
In Vitro–In Vivo Correlation of Tianeptine Sodium Sustained-Release Dual-Layer Tablets


Abstract
:1. Introduction
2. Results and Discussion
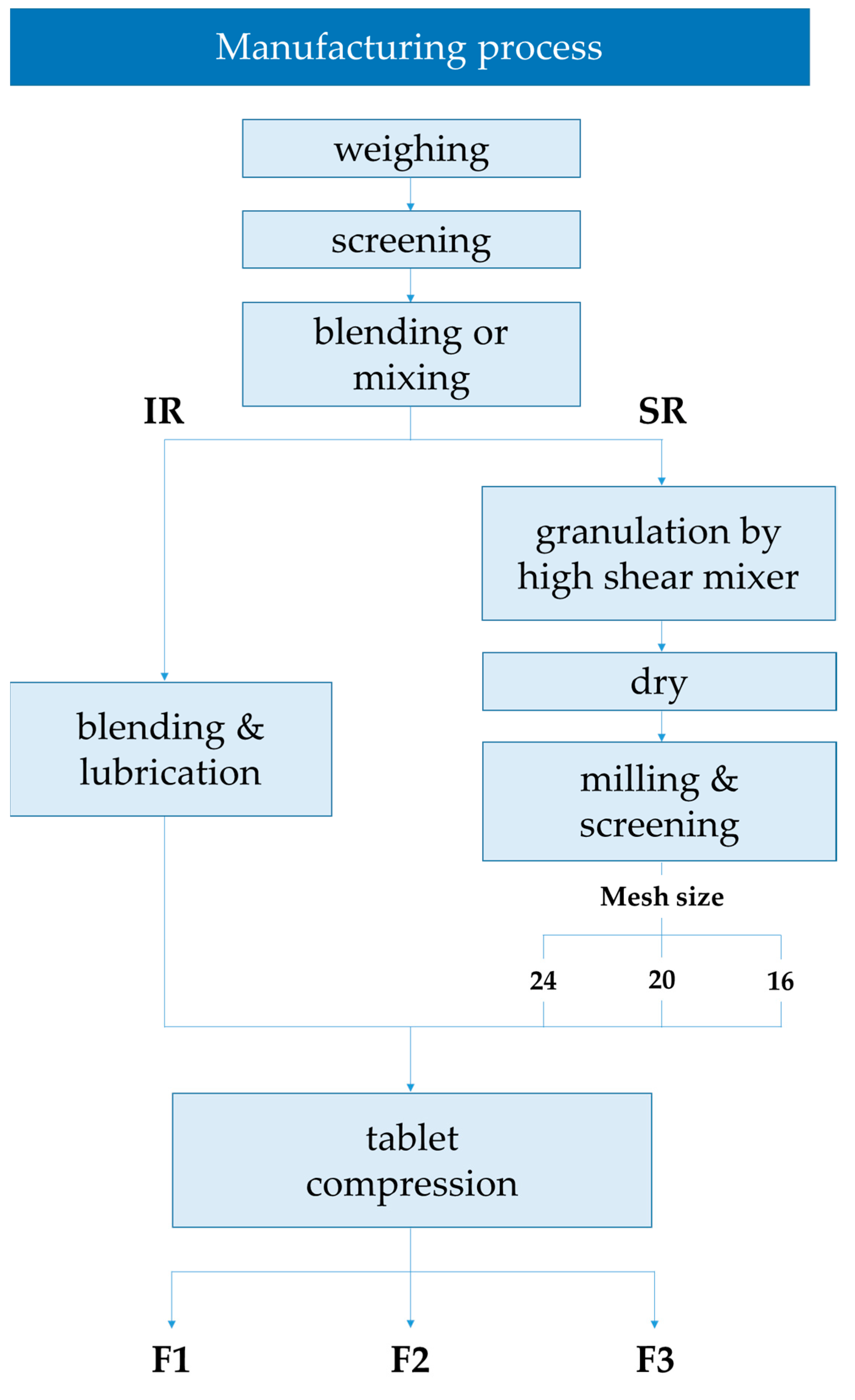
2.1. Characterization according to the Manufacturing Process
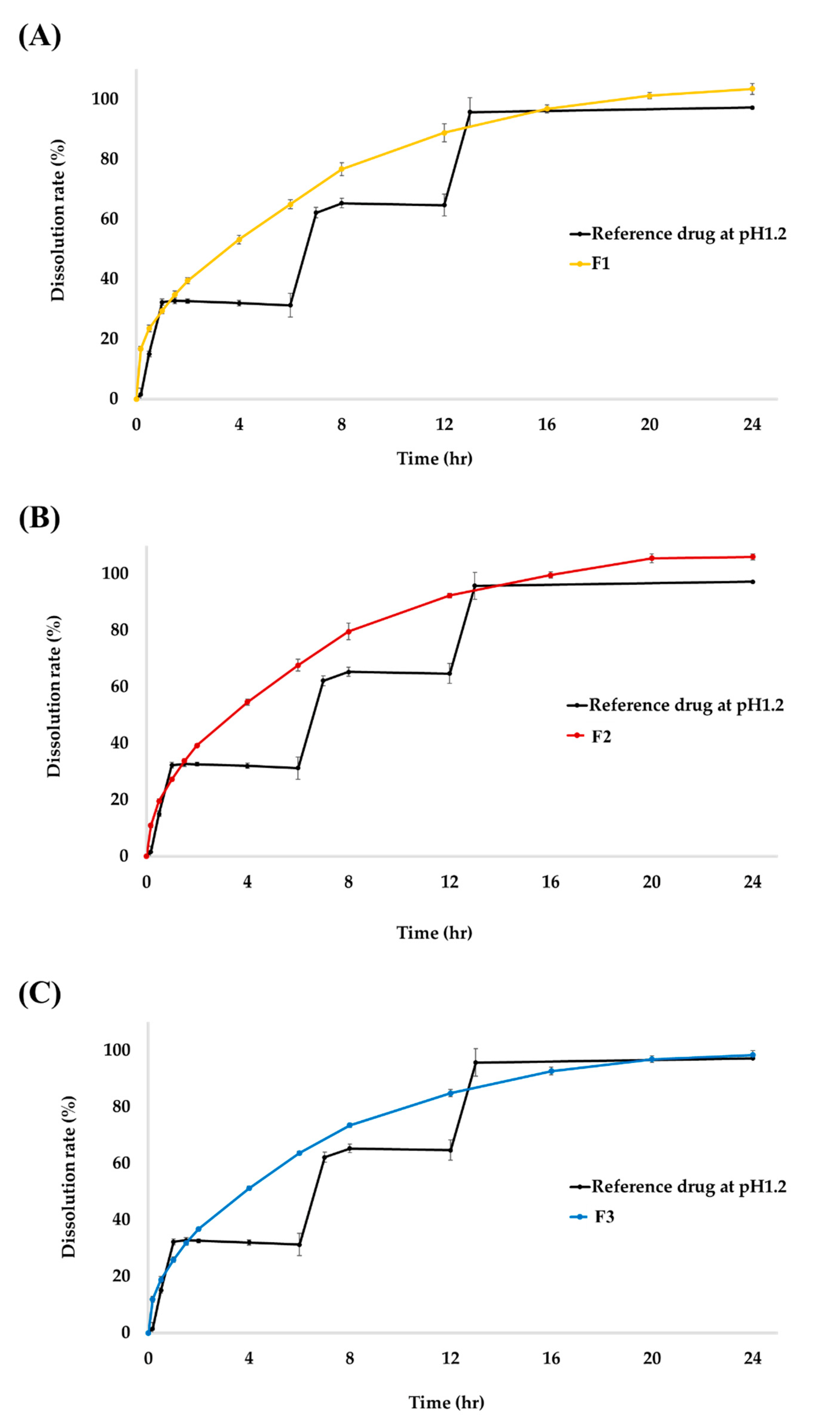
2.2. In Vitro Dissolution Studies
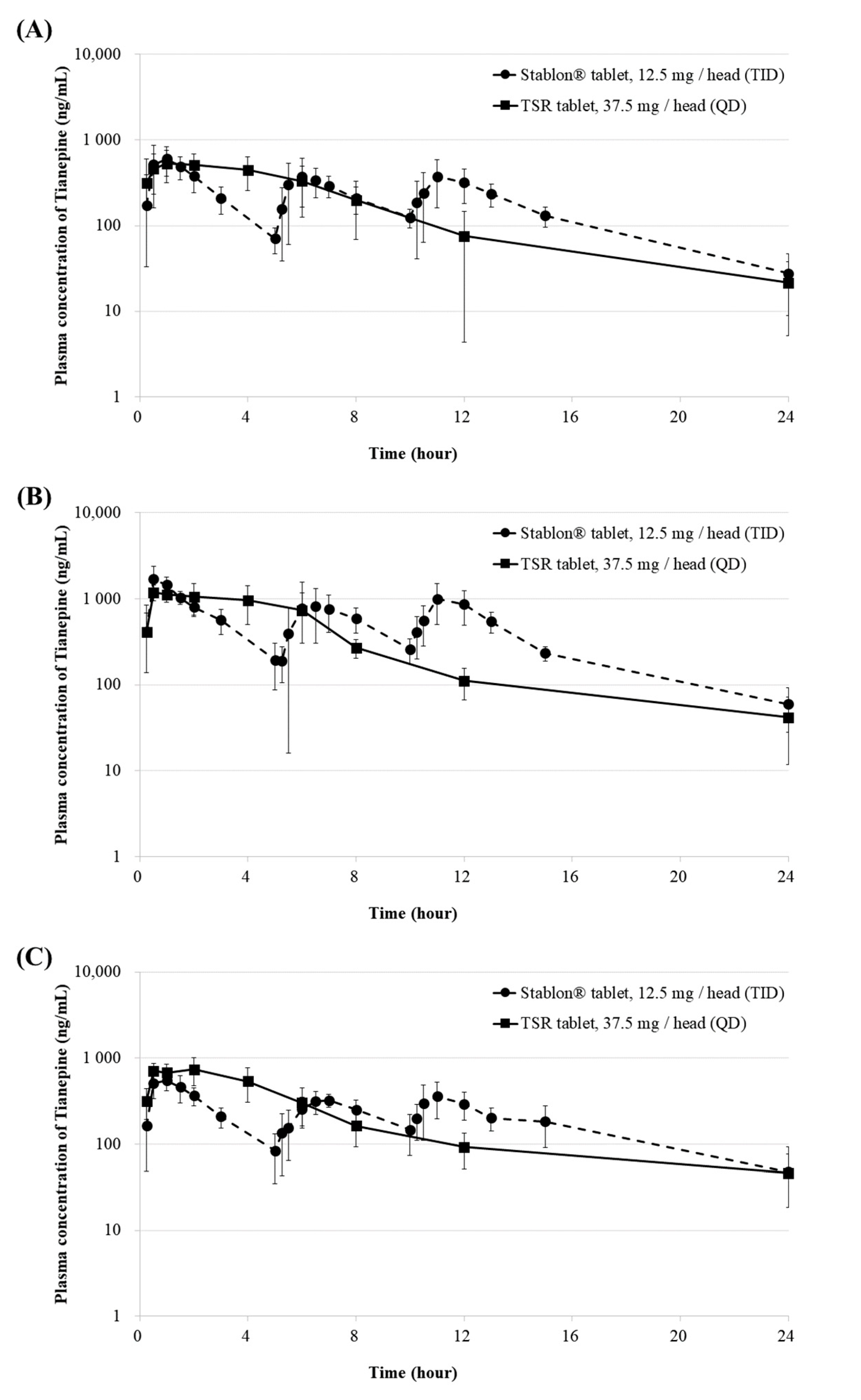
2.3. In Vivo Pharmacokinetic Studies
2.4. IVIVC
3. Materials and Methods
3.1. Materials
3.2. Preparation of TSR Tablets
3.3. Characterization according to the Manufacturing Process
3.4. In Vitro Dissolution Studies
3.5. In Vivo Pharmacokinetic Studies
3.5.1. Animals
3.5.2. Drug Administration and Blood Sampling
3.5.3. Sample Analysis
3.5.4. Statistical Analysis
3.6. IVIVC
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emami, J. In vitro—in vivo correlation: From theory to applications. J. Pharm. Pharm. Sci. 2006, 9, 169–189. [Google Scholar] [PubMed]
- Lee, S.-H.; Kim, J.-E. Quality by Design Applied Development of Immediate-Release Rabeprazole Sodium Dry-Coated Tablet. Pharmaceutics 2021, 13, 259. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Shin, S.; Jeong, S.W.; Lee, J.B.; Shin, B.S. Physiologically relevant in vitro-in vivo correlation (ivivc) approach for sildenafil with site-dependent dissolution. Pharmaceutics 2019, 11, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, W.R. Convolution-Based Approaches for in Vivo-in Vitro Correlation Modeling. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1997; Volume 423, pp. 53–65. [Google Scholar] [CrossRef]
- Kaur, P.; Jiang, X.; Duan, J.; Stier, E. Applications of In Vitro–In Vivo Correlations in Generic Drug Development: Case Studies. AAPS J. 2015, 17, 1035–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stillhart, C.; Pepin, X.; Tistaert, C.; Good, D.; Van Den Bergh, A.; Parrott, N.; Kesisoglou, F. PBPK absorption modeling: Establishing the in vitro–in vivo link—industry perspective. AAPS J. 2019, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wilde, M.I.; Benfield, P. Tianeptine. Drugs 1995, 49, 411–439. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Ruiz, J.; Montes, J.; Giner, J.; Guerrero, J.; Seva, A.; Dourdil, F. Tianeptine therapy for depression in the elderly. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1998, 22, 319–329. [Google Scholar] [CrossRef]
- Brink, C.B.; Harvey, B.H.; Brand, L. Tianeptine: A novel atypical antidepressant that may provide new insights into the biomolecular basis of depression. Recent Pat. CNS Drug Discov. 2006, 1, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Dresse, A.; Rosen, J.M.; Brems, H.; Masset, H.; Defrance, R.; Salvadori, C. Influence of Food on Tianeptine and Its Main Metabolite Kinetics. J. Clin. Pharmacol. 1988, 28, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Royer, R.J.; Royer-Morrot, M.J.; Paille, F.; Barrucand, D.; Schmitt, J.; Defrance, R.; Salvadori, C. Tianeptine and its main metabolite pharmacokinetics in chronic alcoholism and cirrhosis. Clin. Pharmacokinet. 1989, 16, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Eisen, S.A.; Miller, D.K.; Woodward, R.S.; Spitznagel, E.; Przybeck, T.R. The effect of prescribed daily dose frequency on patient medication compliance. Arch. Intern. Med. 1990, 150, 1881–1884. [Google Scholar] [CrossRef] [PubMed]
- Lindeboom, N.; Chang, P.R.; Tyler, R.T. Analytical, Biochemical and Physicochemical Aspects of Starch Granule Size, with Emphasis on Small Granule Starches: A Review. Starch-Stärke 2004, 56, 89–99. [Google Scholar] [CrossRef]
- Kim, T.H.; Shin, S.; Bulitta, J.B.; Youn, Y.S.; Yoo, S.D.; Shin, B.S. Development of a physiologically relevant population pharmacokinetic in vitro–in vivo correlation approach for designing extended-release oral dosage formulation. Mol. Pharm. 2017, 14, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Gray, V.A.; Thomas, W. Rosanske. “Dissolution”. In Specification of Drug Substances and Products; Elsevier: Amsterdam, The Netherlands, 2020; pp. 481–503. [Google Scholar]
- McKnight, P.E.; Najab, J. Mann-Whitney U Test. Corsini Encycl. Psychol. 2010. [Google Scholar] [CrossRef]
- Lee, T.; Hsu, F.B. A cross-performance relationship between Carr’s index and dissolution rate constant: The study of acetaminophen batches. Drug Dev. Ind. Pharm. 2007, 33, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-W.; Kim, J.-E.; Park, Y.-J. Nanoporous Silica Entrapped Lipid-Drug Complexes for the Solubilization and Absorption Enhancement of Poorly Soluble Drugs. Pharmaceutics 2021, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.; Suckow, M.A.; Murthy, S. The IACUC Handbook; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Sakore, S.; Chakraborty, B. In vitro–in vivo correlation (IVIVC): A strategic tool in drug development. J. Bioequiv. Availab. 2011, 3, 1–12. [Google Scholar]
- Oh, G.-H.; Kim, J.-E.; Park, Y.-J. Development of stabilized tenofovir disoproxil tablet: Degradation profile, stabilization, and bioequivalence in beagle dogs. Drug Dev. Ind. Pharm. 2017, 44, 757–766. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characterization | SR Formulation | ||
|---|---|---|---|
| F1 | F2 | F3 | |
| Mesh size | 24 | 20 | 16 |
| Bulk density | 0.43 ± 0.01 | 0.39 ± 0.01 | 0.34 ± 0.02 |
| Tap density | 0.57 ± 0.02 | 0.50 ± 0.02 | 0.42 ± 0.01 |
| Carr’s Index (%) | 23.4 ± 1.60 | 22.2 ± 2.10 | 19.5 ± 1.46 |
| Hardness (kp) | 16.1 ± 0.01 | 15.7 ± 0.01 | 15.0 ± 0.02 |
| Tableting pressure (kN) | 18.2 ± 0.02 | 17.4 ± 0.01 | 16.5 ± 0.01 |
| PK Parameter * | F1 | F2 | F3 | |||
|---|---|---|---|---|---|---|
| Reference Drug (12.5 mg) | Test Drug (37.5 mg) | Reference Drug (12.5 mg) | Test Drug (37.5 mg) | Reference Drug (12.5 mg) | Test Drug (37.5 mg) | |
| Cmax (ng/mL) | 745.25 ± 238.75 | 645.02 ± 190.15 | 1796.0 ± 592.7 | 1310.8 ± 213.1 | 620.8 ± 111.4 | 823.8 ± 217.2 |
| Tmax (h) ** | 1.0 [0.5–6] | 2.0 [0.25–4] | 0.5 [0.5–11] | 1.5 [0.5–4] | 0.8 [0.5–1] | 2.0 [1–4] |
| AUClast (ng·h/mL) | 4559.86 ± 1064.77 | 4284.49 ± 1338.96 | 10607.6 ± 2764.6 | 8269.0 ± 2800.3 | 4801.5 ± 923.8 | 5130.3 ± 714.9 |
| AUCinf (ng·h/mL) | 4724.20 ± 1093.57 | 4465.22 ± 1409.58 | 10959.8 ± 2706.8 | 8664.0 ± 2961.9 | 5285.3 ± 1246.1 | 5814.7 ± 1364.0 |
| t1/2 (h) | 3.61 ± 1.16 | 4.88 ± 1.79 | 3.7 ± 1.2 | 5.5 ± 2.3 | 5.6 ± 3.3 | 7.6 ± 6.0 |
| Statistical BE test of F1 | Statistical BE test of F2 | Statistical BE test of F3 | ||||
| Bioequivalence | Cmax (ng/mL) | AUClast (ng·h/mL) | Cmax (ng/mL) | AUClast (ng·h/mL) | Cmax (ng/mL) | AUClast (ng·h/mL) |
| Geometric mean ratio (Test/Ref, %) | 87.93 | 92.28 | 75.48 | 75.46 | 130.92 | 107.82 |
| 90% CI (Lower) | 77.50 | 79.28 | 57.93 | 51.87 | 105.05 | 89.32 |
| 90% CI (Upper) | 99.76 | 107.42 | 98.35 | 109.78 | 163.17 | 130.16 |
| Ingredient | Formulation | |
|---|---|---|
| IR (Immediate-Release) | SR (Sustained-Release) | |
| Tianeptine sodium | 4.3 (6.5 mg) | 5.2 (31 mg) |
| MCC 1 | 64.4 | - |
| D-mannitol | 26.8 | 59.2 |
| HPMC 2 100M cp | - | 30.0 |
| PEO 3 | - | 3.3 |
| Mg stearate | 1.2 | 2.0 |
| colloidal silicon dioxide | 3.3 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-J.; Kim, J.-E. In Vitro–In Vivo Correlation of Tianeptine Sodium Sustained-Release Dual-Layer Tablets. Molecules 2022, 27, 2828. https://doi.org/10.3390/molecules27092828
Lee Y-J, Kim J-E. In Vitro–In Vivo Correlation of Tianeptine Sodium Sustained-Release Dual-Layer Tablets. Molecules. 2022; 27(9):2828. https://doi.org/10.3390/molecules27092828
Chicago/Turabian StyleLee, Ye-Ji, and Joo-Eun Kim. 2022. "In Vitro–In Vivo Correlation of Tianeptine Sodium Sustained-Release Dual-Layer Tablets" Molecules 27, no. 9: 2828. https://doi.org/10.3390/molecules27092828
APA StyleLee, Y.-J., & Kim, J.-E. (2022). In Vitro–In Vivo Correlation of Tianeptine Sodium Sustained-Release Dual-Layer Tablets. Molecules, 27(9), 2828. https://doi.org/10.3390/molecules27092828