
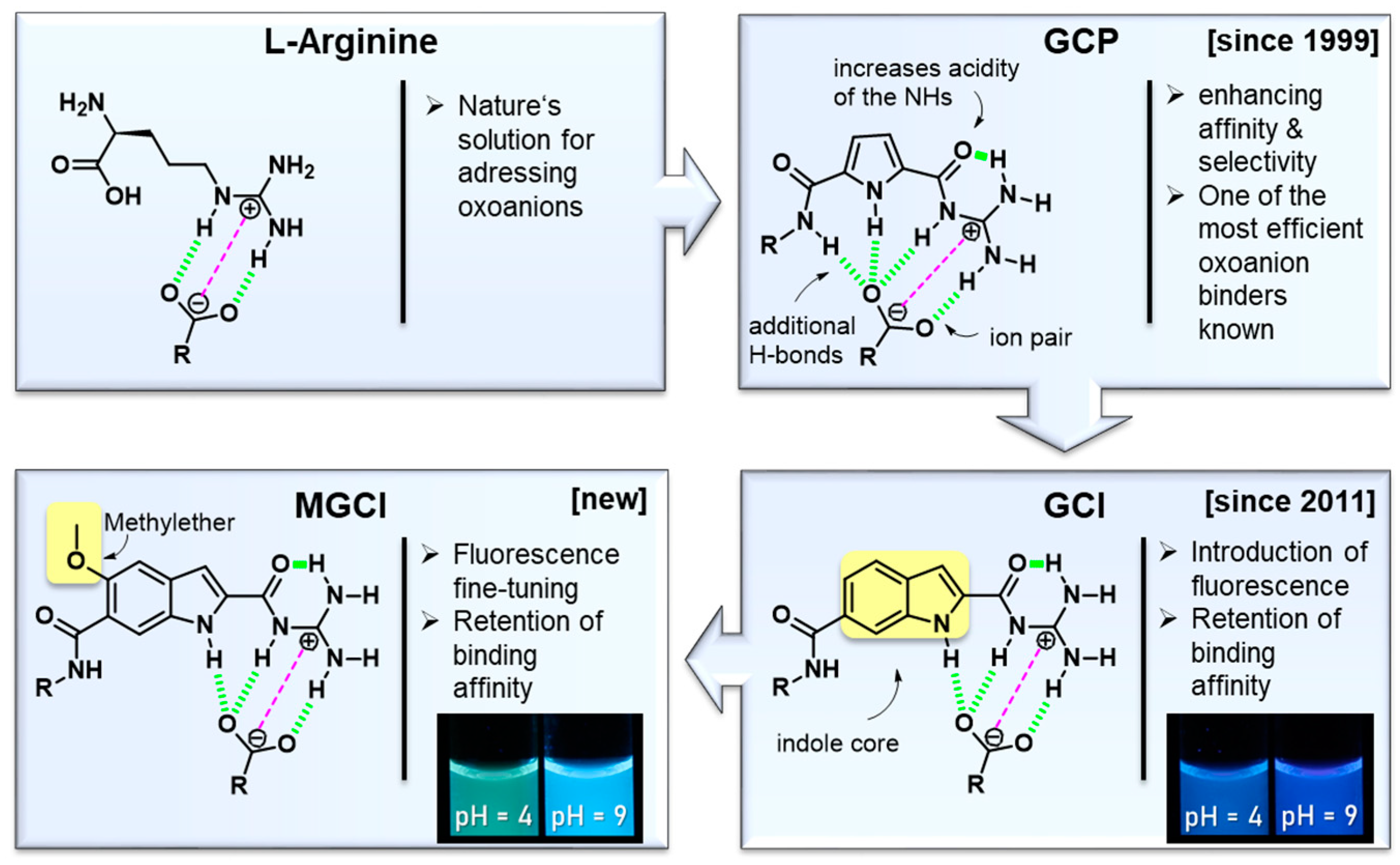
Evolution of Artificial Arginine Analogues—Fluorescent Guanidiniocarbonyl-Indoles as Efficient Oxo-Anion Binders

 and
and
Abstract
:
1. Introduction
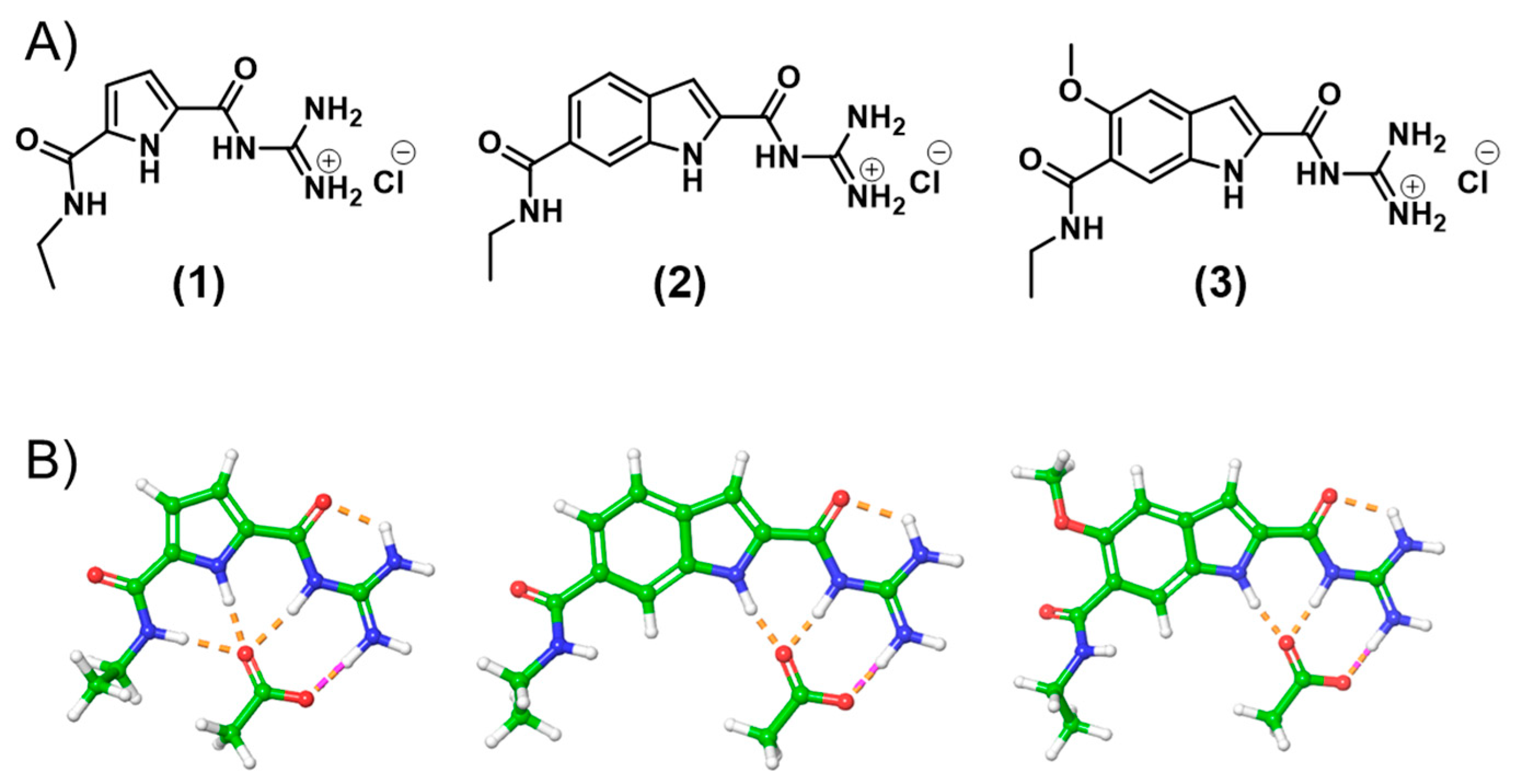
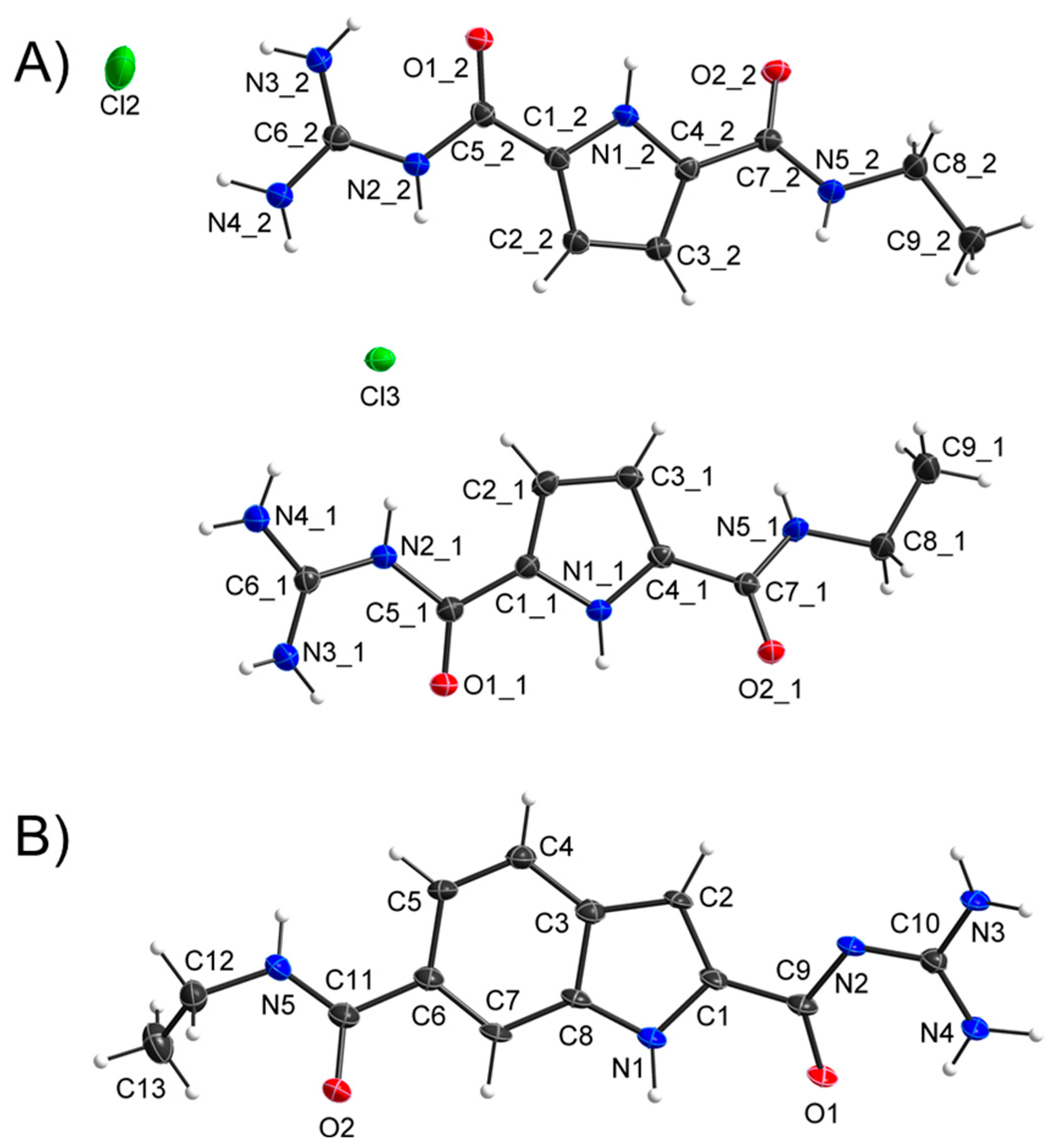
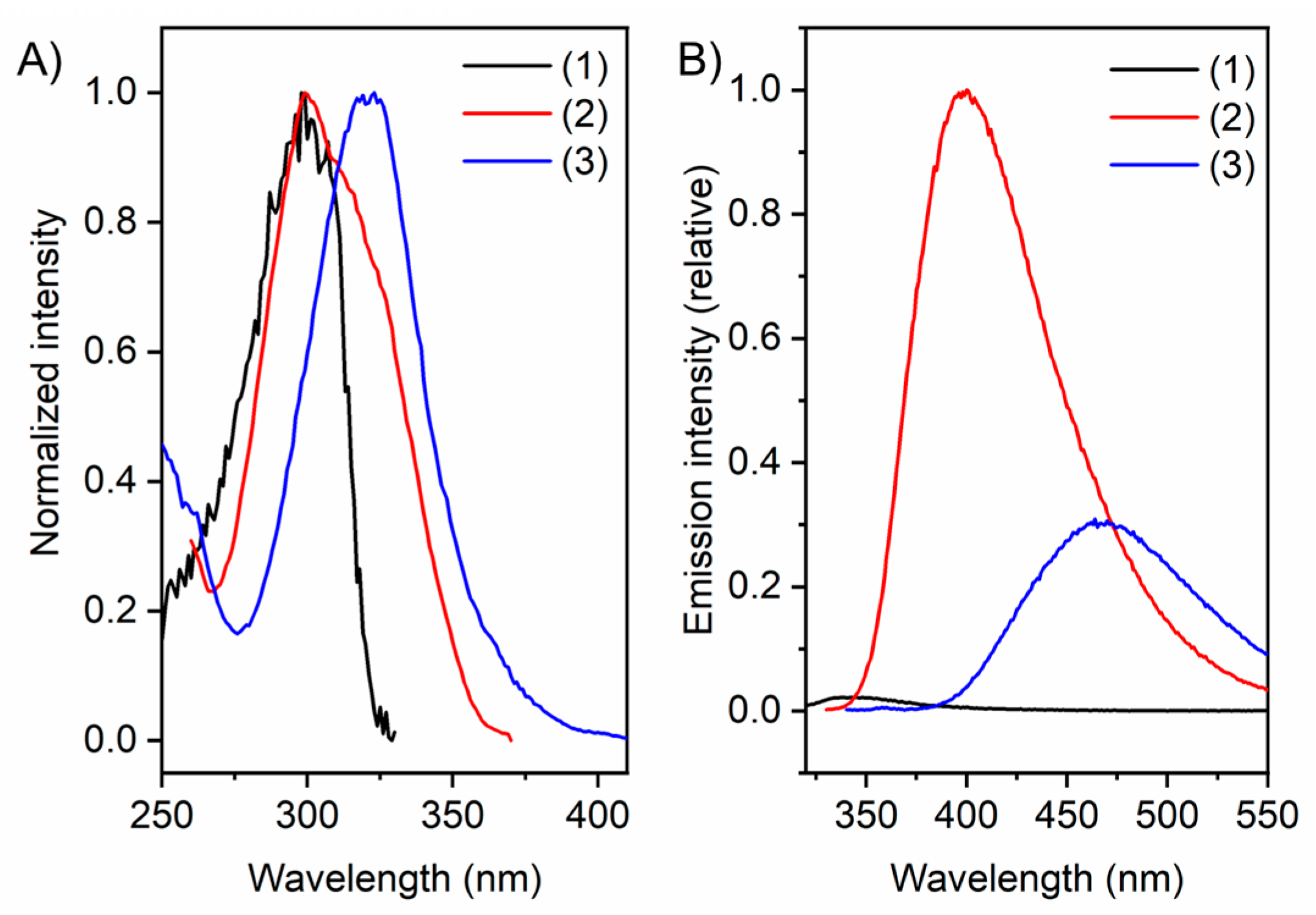
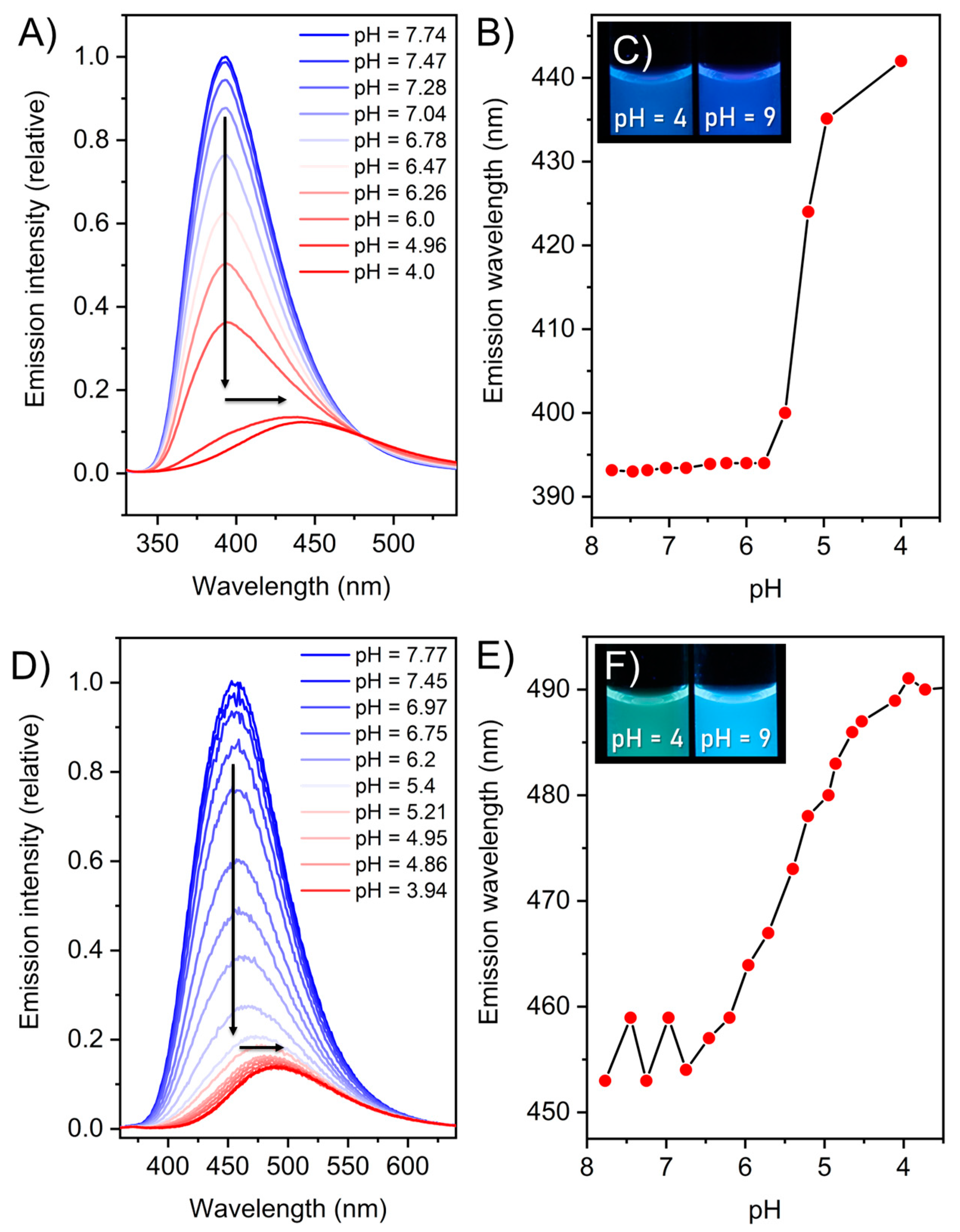
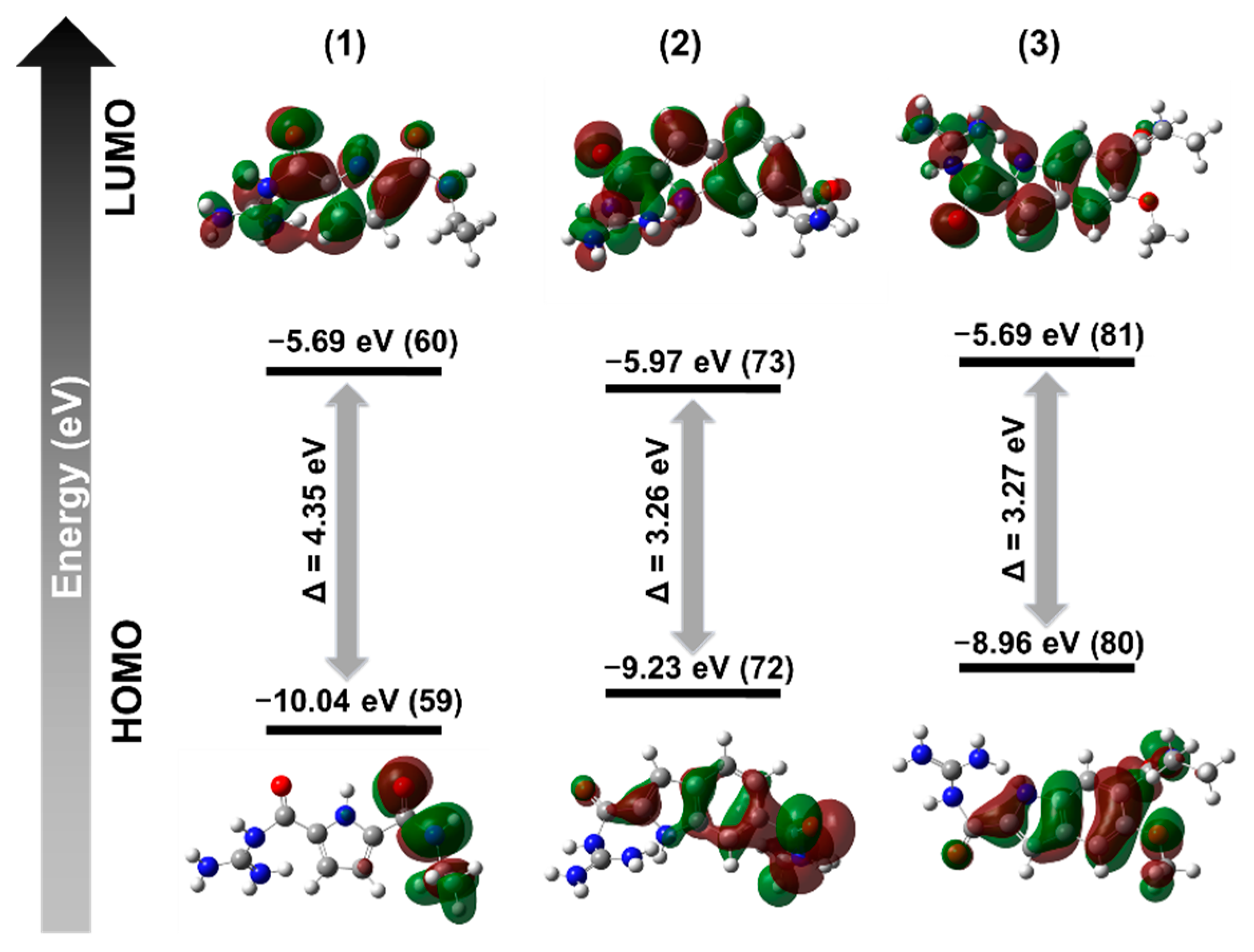
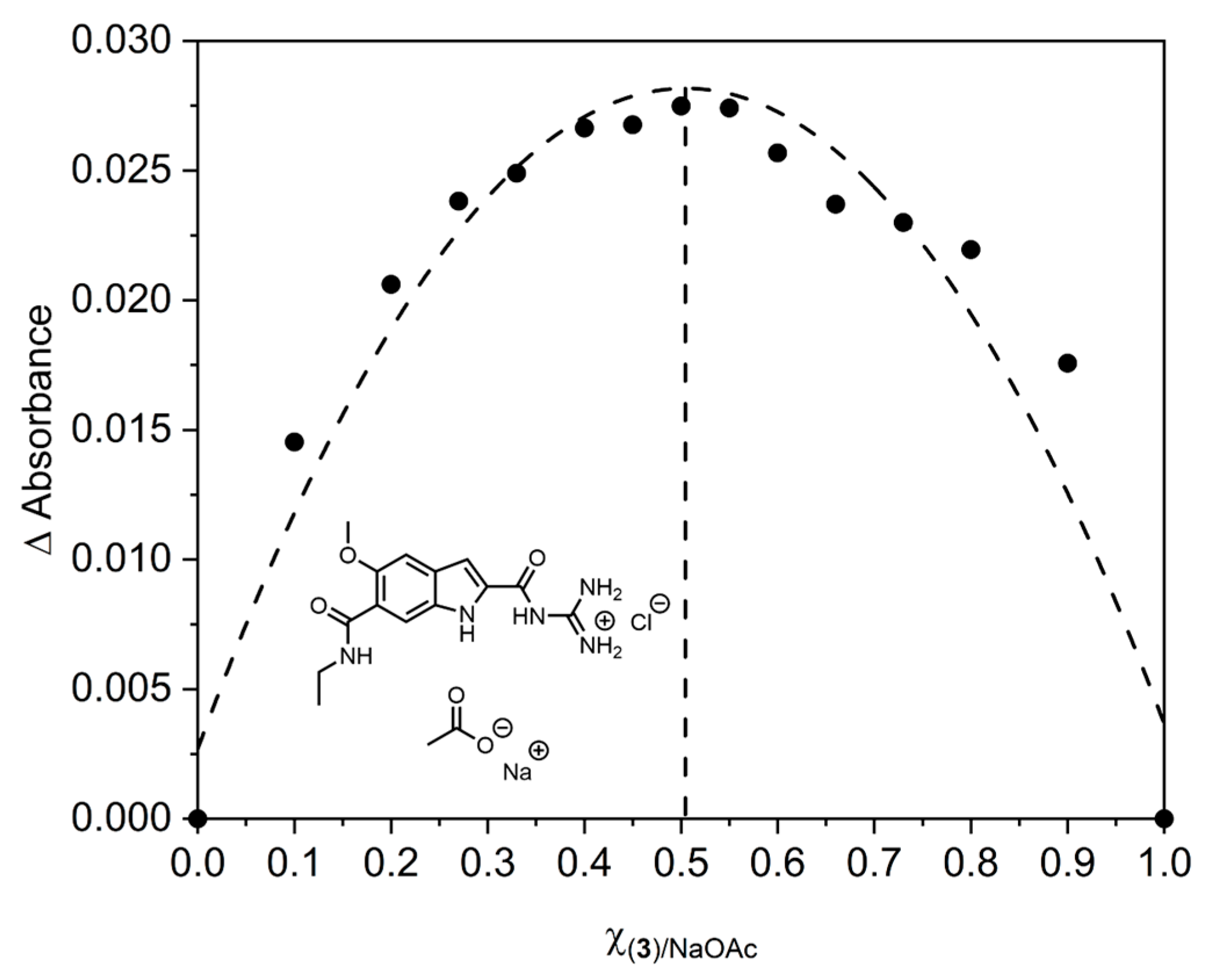
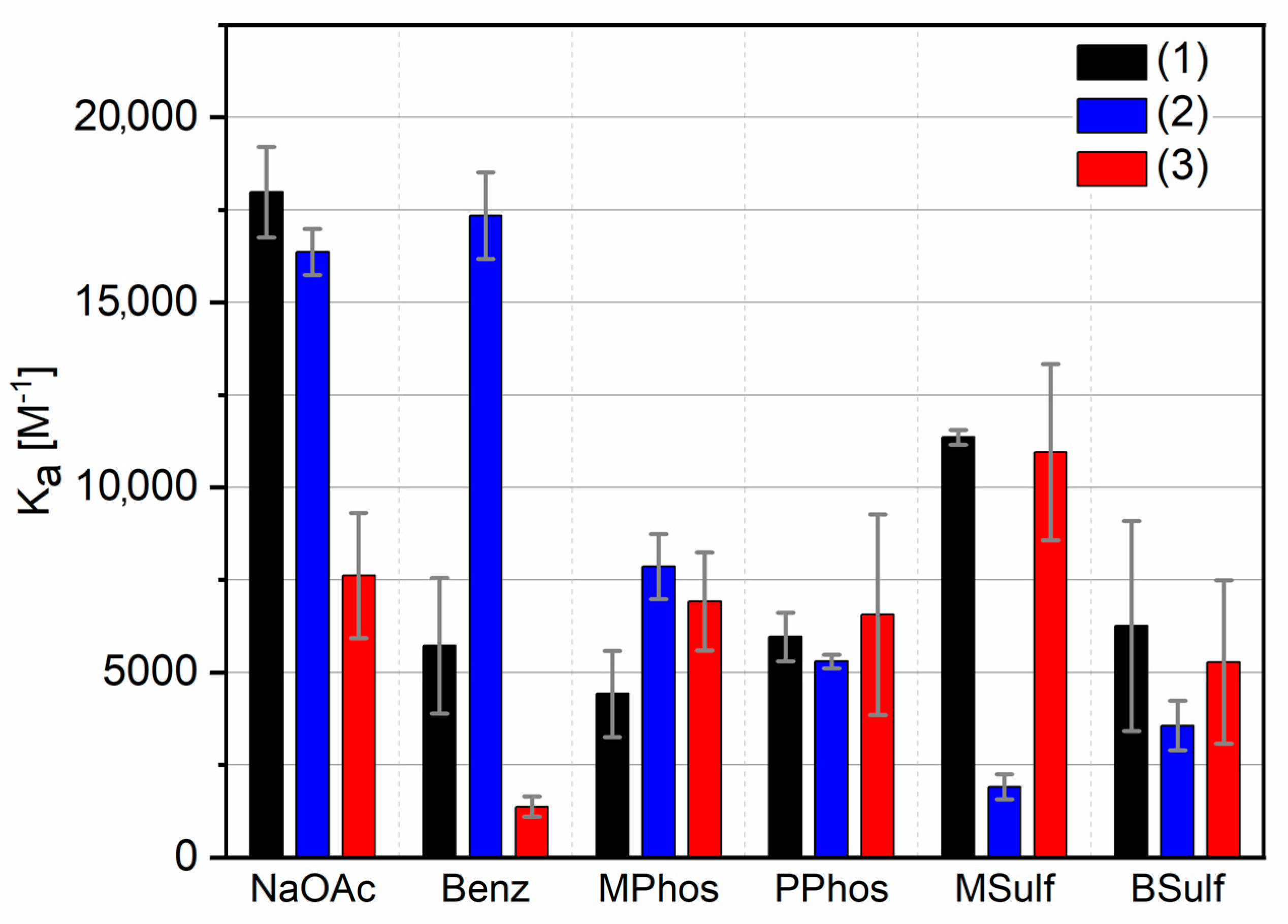
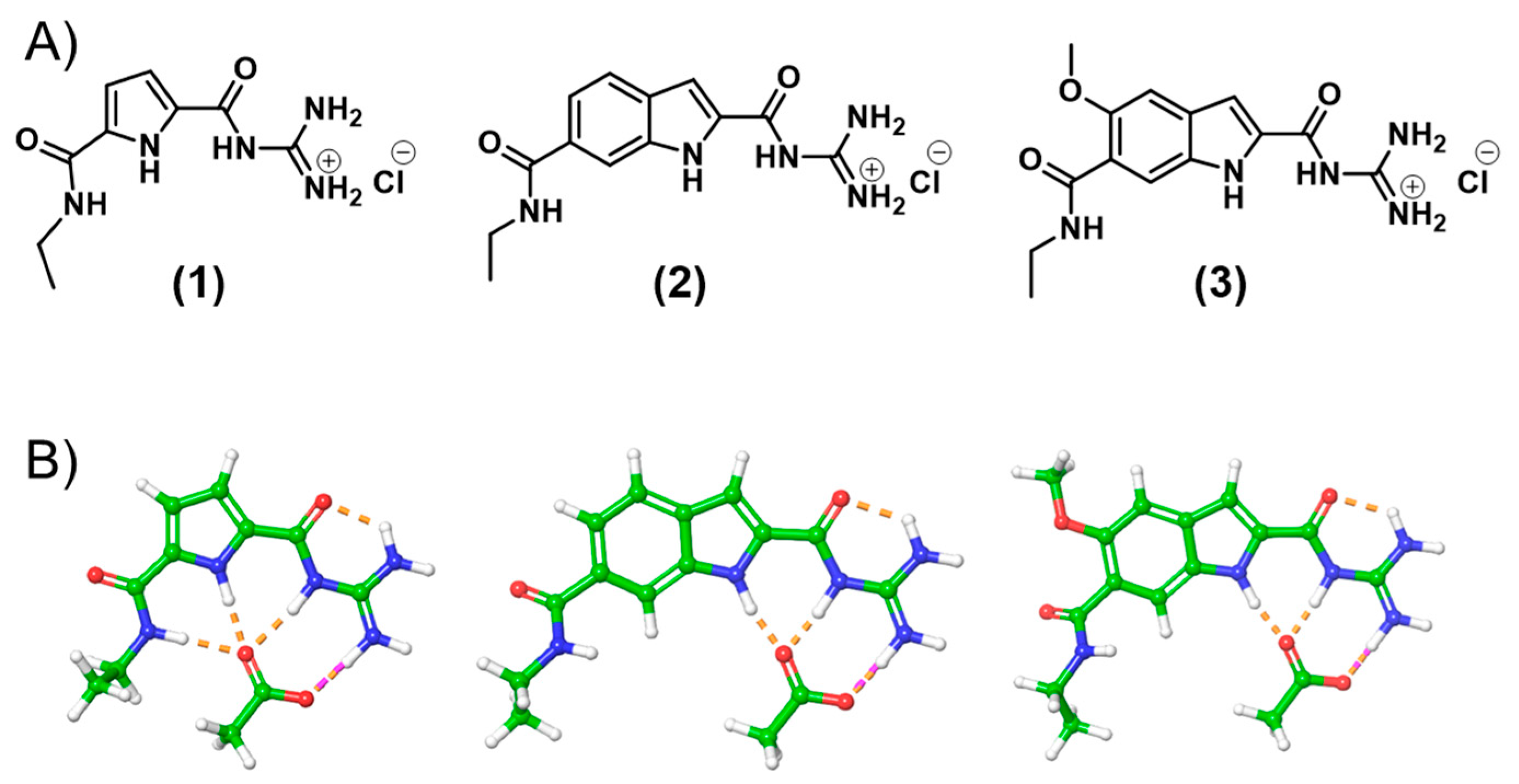
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Evans, N.H.; Beer, P.D. Advances in Anion Supramolecular Chemistry: From Recognition to Chemical Applications. Angew. Chem. Int. Ed. 2014, 53, 11716–11754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langton, M.J.; Serpell, C.J.; Beer, P.D. Anion Recognition in Water: Recent Advances from a Supramolecular and Macromolecular Perspective. Angew. Chem. Int. Ed. 2016, 55, 1974–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmuck, C. How to improve guanidinium cations for oxoanion binding in aqueous solution? The design of artificial peptide receptors. Coord. Chem. Rev. 2006, 250, 3053–3067. [Google Scholar] [CrossRef]
- Hannon, C.L.; Anslyn, E.V. The Guanidinium Group: Its Biological Role and Synthetic Analogs. In Bioorganic Chemistry Frontiers; Dugas, H., Schmidtchen, F.P., Eds.; Springer: Berlin/Heidelberg, Germany, 1993; pp. 193–255. [Google Scholar]
- Cotton, F.A.; Hazen, E.E.; Legg, M.J. Staphylococcal nuclease: Proposed mechanism of action based on structure of enzyme—thymidine 3′,5′-bisphosphate—Calcium ion complex at 1.5-Å resolution. Proc. Natl. Acad. Sci. USA 1979, 76, 2551–2555. [Google Scholar] [CrossRef] [Green Version]
- Christianson, D.W.; Lipscomb, W.N. Carboxypeptidase A. Acc. Chem. Res. 1989, 22, 62–69. [Google Scholar] [CrossRef]
- McKay, A.F.; Kreling, M.E. Preparation and Chemistry of Δ8-Hexahydro-1,4,8-Pyrimidazole, Δ9-1,5,9-Triazabicyclo(4.4.0)Decene, and Δ9-1,4,9-Triazabicyglo(5.3.0)Decene. Can. J. Chem. 1957, 35, 1438–1445. [Google Scholar] [CrossRef]
- Dietrich, B.; Fyles, T.M.; Lehn, J.-M.; Pease, L.G.; Fyles, D.L. Anion receptor molecules. Synthesis and some anion binding properties of macrocyclic guanidinium salts. J. Chem. Soc. Chem. Commun. 1978, 21, 934–936. [Google Scholar] [CrossRef]
- Müller, G.; Riede, J.; Schmidtchen, F.P. Host-Guest Bonding of Oxoanions to Guanidinium Anchor Groups. Angew. Chem. Int. Ed. 1988, 27, 1516–1518. [Google Scholar] [CrossRef]
- Echavarren, A.; Galan, A.; Lehn, J.M.; De Mendoza, J. Chiral recognition of aromatic carboxylate anions by an optically active abiotic receptor containing a rigid guanidinium binding subunit. J. Am. Chem. Soc. 2002, 111, 4994–4995. [Google Scholar] [CrossRef]
- Hannon, C.L.; Bell, D.A.; Kelly-Rowley, A.M.; Cabell, L.A.; Anslyn, E.V. Non-Aqueous Titrations as a Tool in the study of Molecular Recognition Phenomena. Uses in Distinguishing Hydrogen Bonding from Proton Transfer, the Measurement of Complex Induced pKa Shifts, and the Ability to Distinguish the Catalytic Roles of General Acids and Bases. J. Phys. Org. Chem. 1997, 10, 396–404. [Google Scholar]
- Blondeau, P.; Segura, M.; Pérez-Fernández, R.; de Mendoza, J. Molecular recognition of oxoanions based on guanidinium receptors. Chem. Soc. Rev. 2007, 36, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.P.; Geib, S.J.; Hamilton, A.D. Molecular recognition: Bis-acylguanidiniums provide a simple family of receptors for phosphodiesters. J. Am. Chem. Soc. 1992, 114, 365–366. [Google Scholar] [CrossRef]
- Linton, B.R.; Goodman, M.S.; Fan, E.; van Arman, S.A.; Hamilton, A.D. Thermodynamic Aspects of Dicarboxylate Recognition by Simple Artificial Receptors. J. Org. Chem. 2001, 66, 7313–7319. [Google Scholar] [CrossRef]
- Best, M.D.; Tobey, S.L.; Anslyn, E.V. Abiotic guanidinium containing receptors for anionic species. Coord. Chem. Rev. 2003, 240, 3–15. [Google Scholar] [CrossRef]
- Fitzmaurice, R.J.; Gaggini, F.; Srinivasan, N.; Kilburn, J.D. Carboxylate binding in polar solvents using pyridylguanidinium salts. Org. Biomol. Chem. 2007, 5, 1706–1714. [Google Scholar] [CrossRef]
- Ariga, K.; Anslyn, E.V. Manipulating the stoichiometry and strength of phosphodiester binding to a bisguanidine cleft in DMSO/water solutions. J. Org. Chem. 1992, 57, 417–419. [Google Scholar] [CrossRef]
- Zepik, H.H.; Benner, S.A. Catalysts, Anticatalysts, and Receptors for Unactivated Phosphate Diesters in Water. J. Org. Chem. 1999, 64, 8080–8083. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhong, C.; Gong, R.; Fu, E. A highly selective fluorescent probe for pyrophosphate in aqueous solution. Org. Biomol. Chem. 2008, 6, 3044–3047. [Google Scholar] [CrossRef]
- Beck, C.L.; Berg, S.A.; Winter, A.H. Pincher ferrocene-derived cation carboxylate ion pairs in aqueous DMSO. Org. Biomol. Chem. 2013, 11, 5827–5835. [Google Scholar] [CrossRef]
- Beck, C.L.; Winter, A.H. Noncovalent Catch and Release of Carboxylates in Water. J. Org. Chem. 2014, 79, 3152–3158. [Google Scholar] [CrossRef] [Green Version]
- Schmuck, C. Side chain selective binding of N-acetyl-α-amino acid carboxylates by a 2-(guanidiniocarbonyl)pyrrole receptor in aqueous solvents. Chem. Commun. 1999, 843–844. [Google Scholar] [CrossRef]
- Schmuck, C. Carboxylate Binding by 2-(Guanidiniocarbonyl)pyrrole Receptors in Aqueous Solvents: Improving the Binding Properties of Guanidinium Cations through Additional Hydrogen Bonds. Chem. Eur. J. 2000, 6, 709–718. [Google Scholar] [CrossRef]
- Giese, M.; Niemeyer, J.; Voskuhl, J. Guanidiniocarbonyl-Pyrroles (GCP)—20 Years of the Schmuck Binding Motif. ChemPlusChem 2020, 85, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Kuchelmeister, H.Y.; Schmuck, C. Nucleotide Recognition in Water by a Guanidinium-Based Artificial Tweezer Receptor. Chem. Eur. J. 2011, 17, 5311–5318. [Google Scholar] [CrossRef]
- Li, M.; Radić Stojković, M.; Ehlers, M.; Zellermann, E.; Piantanida, I.; Schmuck, C. Use of an Octapeptide–Guanidiniocarbonylpyrrole Conjugate for the Formation of a Supramolecular β-Helix That Self-Assembles into pH-Responsive Fibers. Angew. Chem. Int. Ed. 2016, 55, 13015–13018. [Google Scholar] [CrossRef]
- Gigante, A.; Grad, J.N.; Briels, J.; Bartel, M.; Hoffmann, D.; Ottmann, C.; Schmuck, C. A new class of supramolecular ligands stabilizes 14-3-3 protein–protein interactions by up to two orders of magnitude. Chem. Commun. 2019, 55, 111–114. [Google Scholar] [CrossRef]
- Vallet, C.; Aschmann, D.; Beuck, C.; Killa, M.; Meiners, A.; Mertel, M.; Ehlers, M.; Bayer, P.; Schmuck, C.; Giese, M.; et al. Functional Disruption of the Cancer-Relevant Interaction between Survivin and Histone H3 with a Guanidiniocarbonyl Pyrrole Ligand. Angew. Chem. Int. Ed. 2020, 59, 5567–5571. [Google Scholar] [CrossRef]
- Li, M.; Schlesiger, S.; Knauer, S.K.; Schmuck, C. A Tailor-Made Specific Anion-Binding Motif in the Side Chain Transforms a Tetrapeptide into an Efficient Vector for Gene Delivery. Angew. Chem. Int. Ed. 2015, 54, 2941–2944. [Google Scholar] [CrossRef]
- Kuchelmeister, H.Y.; Gutschmidt, A.; Tillmann, S.; Knauer, S.; Schmuck, C. Efficient gene delivery into cells by a surprisingly small three-armed peptide ligand. Chem. Sci. 2012, 3, 996–1002. [Google Scholar] [CrossRef]
- Hatai, J.; Schmuck, C. Diverse Properties of Guanidiniocarbonyl Pyrrole-Based Molecules: Artificial Analogues of Arginine. Acc. Chem. Res. 2019, 52, 1709–1720. [Google Scholar] [CrossRef]
- Rether, C.; Schmuck, C. Carboxylate Binding by Indole-Based Guanidinium Receptors: Acylguanidinium Cations are Better than Aromatic Guanidinium Cations. Eur. J. Org. Chem. 2011, 2011, 1459–1466. [Google Scholar] [CrossRef]
- Ravanat, J.L.; Douki, T.; Cadet, J. Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B Biol. 2001, 63, 88–102. [Google Scholar] [CrossRef]
- Holtum, T.; Kumar, V.; Sebena, D.; Voskuhl, J.; Schlücker, S. UV Resonance Raman Spectroscopy of the Supramolecular Ligand Guanidiniocarbonyl Indole (GCI) with 244 nm Laser Excitation. Beilstein J. Org. Chem. 2020, 16, 2911–2919. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, C.; Wienand, W. Highly Stable Self-Assembly in Water: Ion Pair Driven Dimerization of a Guanidiniocarbonyl Pyrrole Carboxylate Zwitterion. J. Am. Chem. Soc. 2003, 125, 452–459. [Google Scholar] [CrossRef]
- Asokan, A.; Cho, M.J. Exploitation of Intracellular pH Gradients in the Cellular Delivery of Macromolecules. J. Pharm. Sci. 2002, 91, 903–913. [Google Scholar] [CrossRef]
- Schug, K.A.; Lindner, W. Noncovalent Binding between Guanidinium and Anionic Groups: Focus on Biological- and Synthetic-Based Arginine/Guanidinium Interactions with Phosph[on]ate and Sulf[on]ate Residues. Chem. Rev. 2005, 105, 67–114. [Google Scholar] [CrossRef]
- Ravikumar, I.; Ghosh, P. Recognition and separation of sulfate anions. Chem. Soc. Rev. 2012, 41, 3077–3098. [Google Scholar] [CrossRef]
- Job, P. Spectrographic study of the formation of complexes in solution and their stability. Compt. Rend. 1925, 180, 928–930. [Google Scholar]
- MacCarthy, P. Simplified experimental route for obtaining Job’s curves. Anal. Chem. 1978, 50, 2165. [Google Scholar] [CrossRef]
- Available online: http://supramolecular.org (accessed on 25 May 2021).
- Brynn Hibbert, D.; Thordarson, P. The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis. Chem. Commun. 2016, 52, 12792–12805. [Google Scholar] [CrossRef] [Green Version]
- Schmuck, C.; Schwegmann, M. A Molecular Flytrap for the Selective Binding of Citrate and Other Tricarboxylates in Water. J. Am. Chem. Soc. 2005, 127, 3373–3379. [Google Scholar] [CrossRef] [PubMed]
- García-España, E.; Díaz, P.; Llinares, J.M.; Bianchi, A. Anion coordination chemistry in aqueous solution of polyammonium receptors. Coord. Chem. Rev. 2006, 250, 2952–2986. [Google Scholar] [CrossRef]
- Gibb, B.C. Hitting the buffers. Nat. Chem. 2021, 13, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Rehm, T.H.; Schmuck, C. Ion-pair induced self-assembly in aqueous solvents. Chem. Soc. Rev. 2010, 39, 3597–3611. [Google Scholar] [CrossRef]
- Tsuji, T.; Onoda, M.; Otani, Y.; Ohwada, T.; Nakajima, T.; Hirao, K. Theoretical study on the excited states of heteroarene chromophores: Comparison of calculated and experimental values. Chem. Phys. Lett. 2009, 473, 196–200. [Google Scholar] [CrossRef]
- Vogrig, A.; Dorr, L.; Bouzidi, N.; Boucherle, B.; Wattiez, A.S.; Cassier, E.; Vallon, G.; Ripoche, I.; Abrunhosa-Thomas, I.; Marin, P. Structure-based design of PDZ ligands as inhibitors of 5-HT(2A) receptor/PSD-95 PDZ1 domain interaction possessing anti-hyperalgesic activity. ACS Chem. Biol. 2013, 8, 2209–2216. [Google Scholar] [CrossRef]
- Schmuck, C.; Bickert, V.; Merschky, M.; Geiger, L.; Rupprecht, D.; Dudaczek, J.; Wich, P.; Rehm, T.; Machon, U. A facile and efficient multi-gram synthesis of N-protected 5-(Guanidinocarbonyl)-1H-pyrrole-2-carboxylic acids. Eur. J. Org. Chem. 2008, 2, 324–329. [Google Scholar] [CrossRef]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef]
- Hirose, K. Determination of Binding Constants. In Analytical Methods in Supramolecular Chemistry; Wiley-VCH: Weinheim, Germany, 2006; pp. 17–54. [Google Scholar]
- C4.2.2.1 Determination of the Acidity Constant of Bromothymol Blue; LD DIDACTIC GmbH: Hürth, Germany, 2014.
- Sheldrick, G.M. Phase Annealing in Shelx-90 - Direct Methods for Larger Structures. Acta Crystallogr. A 1990, 46, 467–473. [Google Scholar] [CrossRef]
- Hubschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta. Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 09, Revision A.02, Gaussian, Inc.: Wallingford, CT, USA.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λabs [a] ± 5 [nm] | λem [a] [nm] | λex [a] [nm] | Stokes Shift [cm−1] | ΦF [d] ± 1 [%] | ΦF [b] ± 1 [%] | ΦF [e] ± 1 [%] | |
|---|---|---|---|---|---|---|---|
| 1 | 296 | 344 | 299 | 4714 | - [c] | - [c] | - [c] |
| 2 | 314 | 400 | 299 | 6847 | 8.9 | 14.6 | 17.1 |
| 3 | 320 | 468 | 321 | 9882 | 2.6 | 7.1 | 15.7 |
| (1) | (2) | (3) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Kass [M−1] | σ [M−1] | log K | Kass [M−1] | σ [M−1] | log K | Kass [M−1] | σ [M−1] | log K | |
| NaOAc | 17,981 | 1220 | 4.25 | 16364 | 626 | 4.21 | 7617 | 1690 | 3.88 |
| Benz | 5727 | 1831 | 3.76 | 17,339 | 1173 | 4.24 | 1376 | 273 | 3.14 |
| Mphos | 4421 | 1164 | 3.65 | 7861 | 881 | 3.90 | 6919 | 1320 | 3.84 |
| PPhos | 5963 | 653 | 3.78 | 5298 | 181 | 3.72 | 6562 | 2712 | 3.82 |
| MSulf | 11,360 | 196 | 4.06 | 1907 | 338 | 3.28 | 10,957 | 2378 | 4.04 |
| BSulf | 6258 | 2841 | 3.80 | 3565 | 665 | 3.55 | 5284 | 2202 | 3.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebena, D.; Rudolph, K.; Roy, B.; Wölper, C.; Nitschke, T.; Lampe, S.; Giese, M.; Voskuhl, J. Evolution of Artificial Arginine Analogues—Fluorescent Guanidiniocarbonyl-Indoles as Efficient Oxo-Anion Binders. Molecules 2022, 27, 3005. https://doi.org/10.3390/molecules27093005
Sebena D, Rudolph K, Roy B, Wölper C, Nitschke T, Lampe S, Giese M, Voskuhl J. Evolution of Artificial Arginine Analogues—Fluorescent Guanidiniocarbonyl-Indoles as Efficient Oxo-Anion Binders. Molecules. 2022; 27(9):3005. https://doi.org/10.3390/molecules27093005
Chicago/Turabian StyleSebena, Daniel, Kevin Rudolph, Bibhisan Roy, Christoph Wölper, Till Nitschke, Sarah Lampe, Michael Giese, and Jens Voskuhl. 2022. "Evolution of Artificial Arginine Analogues—Fluorescent Guanidiniocarbonyl-Indoles as Efficient Oxo-Anion Binders" Molecules 27, no. 9: 3005. https://doi.org/10.3390/molecules27093005
APA StyleSebena, D., Rudolph, K., Roy, B., Wölper, C., Nitschke, T., Lampe, S., Giese, M., & Voskuhl, J. (2022). Evolution of Artificial Arginine Analogues—Fluorescent Guanidiniocarbonyl-Indoles as Efficient Oxo-Anion Binders. Molecules, 27(9), 3005. https://doi.org/10.3390/molecules27093005