
Metabolite Identification of a Novel Anti-Leishmanial Agent OJT007 in Rat Liver Microsomes Using LC-MS/MS

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
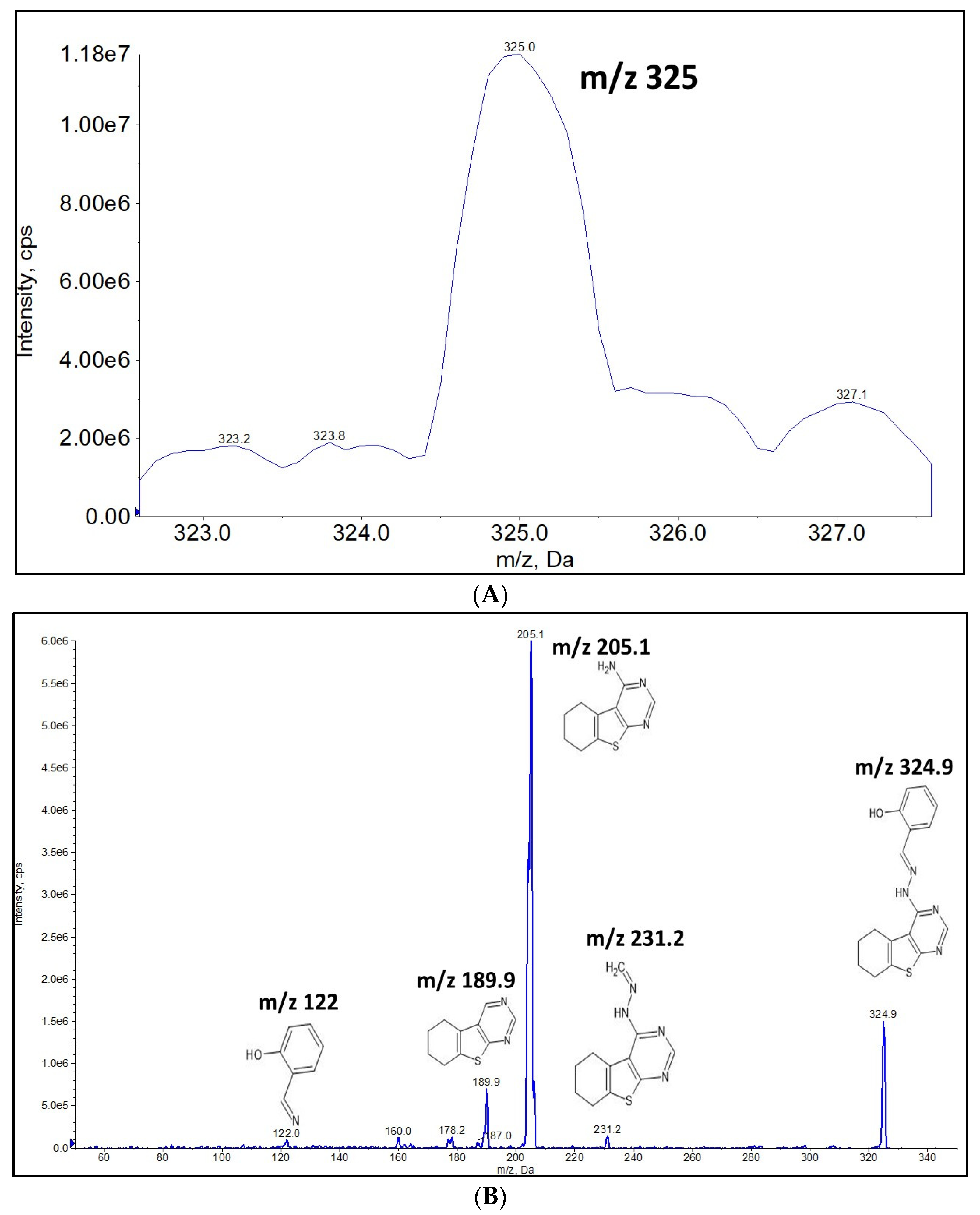
2.1. Full Mass Scan and Product Ion Scan Spectra for Matrix Free OJT007
2.2. Metabolism of OJT007 in CYP and UGT Reaction Systems
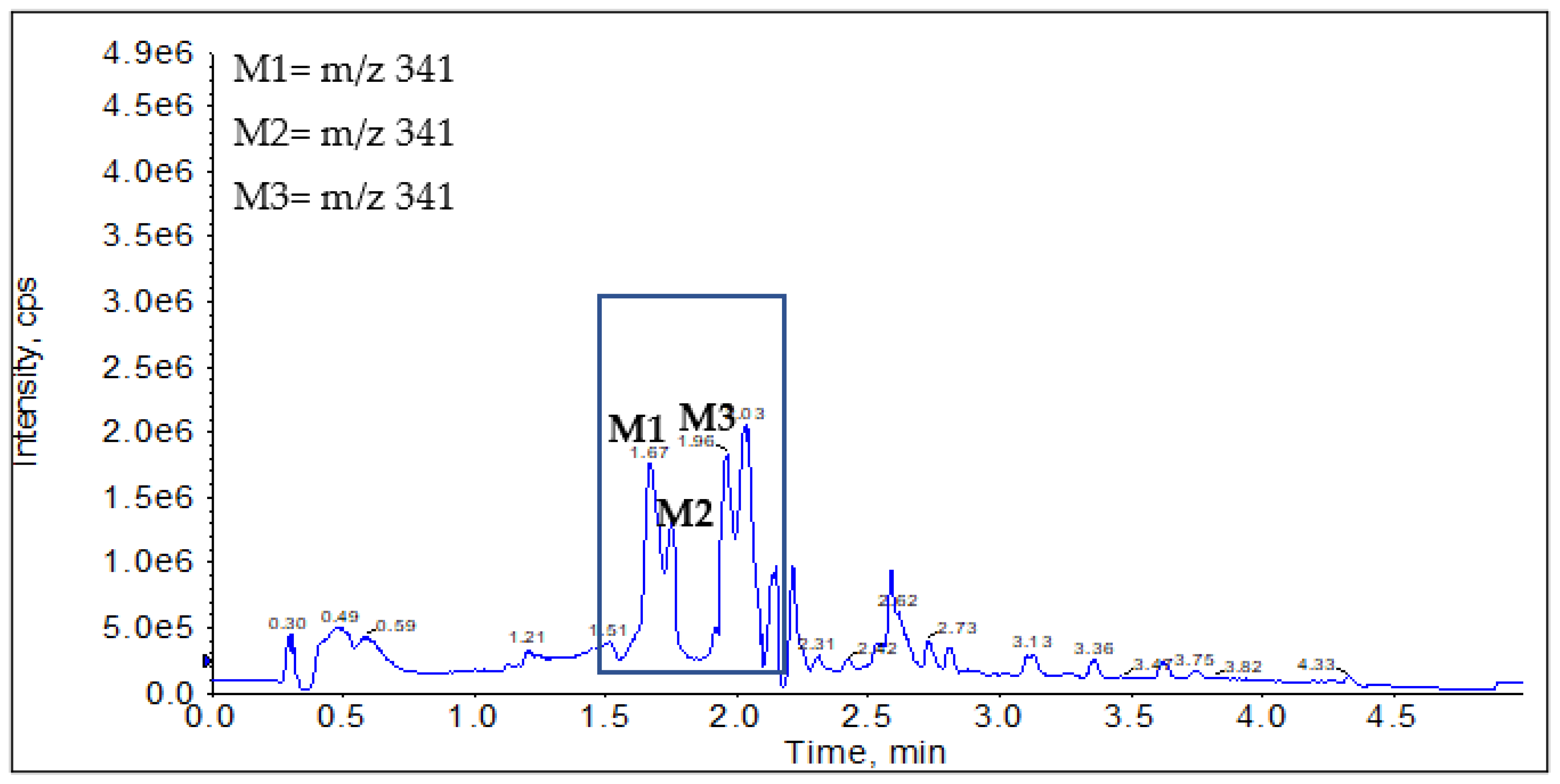
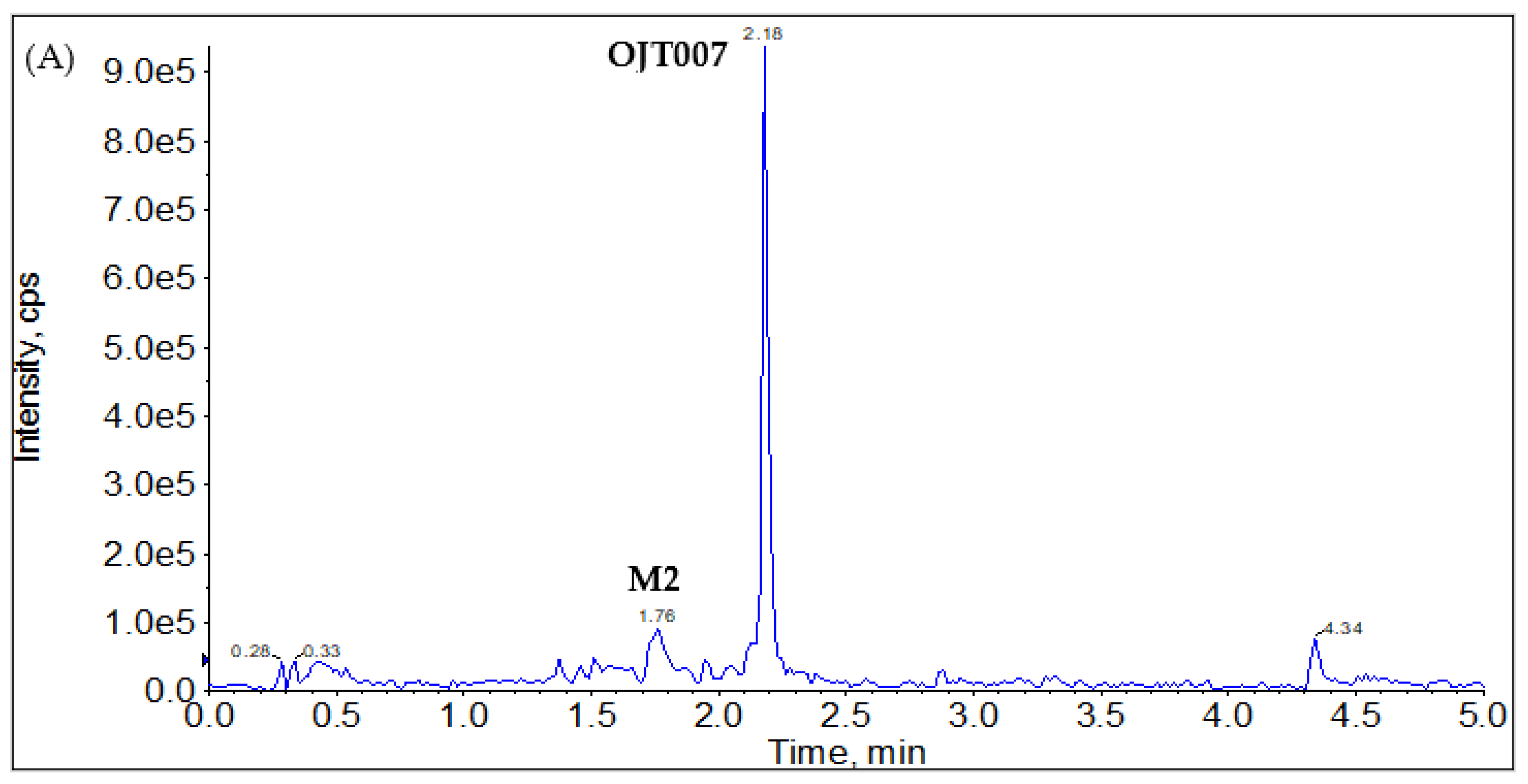
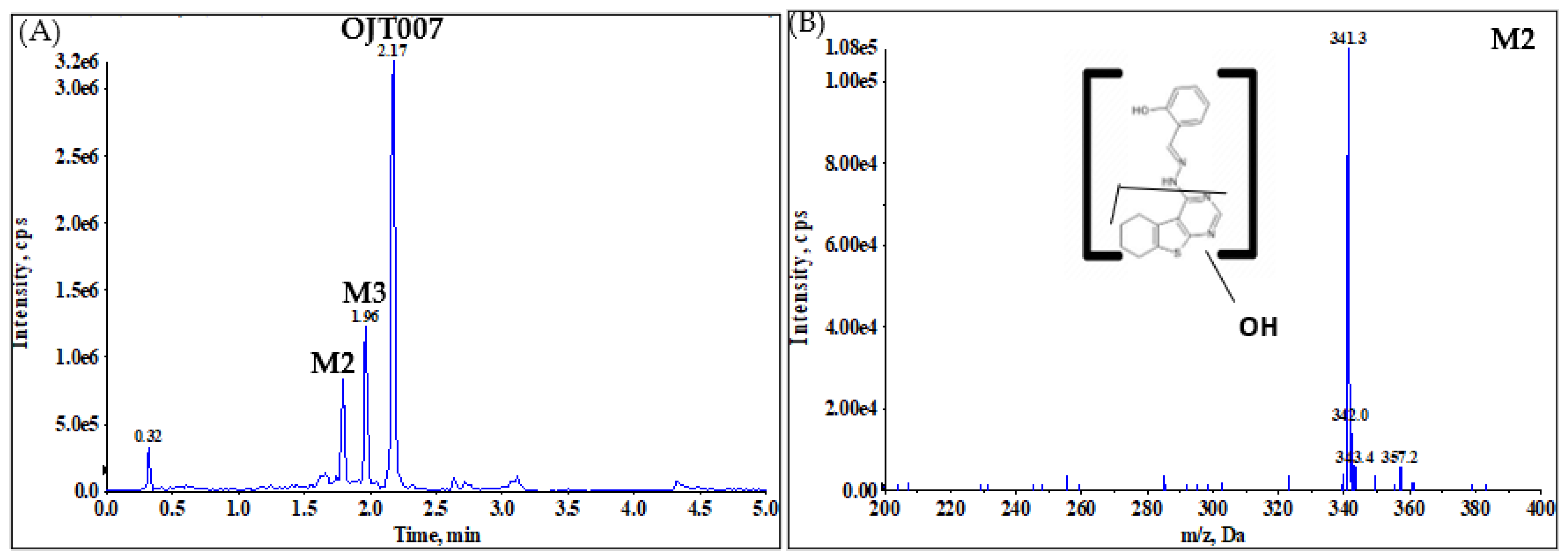
2.2.1. CYP Mediated Phase I Metabolism
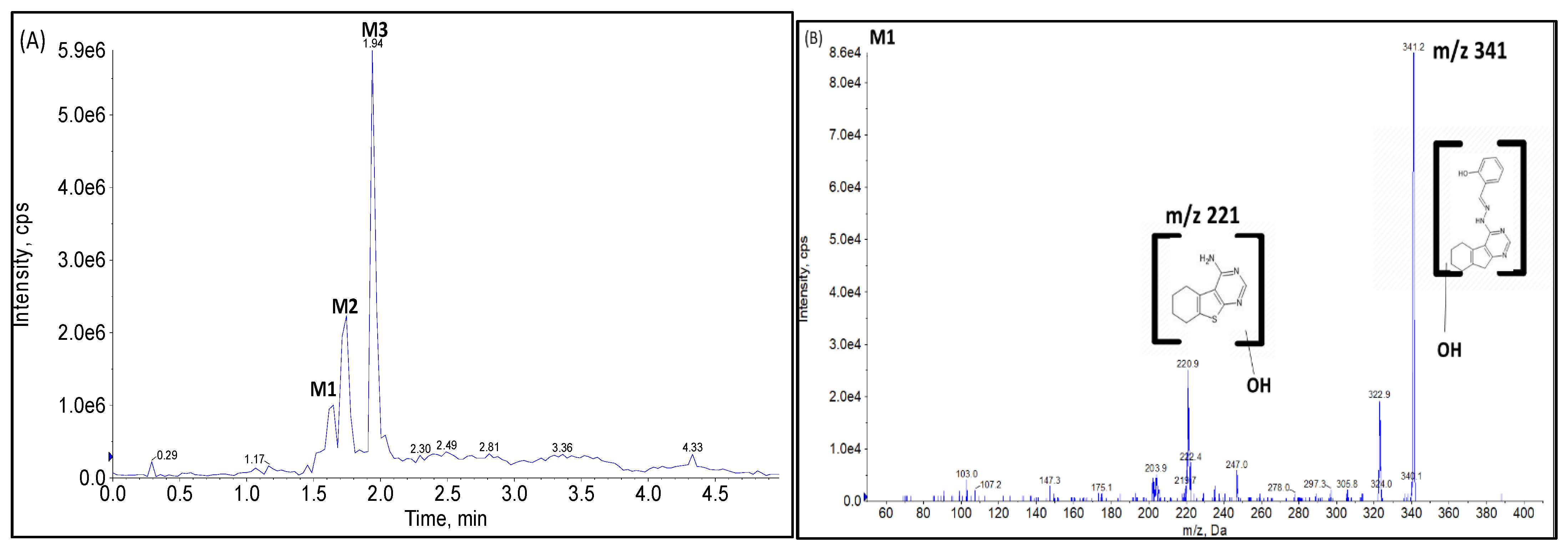
Phase I Metabolites Identification by LC-MS/MS
Metabolic Stability for Oxidation Metabolites
2.2.2. UGT Mediated Glucuronidation Metabolism
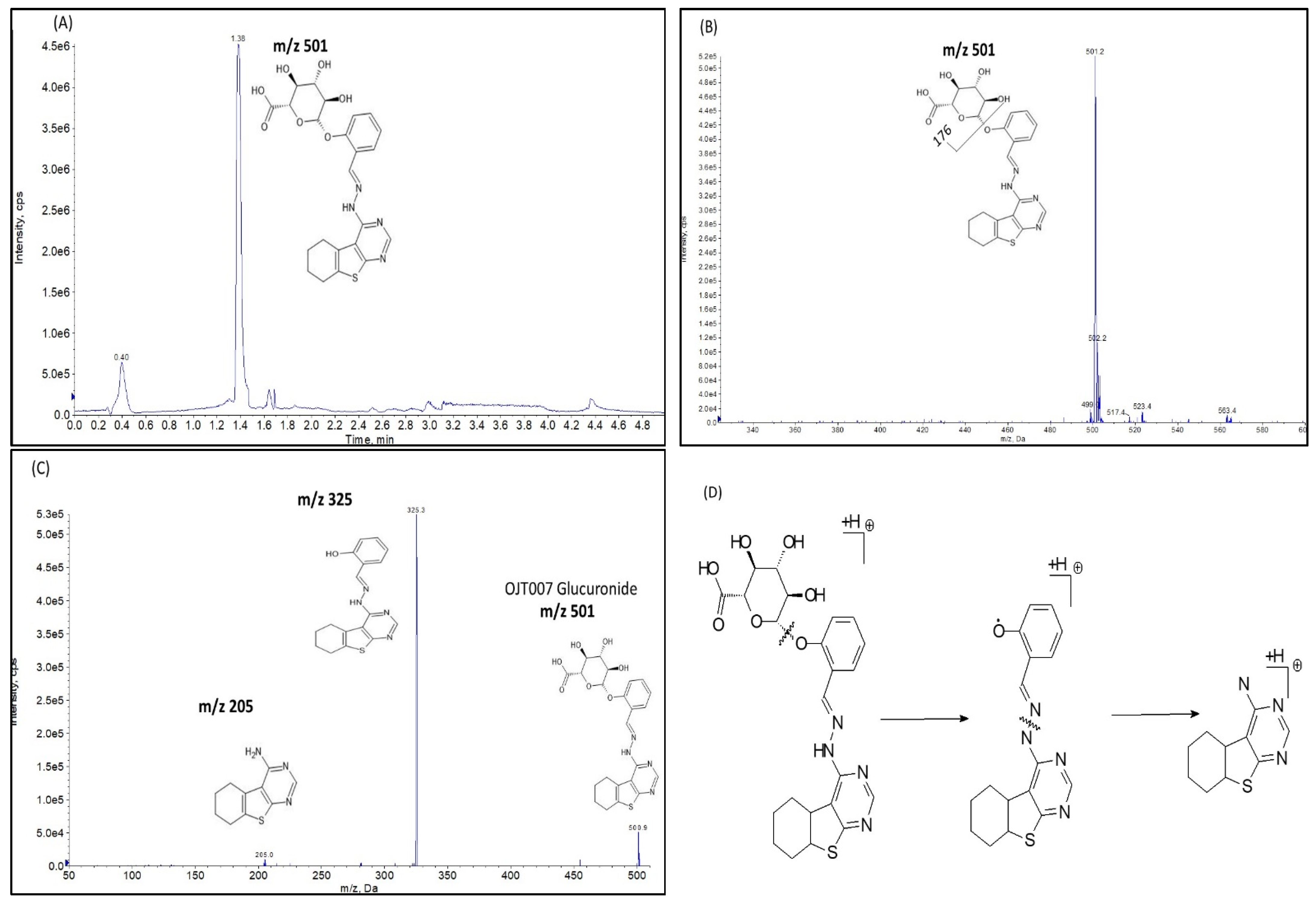
Glucuronide Identification by LC-MS/MS
Glucuronide Hydrolysis by β-Glucuronidase
Conversion Factors (K) of Extinction Coefficient
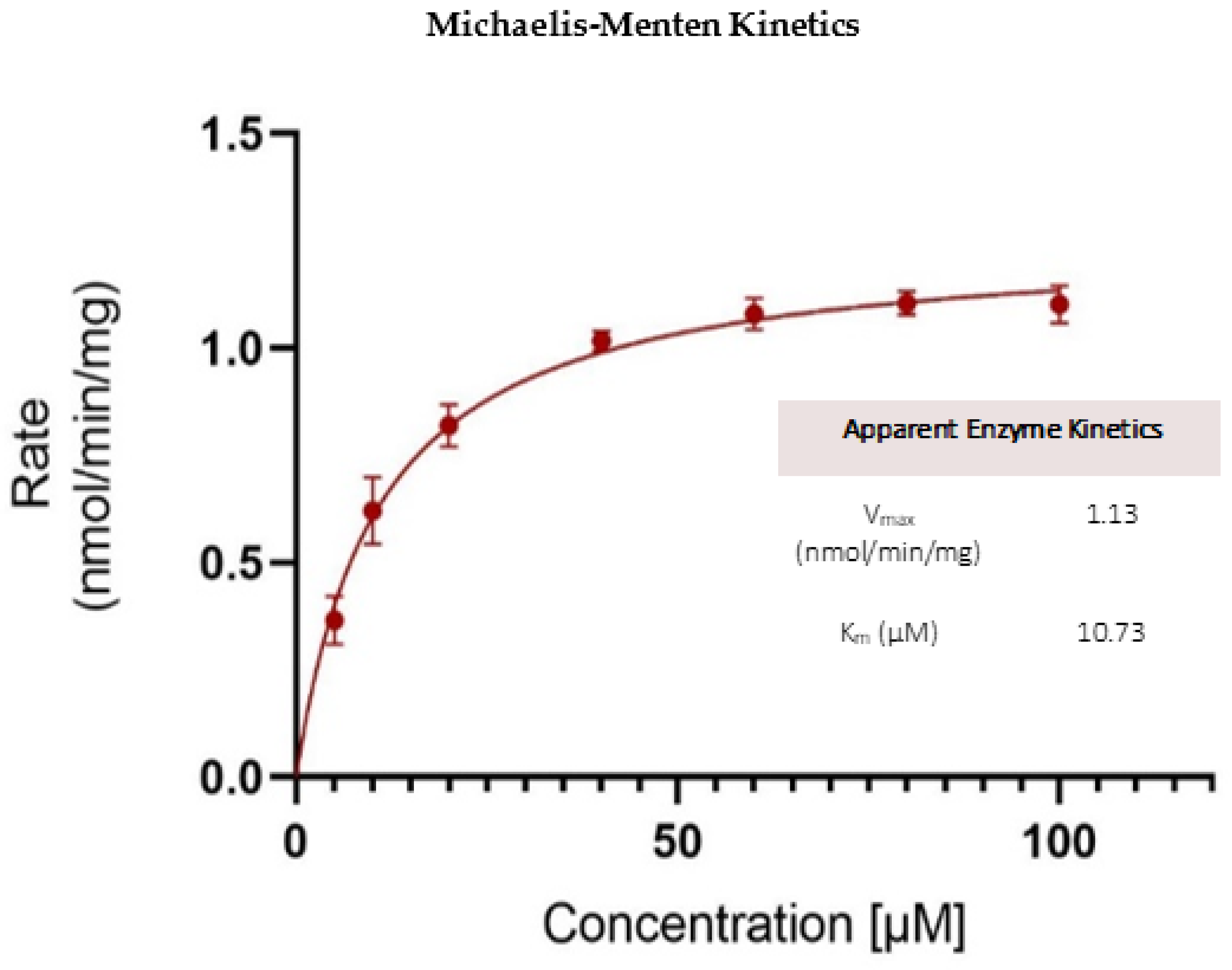
Kinetics of OJT007 Glucuronidation
3. Materials and Methods
3.1. Materials
3.2. Sample Preparation
3.3. LC-MS/MS Method
3.3.1. Chromatography
3.3.2. Mass Spectrometry
3.4. UPLC Method
3.5. In Vitro Metabolic Studies—CYP Reaction System
3.6. In Vitro Metabolic Studies—UGT Reaction System
3.7. Full Mass Scan and Product Scan Spectra for Matrix Free OJT007
3.8. Identification of Phase I Metabolites
LC-MS/MS
3.9. Identification of Phase II Metabolites
3.9.1. LC-MS/MS
3.9.2. Hydrolysis by β-Glucuronidases
3.10. Determination of Molar Extinction Coefficient of OJT007 Glucuronide
3.11. Metabolic Stability Data Analysis
3.12. Glucuronidation Metabolic Kinetics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Leishmaniasis. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 9 March 2022).
- CDC. CDC-Leishmaniasis-General Information-Frequently Asked Questions. 9 March. Available online: https://www.cdc.gov/parasites/leishmaniasis/gen_info/faqs.html (accessed on 9 March 2022).
- Duthie, M.S.; Reed, S.G. Not All Antigens Are Created Equally: Progress, Challenges, and Lessons Associated with Developing a Vaccine for Leishmaniasis. Clin. Vaccine Immunol. 2017, 24, e00108-17. [Google Scholar] [CrossRef] [PubMed]
- Charlton, R.L.; Rossi-Bergmann, B.; Denny, P.W.; Steel, P.G. Repurposing as a strategy for the discovery of new anti-leishmanials: The-state-of-the-art. Parasitology 2018, 145, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.-C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; McGary, E.C.; Chang, S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J. Bacteriol. 1989, 171, 4071–4072. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olaleye, O.; Raghunand, T.R.; Bhat, S.; He, J.; Tyagi, S.; Lamichhane, G.; Gu, P.; Zhou, J.; Zhang, Y.; Grosset, J.; et al. Methionine Aminopeptidases from Mycobacterium tuberculosis as Novel Antimycobacterial Targets. Chem. Biol. 2010, 17, 86–97. [Google Scholar] [CrossRef]
- Giglione, C.; Boularot, A.; Meinnel, T. Protein N-terminal methionine excision. CMLS Cell. Mol. Life Sci. 2004, 61, 1455–1474. [Google Scholar] [CrossRef]
- Giglione, C. Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms. EMBO J. 2000, 19, 5916–5929. [Google Scholar] [CrossRef]
- Giglione, C. Control of protein life-span by N-terminal methionine excision. EMBO J. 2003, 22, 13–23. [Google Scholar] [CrossRef]
- Lowther, W.T.; Matthews, B.W. Structure and function of the methionine aminopeptidases. Biochim. Biophys. Acta—Protein Struct. Mol. Enzymol. 2000, 1477, 157–167. [Google Scholar] [CrossRef]
- Li, X.; Chang, Y.-H. Amino-Terminal Protein Processing in Saccharomyces cerevisiae is an Essential Function that Requires Two Distinct Methionine Aminopeptidases. Proc. Natl. Acad. Sci. USA 1995, 92, 12357–12361. [Google Scholar] [CrossRef]
- Rodriguez, F.; John, S.F.; Iniguez, E.; Montalvo, S.; Michael, K.; White, L.; Liang, D.; Olaleye, O.A.; Maldonado, R.A. In Vitro and In Vivo Characterization of Potent Antileishmanial Methionine Aminopeptidase-1 Inhibitors. Antimicrob. Agents Chemother. 2020, 64, e01422-19. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.T.; Sumner, D.D. The importance of metabolite identification in quantitative risk estimation. J. Toxicol. Clin. Toxicol. 1982, 19, 781–805. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. Metabolite Identification in the Preclinical and Clinical Phase of Drug Development. Curr. Drug Metab. 2021, 22, 838–857. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Safety Testing of Drug Metabolites; US Food and Drug Administration: Silver Spring, MD, USA, 2022.
- Xiao, J.F.; Zhou, B.; Ressom, H.W. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. Trends Analyt Chem. 2012, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gonzalez, F.J.; Idle, J.R. LC-MS-Based Metabolomics in Drug Metabolism. Drug Metab. Rev. 2007, 39, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Dai, W.; Xu, G. Advances of metabolite identification in liquid chromatography-mass spectrometry based metabolomics. Se Pu 2014, 32, 1052–1057. [Google Scholar] [CrossRef]
- Wu, B.; Kulkarni, K.; Basu, S.; Zhang, S.; Hu, M. First-Pass Metabolism via UDP-Glucuronosyltransferase: A Barrier to Oral Bioavailability of Phenolics. J. Pharm. Sci. 2011, 100, 3655–3681. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, L.; Wang, S.; Wu, J.; Shi, J.; Yan, T.; Xie, C.; Li, Q.; Hu, M.; Liu, Z. Chapter 11—Oral Absorption Basics: Pathways and Physicochemical and Biological Factors Affecting Absorption. In Developing Solid Oral Dosage Forms, 2nd ed.; Qiu, Y., Chen, Y., Zhang, G.G.Z., Yu, L., Mantri, R.V., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 297–329. [Google Scholar] [CrossRef]
- Di, L.; Obach, R.S. Addressing the Challenges of Low Clearance in Drug Research. AAPS J. 2015, 17, 352–357. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Gao, N.; Li, S.Q.; Huang, Y.; Bourassa, J.L.; Huryn, D.M. Experimental Design on Single-Time-Point High-Throughput Microsomal Stability Assay. J. Pharm. Sci. 2004, 93, 1537–1544. [Google Scholar] [CrossRef]
- Słoczyńska, K.; Gunia-Krzyżak, A.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Żelaszczyk, D.; Popiół, J.; Pękala, E. Metabolic stability and its role in the discovery of new chemical entities. Acta Pharm. 2019, 69, 345–361. [Google Scholar] [CrossRef]
- Blanz, J.; Williams, G.; Dayer, J.; Délémonté, T.; Gertsch, W.; Ramstein, P.; Aichholz, R.; Trunzer, M.; Pearson, D. Evaluation of relative MS response factors of drug metabolites for semi-quantitative assessment of chemical liabilities in drug discovery. J. Mass Spectrom. 2017, 52, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, X.; Li, J.; Meng, Z.-Y.; Li, S.-C.; Li, Z.-J.; Lu, Y.-Y.; Ren, H.; Lou, Y.-Q.; Lu, C.; et al. Quantitative and qualitative analysis of the novel antitumor 1,3,4-oxadiazole derivative (GLB) and its metabolites using HPLC-UV and UPLC-QTOF-MS. Sci. Rep. 2015, 5, 11906. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yang, Z.; Yin, T.; You, M.; Hu, M. Validated LC-MS/MS method for the determination of maackiain and its sulfate and glucuronide in blood: Application to pharmacokinetic and disposition studies. J. Pharm. Biomed. Anal. 2011, 55, 288–293. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, G.; Zuo, Z. Position preference on glucuronidation of mono-hydroxylflavones in human intestine. Life Sci. 2006, 78, 2772–2780. [Google Scholar] [CrossRef] [PubMed]
- Nigro, M.R.; Ma, J.; Awosemo, O.T.; Xie, H.; Olaleye, O.A.; Liang, D. Development and Validation of a Sensitive, Specific and Reproducible UPLC-MS/MS Method for the Quantification of OJT007, A Novel Anti-Leishmanial Agent: Application to a Pharmacokinetic Study. Int. J. Environ. Res. Public Health 2021, 18, 4624. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, W.; Gao, S.; Xu, H.; Wu, B.; Kulkarni, K.; Singh, R.; Tang, L.; Hu, M. Simultaneous determination of genistein and its four phase II metabolites in blood by a sensitive and robust UPLC–MS/MS method: Application to an oral bioavailability study of genistein in mice. J. Pharm. Biomed. Anal. 2010, 53, 81–89. [Google Scholar] [CrossRef]
- Jiamboonsri, P.; Pithayanukul, P.; Bavovada, R.; Leanpolchareanchai, J.; Gao, S.; Hu, M. In vitro glucuronidation of methyl gallate and pentagalloyl glucopyranose by liver microsomes. Drug Metab. Pharm. 2016, 31, 292–303. [Google Scholar] [CrossRef]
- Sui, Y.; Wu, J.; Chen, J. The Role of Gut Microbial β-Glucuronidase in Estrogen Reactivation and Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 2067. [Google Scholar] [CrossRef]
- Seibert, E.; Tracy, T.S. Fundamentals of Enzyme Kinetics. In Enzyme Kinetics in Drug Metabolism: Fundamentals and Applications; Nagar, S., Argikar, U.A., Tweedie, D.J., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 9–22. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rincon Nigro, M.E.; Du, T.; Gao, S.; Kaur, M.; Xie, H.; Olaleye, O.A.; Liang, D. Metabolite Identification of a Novel Anti-Leishmanial Agent OJT007 in Rat Liver Microsomes Using LC-MS/MS. Molecules 2022, 27, 2854. https://doi.org/10.3390/molecules27092854
Rincon Nigro ME, Du T, Gao S, Kaur M, Xie H, Olaleye OA, Liang D. Metabolite Identification of a Novel Anti-Leishmanial Agent OJT007 in Rat Liver Microsomes Using LC-MS/MS. Molecules. 2022; 27(9):2854. https://doi.org/10.3390/molecules27092854
Chicago/Turabian StyleRincon Nigro, Maria Eugenia, Ting Du, Song Gao, Manvir Kaur, Huan Xie, Omonike Arike Olaleye, and Dong Liang. 2022. "Metabolite Identification of a Novel Anti-Leishmanial Agent OJT007 in Rat Liver Microsomes Using LC-MS/MS" Molecules 27, no. 9: 2854. https://doi.org/10.3390/molecules27092854
APA StyleRincon Nigro, M. E., Du, T., Gao, S., Kaur, M., Xie, H., Olaleye, O. A., & Liang, D. (2022). Metabolite Identification of a Novel Anti-Leishmanial Agent OJT007 in Rat Liver Microsomes Using LC-MS/MS. Molecules, 27(9), 2854. https://doi.org/10.3390/molecules27092854