
Computational-Based Discovery of the Anti-Cancer Activities of Pyrrole-Based Compounds Targeting the Colchicine-Binding Site of Tubulin

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Compounds
2.2. Molecular Modeling Methods
2.3. Cell Lines and Culture Conditions
2.4. Antibodies
2.5. Western Blotting Analysis
2.6. Tubulin Polymerization Assay
2.7. Flow Cytometry
2.8. Statistics
3. Results
3.1. Synthesis of 2-Aminopyrrole Derivatives and Their Characteristics
3.2. EAPCs Reduce the Viability of the Epithelial Cancer Cell Lines In Vitro
3.3. EAPC-67 and -70 Arrest Cancer Cells in M-Phase and Inhibit Tubulin Polymerization
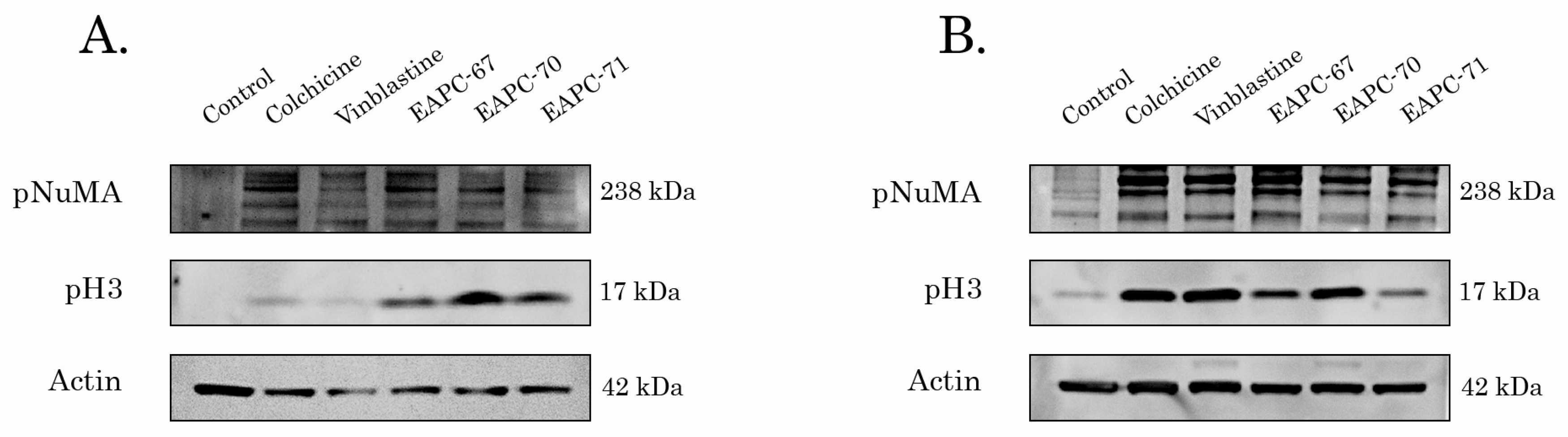
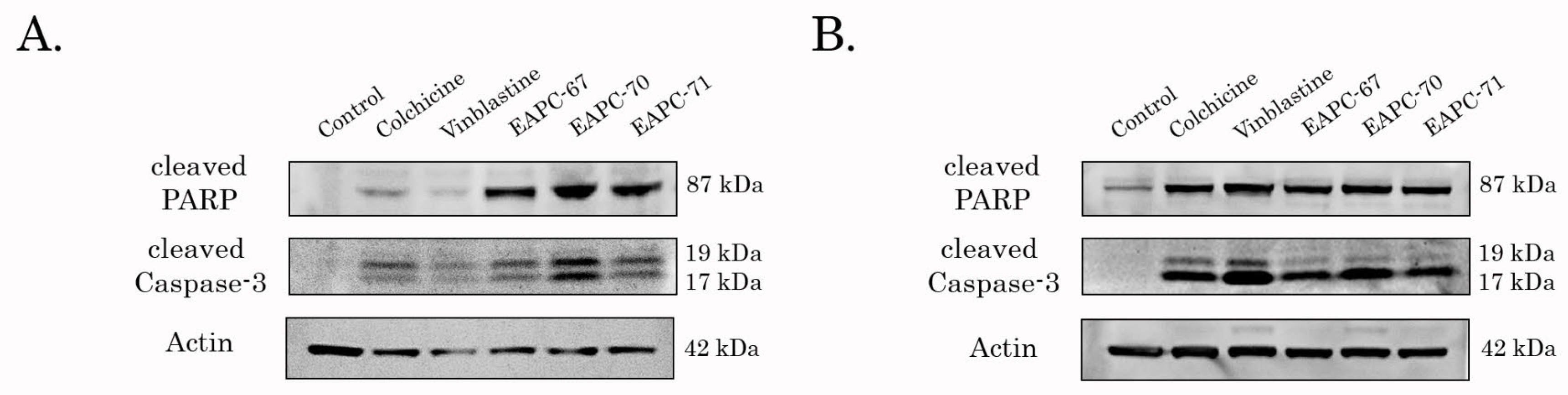
3.4. EAPCs Induce Apoptosis of Breast and Lung Cancer Cells
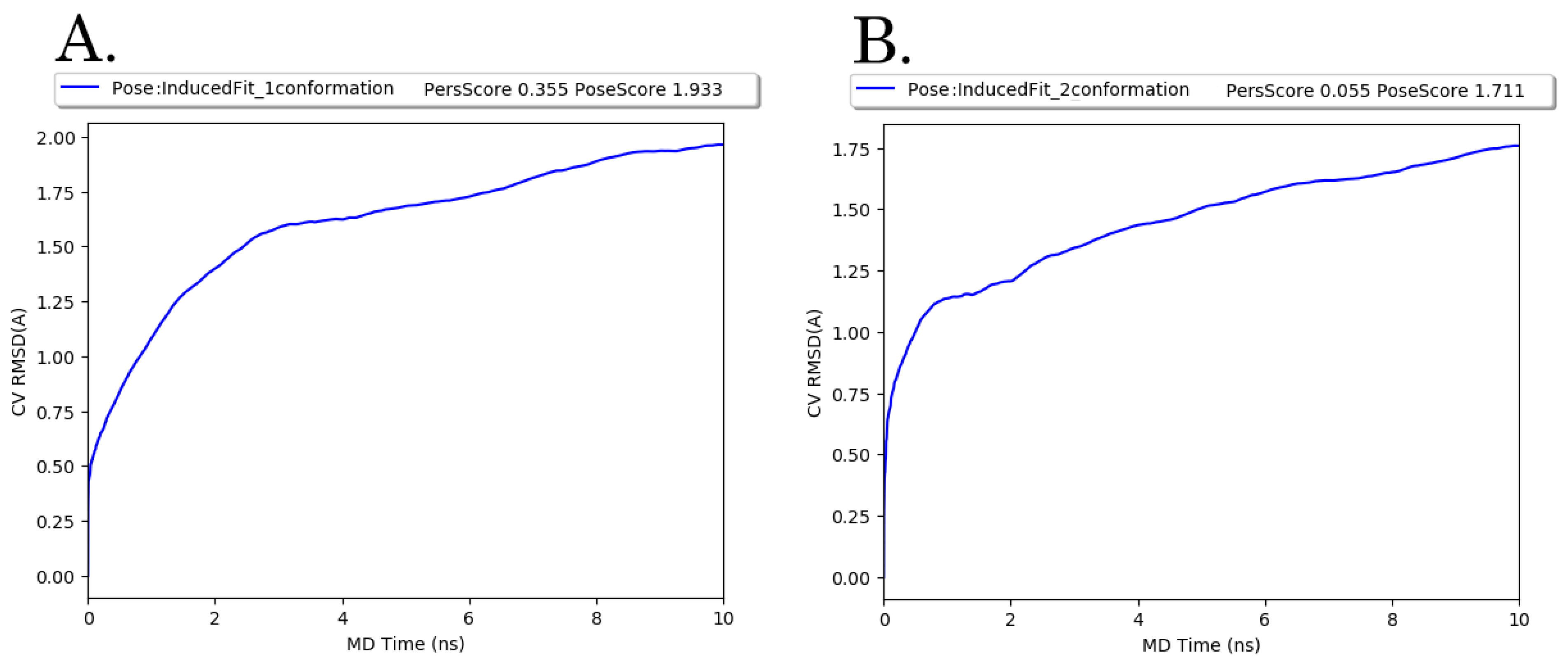
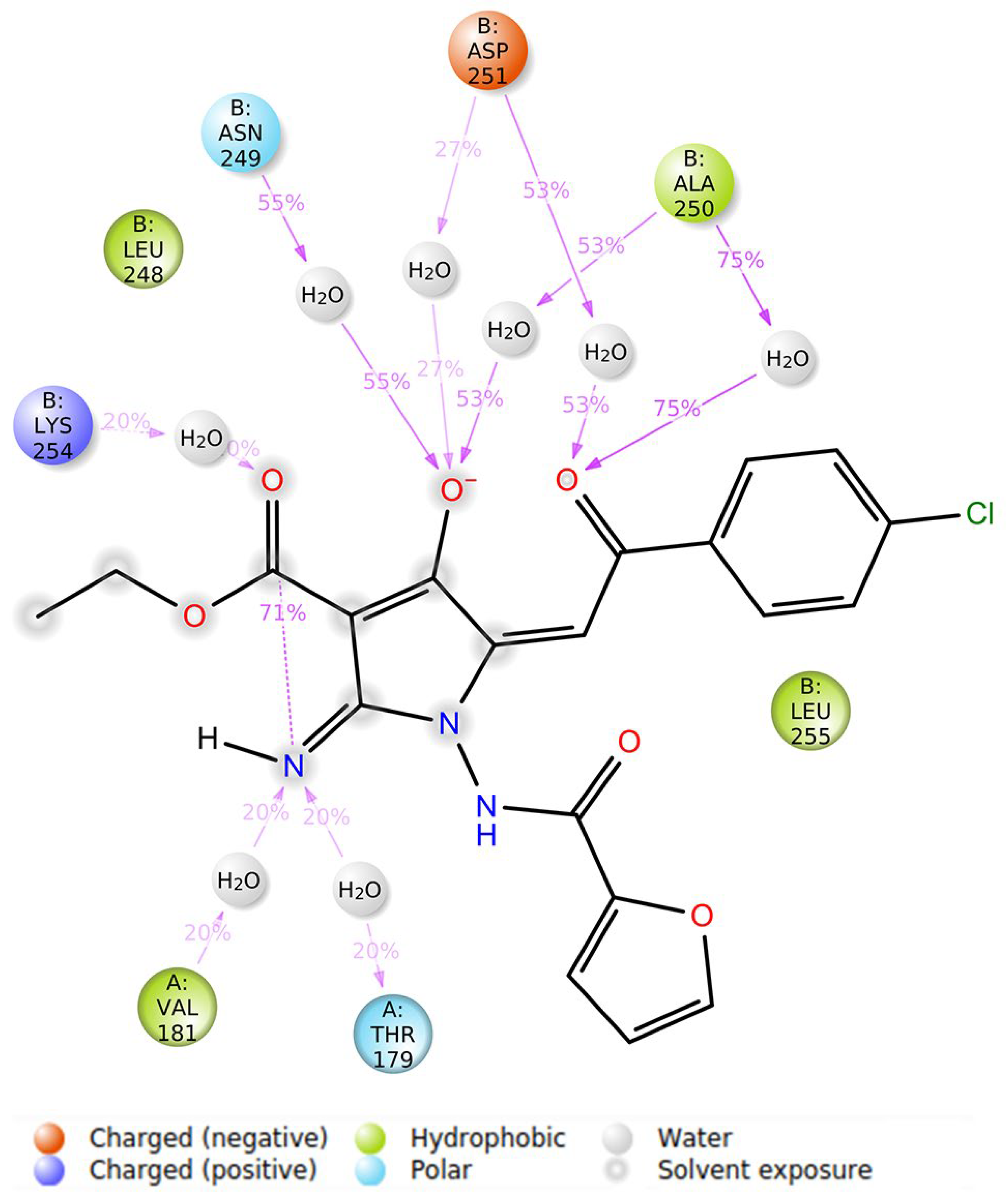
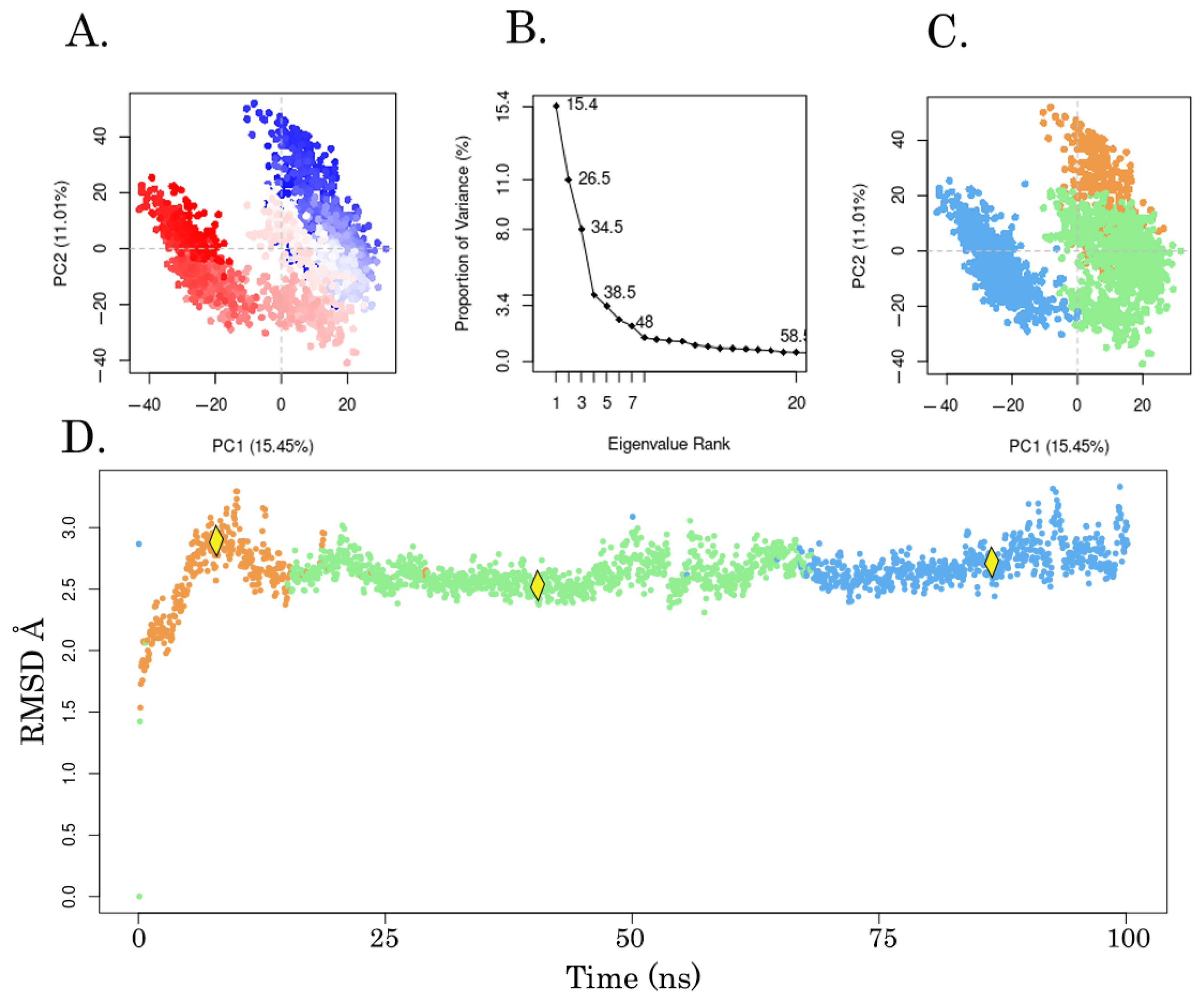
3.5. Molecular Modeling Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Avila, J. Microtubule functions. Life Sci. 1992, 50, 327–334. [Google Scholar] [CrossRef]
- Vukušić, K.; Buđa, R.; Tolić, I.M. Force-generating mechanisms of anaphase in human cells. J. Cell Sci. 2019, 132, jcs231985. [Google Scholar] [CrossRef] [PubMed]
- de Forges, H.; Bouissou, A.; Perez, F. Interplay between microtubule dynamics and intracellular organization. Int. J. Biochem. Cell Biol. 2012, 44, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Neefjes, J. Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol. 2017, 47, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar]
- Hamel, E. Natural products which interact with tubulin in the vinca domain: Maytansine, rhizoxin, phomopsin a, dolastatins 10 and 15 and halichondrin B. Pharmacol. Ther. 1992, 55, 31–51. [Google Scholar] [CrossRef]
- von Hoff, D.D. The taxoids: Same roots, different drugs. Semin. Oncol. 1997, 24 (Suppl. 13), S13-3–S13-10. [Google Scholar]
- Bollag, D.M.; McQueney, P.A.; Zhu, J.; Hensens, O.; Koupal, L.; Liesch, J.; Goetz, M.; Lazarides, E.; Woods, C.M. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef]
- Hastie, S.B. Interactions of colchicine with tubulin. Pharmacol. Ther. 1991, 51, 377–401. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer: Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef]
- Mechetner, E.; Kyshtoobayeva, A.; Zonis, S.; Kim, H.; Stroup, R.; Garcia, R.; Parker, R.J.; Fruehauf, J.P. Levels of multidrug resistance (MDR1) P-glycoprotein expression by human breast cancer correlate with in vitro resistance to taxol and doxorubicin. Clin. Cancer Res. 1998, 4, 389–398. [Google Scholar]
- Kavallaris, M.; Kuo, D.Y.; Burkhart, C.A.; Regl, D.L.; Norris, M.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef]
- Stengel, C.; Newman, S.P.; Leese, M.P.; Potter, B.V.L.; Reed, M.J.; Purohit, A. Class III β-tubulin expression and in vitro resistance to microtubule targeting agents. Br. J. Cancer 2009, 102, 316–324. [Google Scholar] [CrossRef]
- Chaplin, D.J.; Hill, S.A. The development of combretastatin A4 phosphate as a vascular targeting agent. Int. J. Radiat. Oncol. 2002, 54, 1491–1496. [Google Scholar] [CrossRef]
- Siemann, D.W.; Shi, W. Dual targeting of tumor vasculature: Combining Avastin and vascular disrupting agents (CA4P or OXi4503). Anticancer Res. 2008, 28, 2027–2031. [Google Scholar]
- Lindamulage, I.K.; Vu, H.-Y.; Karthikeyan, C.; Knockleby, J.; Lee, Y.-F.; Trivedi, P.; Lee, H. Novel quinolone chalcones targeting colchicine-binding pocket kill multidrug-resistant cancer cells by inhibiting tubulin activity and MRP1 function. Sci. Rep. 2017, 7, 10298. [Google Scholar] [CrossRef]
- Gupta, S.; Banerjee, M.; Poddar, A.; Banerjee, A.; Basu, G.; Roy, D.; Bhattacharyya, B. Biphasic Kinetics of the Colchicine−Tubulin Interaction: Role of Amino Acids Surrounding the A ring of Bound Colchicine Molecule. Biochemistry 2005, 44, 10181–10188. [Google Scholar] [CrossRef][Green Version]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Arnst, K.E.; Banerjee, S.; Chen, H.; Deng, S.; Hwang, D.; Li, W.; Miller, D.D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2019, 39, 1398–1426. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Quinn, F.R.; Neiman, Z.; Beisler, J.A. Toxicity and quantitative structure-activity relationships of colchicines. J. Med. Chem. 1981, 24, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Ringel, I.; Jaffe, D.; Alerhand, S.; Boye, O.; Muzaffar, A.; Brossi, A. Fluorinated colchicinoids: Antitubulin and cytotoxic properties. J. Med. Chem. 1991, 34, 3334–3338. [Google Scholar] [CrossRef]
- Sapra, S.; Bhalla, Y.; Nandani; Sharma, S.; Singh, G.; Nepali, K.; Budhiraja, A.; Dhar, K.L. Colchicine and its various physicochemical and biological aspects. Med. Chem. Res. 2012, 22, 531–547. [Google Scholar] [CrossRef]
- Zykova, S.S.; Kizimova, I.A.; Syutkina, A.I.; Toksarova, Y.S.; Igidov, N.M.; Ibishov, D.F.; Boichuk, S.V.; Dunaev, P.D.; Galembikova, A.R.; Korochkina, R.R. Synthesis and Cytostatic Activity of (E)-Ethyl-2-Amino-5-(3,3-Dimethyl-4-Oxobutyliden)-4-Oxo-1- (2-Phenylaminobenzamido)-4,5-Dihydro-1Hpyrrol-3-Carboxylate. Pharm. Chem. J. 2020, 53, 895–898. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Fusani, L.; Palmer, D.S.; Somers, D.O.; Wall, I.D. Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics. J. Chem. Inf. Model. 2020, 60, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; IEEE: Piscataway, NJ, USA, 2006; p. 43. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: https://www.R-project.org/ (accessed on 28 April 2022).
- Chaimontree, S.; Atkinson, K.; Coenen, F. Best Clustering Configuration Metrics: Towards Multiagent Based Clustering. In International Conference on Advanced Data Mining and Applications, Chongqing, China, 19–21 November 2010; Springer: Berlin/Heidelberg, Germany, 2010; pp. 48–59. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins: Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. Pyrrole: A resourceful small molecule in key medicinal hetero-aromatics. RSC Adv. 2015, 5, 15233–15266. [Google Scholar] [CrossRef]
- DeSimone, R.W.; Currie, K.S.; Mitchell, S.A.; Darrow, J.W.; Pippin, D.A. Privileged Structures: Applications in Drug Discovery. Comb. Chem. High Throughput Screen. 2004, 7, 473–493. [Google Scholar] [CrossRef]
- Duarte, C.D.; Barreiro, E.J.; Fraga, C.A.M. Privileged structures: A useful concept for the rational design of new lead drug candidates. Mini-Rev. Med. Chem. 2007, 7, 1108–1119. [Google Scholar] [CrossRef]
- Petri, G.L.; Spanò, V.; Spatola, R.; Holl, R.; Raimondi, M.V.; Barraja, P.; Montalbano, A. Bioactive pyrrole-based compounds with target selectivity. Eur. J. Med. Chem. 2020, 208, 112783. [Google Scholar] [CrossRef]
- Walsh, C.T.; Garneau-Tsodikova, S.; Howard-Jones, A.R. Biological formation of pyrroles: Nature’s logic and enzymatic machinery. Nat. Prod. Rep. 2006, 23, 517–531. [Google Scholar] [CrossRef]
- Ahmad, S.; Alam, O.; Naim, M.J.; Shaquiquzzaman, M.; Alam, M.M.; Iqbal, M. Pyrrole: An insight into recent pharmacological advances with structure activity relationship. Eur. J. Med. Chem. 2018, 157, 527–561. [Google Scholar] [CrossRef] [PubMed]
- Bianco, M.d.C.A.D.; Marinho, D.I.L.F.; Hoelz, L.V.B.; Bastos, M.M.; Boechat, N. Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors. Pharmaceuticals 2021, 14, 893. [Google Scholar] [CrossRef] [PubMed]
- La Regina, G.; Bai, R.; Coluccia, A.; Famiglini, V.; Pelliccia, S.; Passacantilli, S.; Mazzoccoli, C.; Ruggieri, V.; Sisinni, L.; Bolognesi, A.; et al. New Pyrrole Derivatives with Potent Tubulin Polymerization Inhibiting Activity As Anticancer Agents Including Hedgehog-Dependent Cancer. J. Med. Chem. 2014, 57, 6531–6552. [Google Scholar] [CrossRef] [PubMed]
- Jadala, C.; Prasad, B.; Prasanthi, A.V.G.; Shankaraiah, N.; Kamal, A. Transition metal-free one-pot synthesis of substituted pyrroles by employing aza-Wittig reaction. RSC Adv. 2019, 9, 30659–30665. [Google Scholar] [CrossRef]
- Tang, S.; Zhou, Z.; Jiang, Z.; Zhu, W.; Qiao, D. Indole-Based Tubulin Inhibitors: Binding Modes and SARs Investigations. Molecules 2022, 27, 1587. [Google Scholar] [CrossRef]
- Romagnoli, R.; Oliva, P.; Salvador, M.K.; Manfredini, S.; Padroni, C.; Brancale, A.; Ferla, S.; Hamel, E.; Ronca, R.; Maccarinelli, F.; et al. A facile synthesis of diaryl pyrroles led to the discovery of potent colchicine site antimitotic agents. Eur. J. Med. Chem. 2021, 214, 113229. [Google Scholar] [CrossRef]
- Sun, J.; Chen, L.; Liu, C.; Wang, Z.; Zuo, D.; Pan, J.; Qi, H.; Bao, K.; Wu, Y.; Zhang, W. Synthesis and biological evaluations of 1,2-diaryl pyrroles as analogues of combretastatin A-4. Chem. Biol. Drug Des. 2015, 86, 1541–1547. [Google Scholar] [CrossRef]
- Ma, Z.; Ma, Z.; Zhang, D. Synthesis of Multi-Substituted Pyrrole Derivatives Through [3+2] Cycloaddition with Tosylmethyl Isocyanides (TosMICs) and Electron-Deficient Compounds. Molecules 2018, 23, 2666. [Google Scholar] [CrossRef]
- Mowery, P.; Mejia, F.B.; Franceschi, C.L.; Kean, M.H.; Kwansare, D.O.; Lafferty, M.M.; Neerukonda, N.D.; Rolph, C.E.; Truax, N.J.; Pelkey, E.T. Synthesis and evaluation of the anti-proliferative activity of diaryl-3-pyrrolin-2-ones and fused analogs. Bioorganic Med. Chem. Lett. 2017, 27, 191–195. [Google Scholar] [CrossRef]
- Boichuk, S.; Galembikova, A.; Syuzov, K.; Dunaev, P.; Bikinieva, F.; Aukhadieva, A.; Zykova, S.; Igidov, N.; Gankova, K.; Novikova, M.; et al. The Design, Synthesis, and Biological Activities of Pyrrole-Based Carboxamides: The Novel Tubulin Inhibitors Targeting the Colchicine-Binding Site. Molecules 2021, 26, 5780. [Google Scholar] [CrossRef]
- Mohammad, T.; Siddiqui, S.; Shamsi, A.; Alajmi, M.F.; Hussain, A.; Islam, A.; Ahmad, F.; Hassan, M.I. Virtual Screening Approach to Identify High-Affinity Inhibitors of Serum and Glucocorticoid-Regulated Kinase 1 among Bioactive Natural Products: Combined Molecular Docking and Simulation Studies. Molecules 2020, 25, 823. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Shamsi, A.; Shahbaaz, M.; Queen, A.; Khan, P.; Hasan, G.M.; Islam, A.; Alajmi, M.F.; Hussain, A.; Ahmad, F.; et al. Rosmarinic Acid Exhibits Anticancer Effects via MARK4 Inhibition. Sci. Rep. 2020, 10, 10300. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; DasGupta, D.; Shafie, A.; Alhumaydhi, F.A.; Alsagaby, S.A.; Shahwan, M.; Anjum, F.; Al Abdulmonem, W.; Sharaf, S.E.; Hassan, I. Implications of tempol in pyruvate dehydrogenase kinase 3 targeted anticancer therapeutics: Computational, spectroscopic, and calorimetric studies. J. Mol. Liq. 2022, 350, 118581. [Google Scholar] [CrossRef]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The Novel Microtubule-Destabilizing Drug BAL27862 Binds to the Colchicine Site of Tubulin with Distinct Effects on Microtubule Organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhou, C.; Guan, Z.-Y.; Yin, P.; Chen, F.; Tang, Y.-J. Structural Insights into the Inhibition of Tubulin by the Antitumor Agent 4β-(1,2,4-triazol-3-ylthio)-4-deoxypodophyllotoxin. ACS Chem. Biol. 2017, 12, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Wang, Y.; Wang, C.; Wang, Y.; Jiang, X.; Ma, L.; Wu, C.; Yu, Y.; Chen, Q. Structure of 4′-demethylepipodophyllotoxin in complex with tubulin provides a rationale for drug design. Biochem. Biophys. Res. Commun. 2017, 493, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Arnst, K.E.; Wang, Y.; Hwang, D.-J.; Xue, Y.; Costello, T.; Hamilton, D.; Chen, Q.; Yang, J.; Park, F.; Dalton, J.T.; et al. A Potent, Metabolically Stable Tubulin Inhibitor Targets the Colchicine Binding Site and Overcomes Taxane Resistance. Cancer Res. 2017, 78, 265–277. [Google Scholar] [CrossRef]
- Pallante, L.; Rocca, A.; Klejborowska, G.; Huczynski, A.; Grasso, G.; Tuszynski, J.A.; Deriu, M.A. In silico Investigations of the Mode of Action of Novel Colchicine Derivatives Targeting β-Tubulin Isotypes: A Search for a Selective and Specific β-III Tubulin Ligand. Front. Chem. 2020, 8, 108. [Google Scholar] [CrossRef]
- Gallego-Yerga, L.; Ochoa, R.; Lans, I.; Peña-Varas, C.; Alegría-Arcos, M.; Cossio, P.; Ramírez, D.; Peláez, R. Application of ensemble pharmacophore-based virtual screening to the discovery of novel antimitotic tubulin inhibitors. Comput. Struct. Biotechnol. J. 2021, 19, 4360–4372. [Google Scholar] [CrossRef]
- Hagras, M.; El Deeb, M.A.; Elzahabi, H.S.A.; Elkaeed, E.B.; Mehany, A.B.M.; Eissa, I.H. Discovery of new quinolines as potent colchicine binding site inhibitors: Design, synthesis, docking studies, and anti-proliferative evaluation. J. Enzym. Inhib. Med. Chem. 2021, 36, 640–658. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Q.; Li, W. Recent advances in heterocyclic tubulin inhibitors targeting the colchicine binding site. Anticancer Agents Med. Chem. 2016, 16, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Sun, Z.; Zhao, F.; Shan, G.; Meng, Q. Recent advances in research of colchicine binding site inhibitors and their interaction modes with tubulin. Futur. Med. Chem. 2021, 13, 839–858. [Google Scholar] [CrossRef]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Futur. Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Dorléans, A.; Gigant, B.; Ravelli, R.B.G.; Mailliet, P.; Mikol, V.; Knossow, M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc. Natl. Acad. Sci. USA 2009, 106, 13775–13779. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.X.; Hsu, M.T.; Bonomi, M.; Agard, D.A.; Jacobson, M.P. The Free Energy Profile of Tubulin Straight-Bent Conformational Changes, with Implications for Microtubule Assembly and Drug Discovery. PLoS Comput. Biol. 2014, 10, e1003464. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of EAPC | Cell Lines | ||
|---|---|---|---|
| MDA-MB 231 | H1299 | HCC 1806 | |
| 64 | 21.5 ± 2.01 | 28.7 ± 1.54 | 23.19 ± 3.72 |
| 65 | 28.2 ± 1.12 | 17.5 ± 2.31 | 14.13 ± 2.91 |
| 66 | 40.2 ± 3.42 | 46.3 ± 4.21 | 54.2 ± 4.87 |
| 67 | 2.9 ± 0.21 | 6.7 ± 0.53 | 9.4 ± 0.64 |
| 68 | >100 | >100 | 56.16 ± 3.2 |
| 69 | 26.1 ± 2.56 | 21.5 ± 1.5 | 29.50 ± 1.43 |
| 70 | 6.7 ± 0.52 | 2.2 ± 0.25 | 25.35 ± 2.12 |
| 71 | 10.24 ± 1.36 | 2.5 ± 0.42 | 29.73 ± 1.91 |
| 72 | 85.1 ± 1.25 | 79.6 ± 2.5 | 68.63 ± 4.17 |
| 73 | 39.3 ± 2.97 | >100 | >100 |
| Medoids of Cluster |
MM-GBSA ΔG
bind (kcal/mol) |
Ligand Strain Energy (kcal/mol) |
|---|---|---|
| Snapshot 834 | −24.2 | 11.4 |
| Snapshot 1556 | −31.1 | 9.3 |
| Snapshot 1987 | −25.6 | 10.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boichuk, S.; Syuzov, K.; Bikinieva, F.; Galembikova, A.; Zykova, S.; Gankova, K.; Igidov, S.; Igidov, N. Computational-Based Discovery of the Anti-Cancer Activities of Pyrrole-Based Compounds Targeting the Colchicine-Binding Site of Tubulin. Molecules 2022, 27, 2873. https://doi.org/10.3390/molecules27092873
Boichuk S, Syuzov K, Bikinieva F, Galembikova A, Zykova S, Gankova K, Igidov S, Igidov N. Computational-Based Discovery of the Anti-Cancer Activities of Pyrrole-Based Compounds Targeting the Colchicine-Binding Site of Tubulin. Molecules. 2022; 27(9):2873. https://doi.org/10.3390/molecules27092873
Chicago/Turabian StyleBoichuk, Sergei, Kirill Syuzov, Firuza Bikinieva, Aigul Galembikova, Svetlana Zykova, Ksenia Gankova, Sergei Igidov, and Nazim Igidov. 2022. "Computational-Based Discovery of the Anti-Cancer Activities of Pyrrole-Based Compounds Targeting the Colchicine-Binding Site of Tubulin" Molecules 27, no. 9: 2873. https://doi.org/10.3390/molecules27092873
APA StyleBoichuk, S., Syuzov, K., Bikinieva, F., Galembikova, A., Zykova, S., Gankova, K., Igidov, S., & Igidov, N. (2022). Computational-Based Discovery of the Anti-Cancer Activities of Pyrrole-Based Compounds Targeting the Colchicine-Binding Site of Tubulin. Molecules, 27(9), 2873. https://doi.org/10.3390/molecules27092873