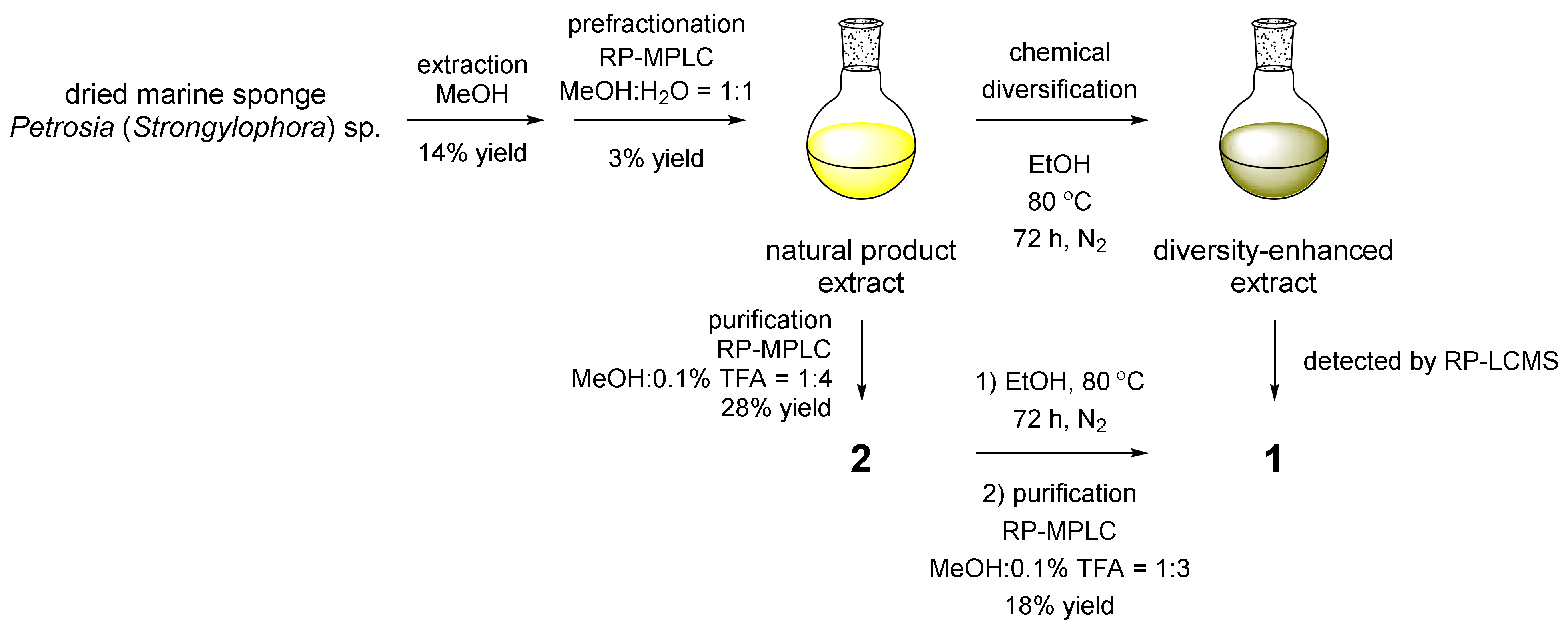
3.3. Extraction and Prefractionation of the MeOH Crude Extract
3.4. Chemical Diversification of Substances Present in NP Extracts to Prepare the Diversity-Enhanced Extracts
3.5. Larger-Scale Extraction, Prefractionation of the MeOH Crude Extract, and Purification of Naturally Occurring Compounds 2–4
3.6. Chemical Conversion of 2 with Heat-Treated EtOH and Purification of 1
3.7. Experiments to Investigate the Possible Reaction Mechanisms
3.8. Anticancer Assay
3.9. Antimycobacterial Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar]
- Butler, M.S.; Robertson, A.A.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; The International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar]
- Li, G.; Lou, H.X. Strategies to diversify natural products for drug discovery. Med. Res. Rev. 2018, 38, 1255–1294. [Google Scholar]
- Lopez, S.N.; Ramallo, I.A.; Sierra, M.G.; Zacchino, S.A.; Furlan, R.L. Chemically engineered extracts as an alternative source of bioactive natural product-like compounds. Proc. Natl. Acad. Sci. USA 2007, 104, 441–444. [Google Scholar]
- Wu, T.; Jiang, C.; Wang, L.; Morris-Natschke, S.L.; Miao, H.; Gu, L.; Xu, J.; Lee, K.H.; Gu, Q. 3,5-Diarylpyrazole derivatives obtained by ammonolysis of the total flavonoids from Chrysanthemum indicum extract show potential for the treatment of Alzheimer’s disease. J. Nat. Prod. 2015, 78, 1593–1599. [Google Scholar]
- Kawamura, T.; Matsubara, K.; Otaka, H.; Tashiro, E.; Shindo, K.; Yanagita, R.C.; Irie, K.; Imoto, M. Generation of ‘unnatural natural product’ library and identification of a small molecule inhibitor of XIAP. Bioorg. Med. Chem. 2011, 19, 4377–4385. [Google Scholar]
- Masuda, T.; Nojima, S.; Miura, Y.; Honda, S.; Masuda, A. An oxidative coupling product of luteolin with cysteine ester and its enhanced inhibitory activity for xanthine oxidase. Bioorg. Med. Chem. Lett. 2015, 25, 3117–3119. [Google Scholar]
- Salazar, M.O.; Micheloni, O.; Escalante, A.M.; Furlan, R.L. Discovery of a beta-glucosidase inhibitor from a chemically engineered extract prepared through sulfonylation. Mol. Divers. 2011, 15, 713–719. [Google Scholar]
- Mendez, L.; Salazar, M.O.; Ramallo, I.A.; Furlan, R.L. Brominated extracts as source of bioactive compounds. ACS Comb. Sci 2011, 13, 200–204. [Google Scholar]
- Garcia, P.; Salazar, M.O.; Ramallo, I.A.; Furlan, R.L. A new fluorinated tyrosinase inhibitor from a chemically engineered essential oil. ACS Comb. Sci. 2016, 18, 283–286. [Google Scholar]
- Kikuchi, H.; Sakurai, K.; Oshima, Y. Development of diversity-enhanced extracts of Curcuma zedoaria and their new sesquiterpene-like compounds. Org. Lett. 2014, 16, 1916–1919. [Google Scholar]
- Kikuchi, H.; Ichinohe, K.; Kida, S.; Murase, S.; Yamada, O.; Oshima, Y. Monoterpene indole alkaloid-like compounds based on diversity-enhanced extracts of iridoid-containing plants and their immune checkpoint inhibitory activity. Org. Lett. 2016, 18, 5948–5951. [Google Scholar]
- Kikuchi, H.; Kawai, K.; Nakashiro, Y.; Yonezawa, T.; Kawaji, K.; Kodama, E.N.; Oshima, Y. Construction of a meroterpenoid-like compounds library based on diversity-enhanced extracts. Chem. Eur. J. 2019, 25, 1106–1112. [Google Scholar]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar]
- Haque, N.; Parveen, S.; Tang, T.; Wei, J.; Huang, Z. Marine natural products in clinical use. Mar. Drugs 2022, 20, 528. [Google Scholar]
- Lee, Y.J.; Cho, Y.; Tran, H.N.K. Secondary metabolites from the marine sponges of the genus Petrosia: A literature review of 43 years of research. Mar. Drugs 2021, 19, 122. [Google Scholar]
- Kuramoto, M.; Miyake, N.; Ishimaru, Y.; Ono, N.; Uno, H. Cylindradines A and B: Novel bromopyrrole alkaloids from the marine sponge Axinella cylindratus. Org. Lett. 2008, 10, 5465–5468. [Google Scholar]
- Tasdemir, D.; Topaloglu, B.; Perozzo, R.; Brun, R.; O’Neill, R.; Carballeira, N.M.; Zhang, X.; Tonge, P.J.; Linden, A.; Ruedi, P. Marine natural products from the Turkish sponge Agelas oroides that inhibit the enoyl reductases from Plasmodium falciparum, Mycobacterium tuberculosis and Escherichia coli. Bioorg. Med. Chem. 2007, 15, 6834–6845. [Google Scholar]
- Meyer, S.W.; Köck, M. NMR studies of phakellins and isophakellins. J. Nat. Prod. 2008, 71, 1524–1529. [Google Scholar]
- Sharma, G.M.; Burkholder, P.R. Structure of dibromophakellin, a new bromine-containing alkaloid from the marine sponge Phakellia flabellata. J. Chem. Soc. D 1971, 3, 151–152. [Google Scholar]
- Forenza, S.; Minale, L.; Riccio, R.; Fattorusso, E. New bromo-pyrrole derivatives from the sponge Agelas oroides. J. Chem. Soc. D 1971, 18, 1129–1130. [Google Scholar]
- Lindel, T. Chemistry and biology of the pyrrole-imidazole alkaloids. Alkaloids Chem. Biol. 2017, 77, 117–219. [Google Scholar]
- Iwata, M.; Kanoh, K.; Imaoka, T.; Nagasawa, K. Total synthesis of (+)-cylindradine A. Chem. Commun. 2014, 50, 6991–6994. [Google Scholar]
- Lopes, N.; Ray, S.; Espada, S.F.; Bomfim, W.A.; Ray, B.; Faccin-Galhardi, L.C.; Linhares, R.E.C.; Nozawa, C. Green seaweed Enteromorpha compressa (Chlorophyta, Ulvaceae) derived sulphated polysaccharides inhibit herpes simplex virus. Int. J. Biol. Macromol. 2017, 102, 605–612. [Google Scholar]
- Kamauchi, H.; Noji, M.; Kinoshita, K.; Takanami, T.; Koyama, K. Coumarins with an unprecedented tetracyclic skeleton and coumarin dimers from chemically engineered extracts of a marine-derived fungus. Tetrahedron 2018, 74, 2846–2856. [Google Scholar]
- Du, Y.; Sun, J.; Gong, Q.; Wang, Y.; Fu, P.; Zhu, W. New alpha-pyridones with quorum-sensing inhibitory activity from diversity-enhanced extracts of a Streptomyces sp. derived from marine algae. J. Agric. Food Chem. 2018, 66, 1807–1812. [Google Scholar]
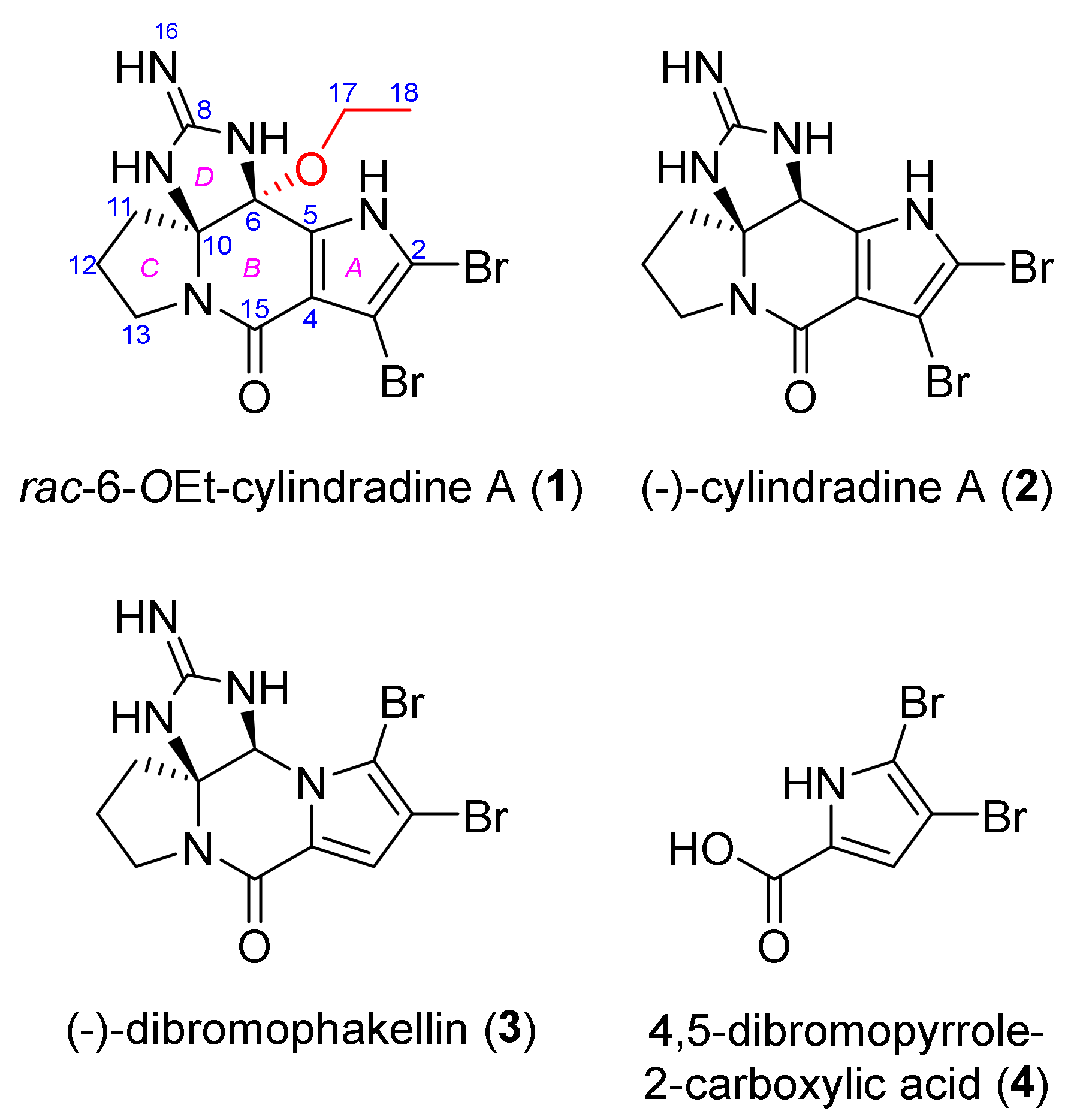
 ) and 2D-NOESY (
) and 2D-NOESY (  ) correlations of rac-6-OEt-cylindradine A (1).
) correlations of rac-6-OEt-cylindradine A (1).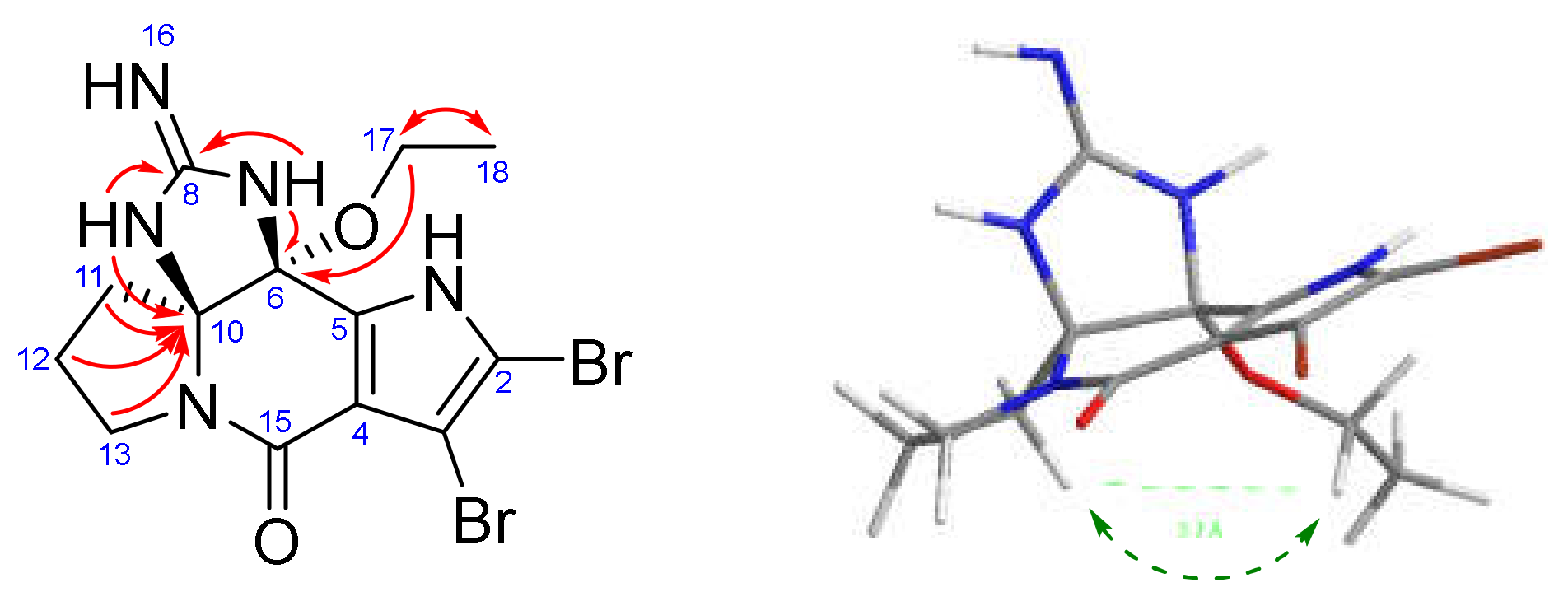

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH (Multiplicity) | δC (Type) | HMBC Correlations from H to C |
|---|---|---|---|
| 1 NH | 13.46 (1H, s) | C-2, C-3, C-4, C-5 | |
| 2 | 106.6 (C) | ||
| 3 | 95.5 (C) | ||
| 4 | 114.0 (C) | ||
| 5 | 131.5 (C) | ||
| 6 | 86.7 (C) | ||
| 7 NH | 10.03 (s) | C-6, C-8 | |
| 8 | 156.0 (C) | ||
| 9 NH | 9.52 (s) | C-8, C-10 | |
| 10 | 86.0 (C) | ||
| 11 | 2.07 (1H, m) 2.27 (1H, m) | 34.3 (CH2) | C-10, C-12, C-13 |
| 12 | 2.04 (2H, m) | 19.2 (CH2) | C-10, C-11, C-13 |
| 13 | 3.46 (1H, m) 3.57 (1H, m) | 44.3 (CH2) | C-10, C-11, C-12 |
| 14 N | |||
| 15 | 157.2 (C) | ||
| 16 NH2 | 8.46 (s) | - | |
| 17 | 3.29 (1H, dq, J = 8.7, 7.0) 3.38 (1H, dq, J = 8.7, 7.0) | 61.3 (CH2) | C-6, C-18 |
| 18 | 1.10 (3H, t, J = 7.0 Hz) | 14.8 (CH3) | C-17 |
| Compound | Anticancer Activity IC50 ± SD (μM) | Antimycobacterial Activity MIC (μM) | ||||
|---|---|---|---|---|---|---|
| Jurkat | HT-29 | PANC-1 | HeLa | Mycobacterium smegmatis | M. bovis | |
| 1 | >100 | >100 | >100 | >100 | >100 | >100 |
| 2 | ||||||
| 3 | 53 ± 8 | |||||
| 4 | 87 ± 9 | 50 | ||||
| cisplatin | 0.2 ± 0.02 | - | - | - | - | |
| isoniazid | - | 3.1 | 0.1 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirimangkalakitti, N.; Harada, K.; Yamada, M.; Arai, M.; Arisawa, M. A New Tetracyclic Bromopyrrole-Imidazole Derivative through Direct Chemical Diversification of Substances Present in Natural Product Extract from Marine Sponge Petrosia (Strongylophora) sp. Molecules 2023, 28, 143. https://doi.org/10.3390/molecules28010143
Sirimangkalakitti N, Harada K, Yamada M, Arai M, Arisawa M. A New Tetracyclic Bromopyrrole-Imidazole Derivative through Direct Chemical Diversification of Substances Present in Natural Product Extract from Marine Sponge Petrosia (Strongylophora) sp. Molecules. 2023; 28(1):143. https://doi.org/10.3390/molecules28010143
Chicago/Turabian StyleSirimangkalakitti, Natchanun, Kazuo Harada, Makito Yamada, Masayoshi Arai, and Mitsuhiro Arisawa. 2023. "A New Tetracyclic Bromopyrrole-Imidazole Derivative through Direct Chemical Diversification of Substances Present in Natural Product Extract from Marine Sponge Petrosia (Strongylophora) sp." Molecules 28, no. 1: 143. https://doi.org/10.3390/molecules28010143
APA StyleSirimangkalakitti, N., Harada, K., Yamada, M., Arai, M., & Arisawa, M. (2023). A New Tetracyclic Bromopyrrole-Imidazole Derivative through Direct Chemical Diversification of Substances Present in Natural Product Extract from Marine Sponge Petrosia (Strongylophora) sp. Molecules, 28(1), 143. https://doi.org/10.3390/molecules28010143