
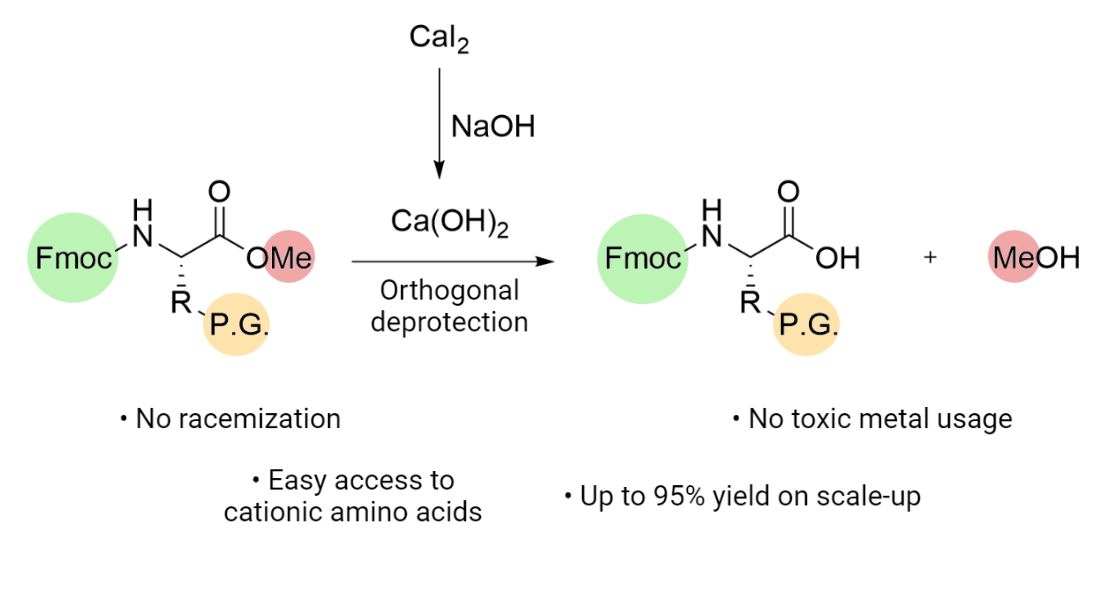
Efficient Fmoc-Protected Amino Ester Hydrolysis Using Green Calcium(II) Iodide as a Protective Agent


Abstract

1. Introduction
2. Material and Methods
2.1. Ester Synthesis
2.1.1. General Esterification for Acid-Resistant Fmoc Amino Esters
2.1.2. General Esterification for Acid-Sensitive Fmoc Amino Methyl Esters
2.1.3. Fmoc-Gly-OtBu Synthesis
2.1.4. Phth-Gly-OMe Synthesis
2.1.5. Fmoc-Gly-OBn Synthesis
2.2. Calcium Iodide 5 M Solution Preparation
2.3. Standard Conditions for Ester Hydrolysis
2.4. Reaction Quench for Sampling
2.5. UPLC-MS Analysis of Crude Material
2.6. NMR Data Acquisition
2.7. Extraction of the Final Product
2.8. Chiral HPLC Methodology
3. Results and Discussion
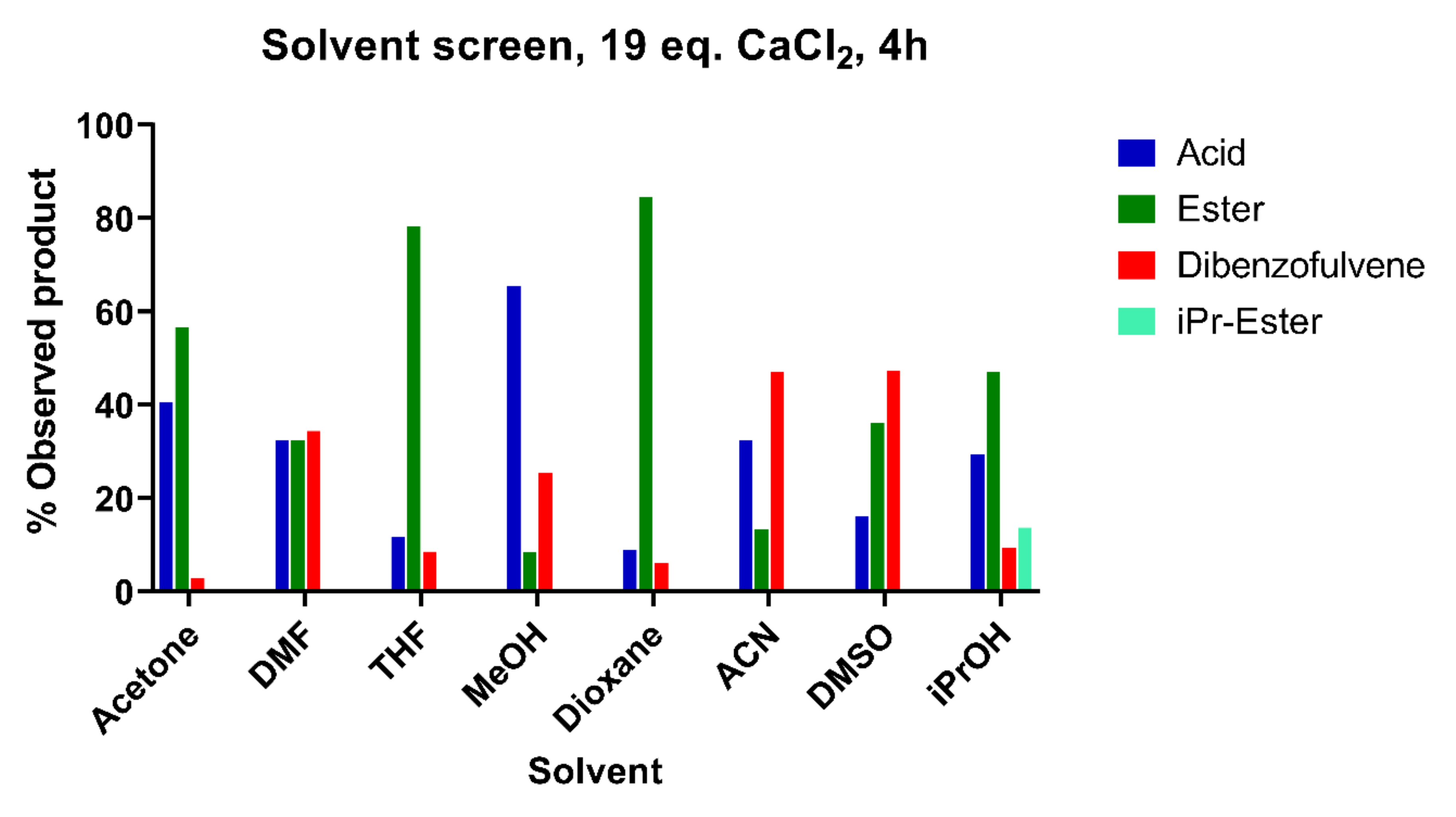
3.1. Nature of the Organic Solvent
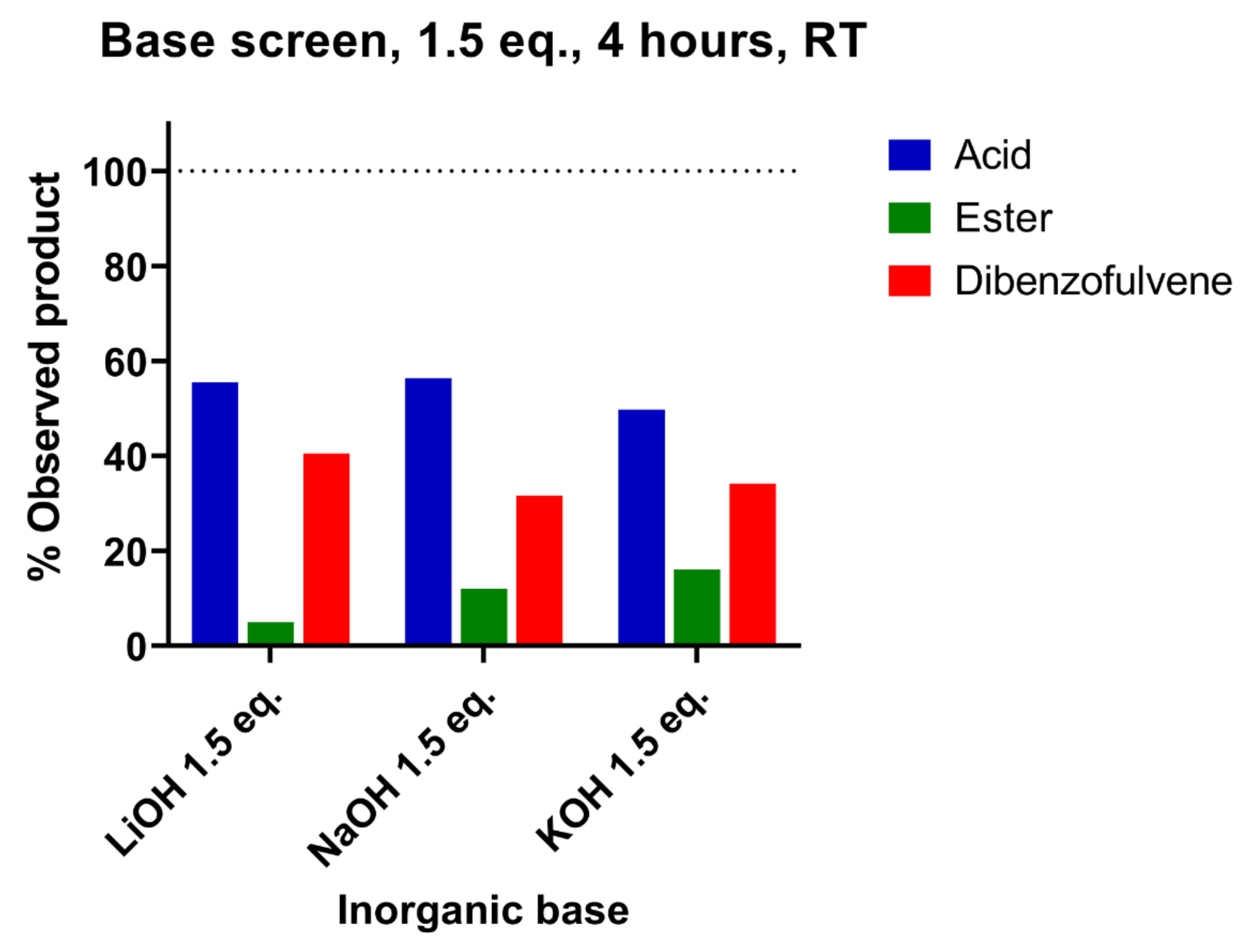
3.2. Nature and Concentration of the Inorganic Base
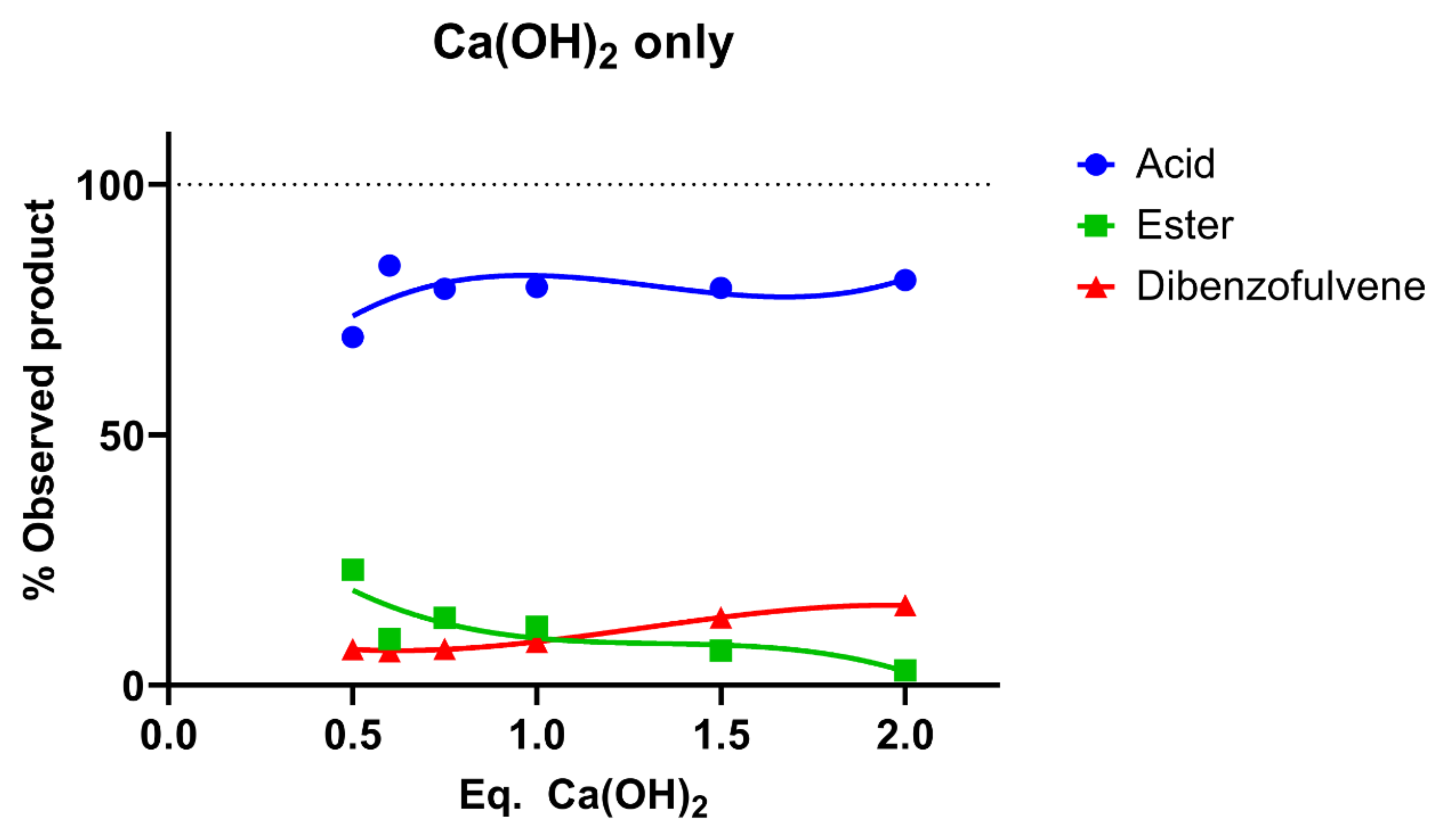
3.3. Initial Considerations of Ca(OH)2
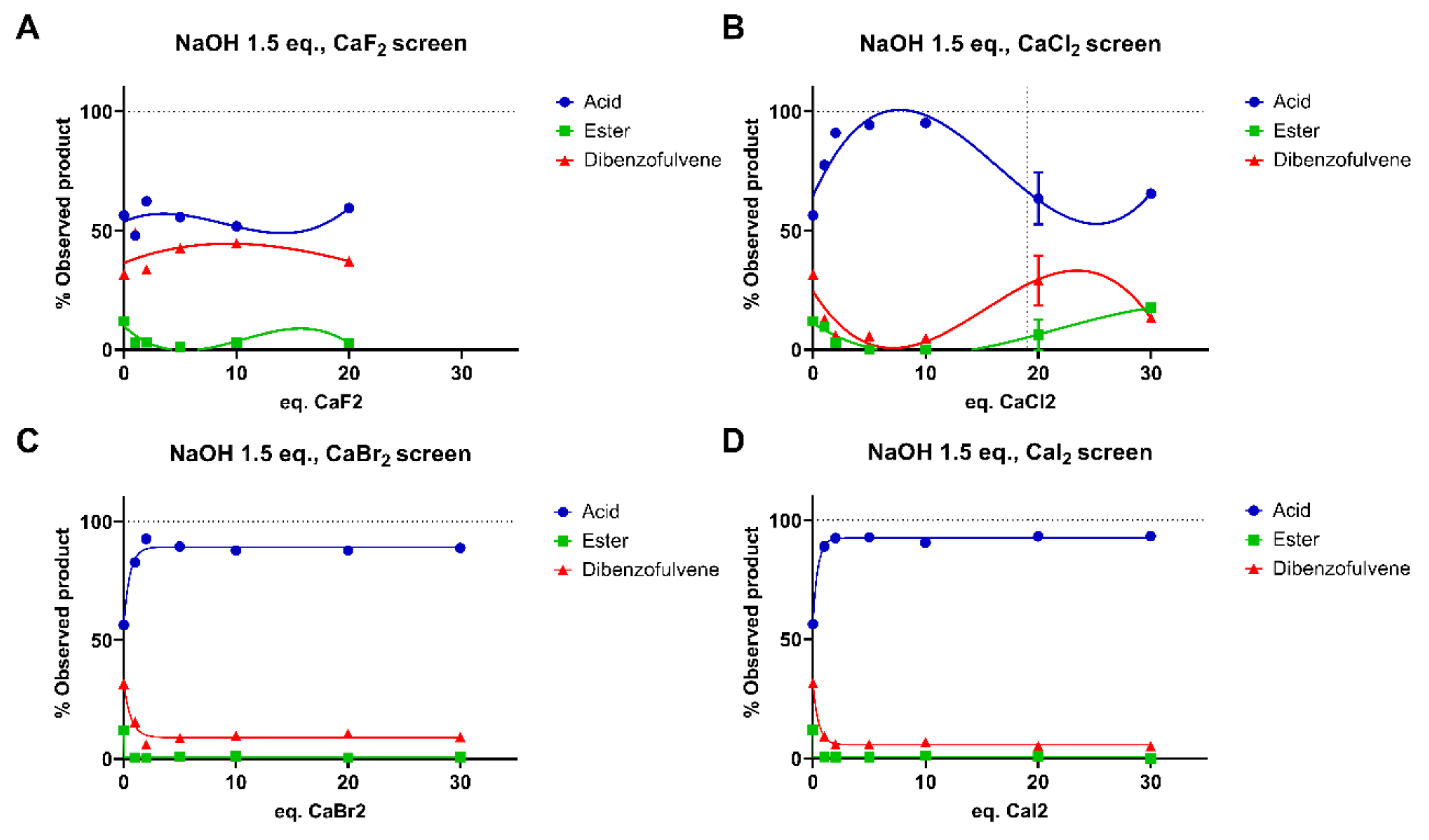
3.4. Nature of the Salt
3.4.1. Nature of the Cation Using Salt Chlorides
3.4.2. Nature and Concentration of the Anion Using Calcium Salts
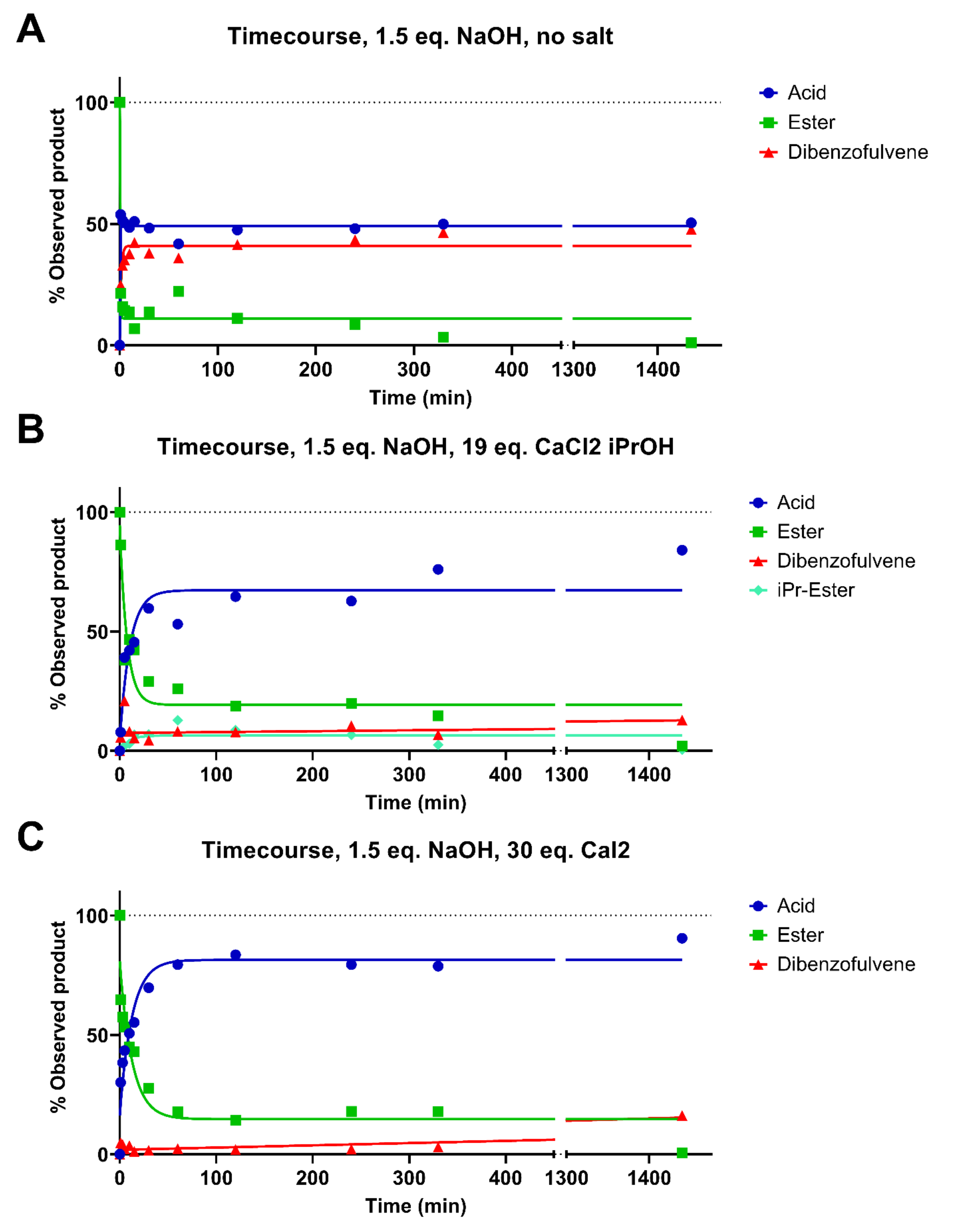
3.5. Reaction Kinetics
3.6. Scope of the Reaction
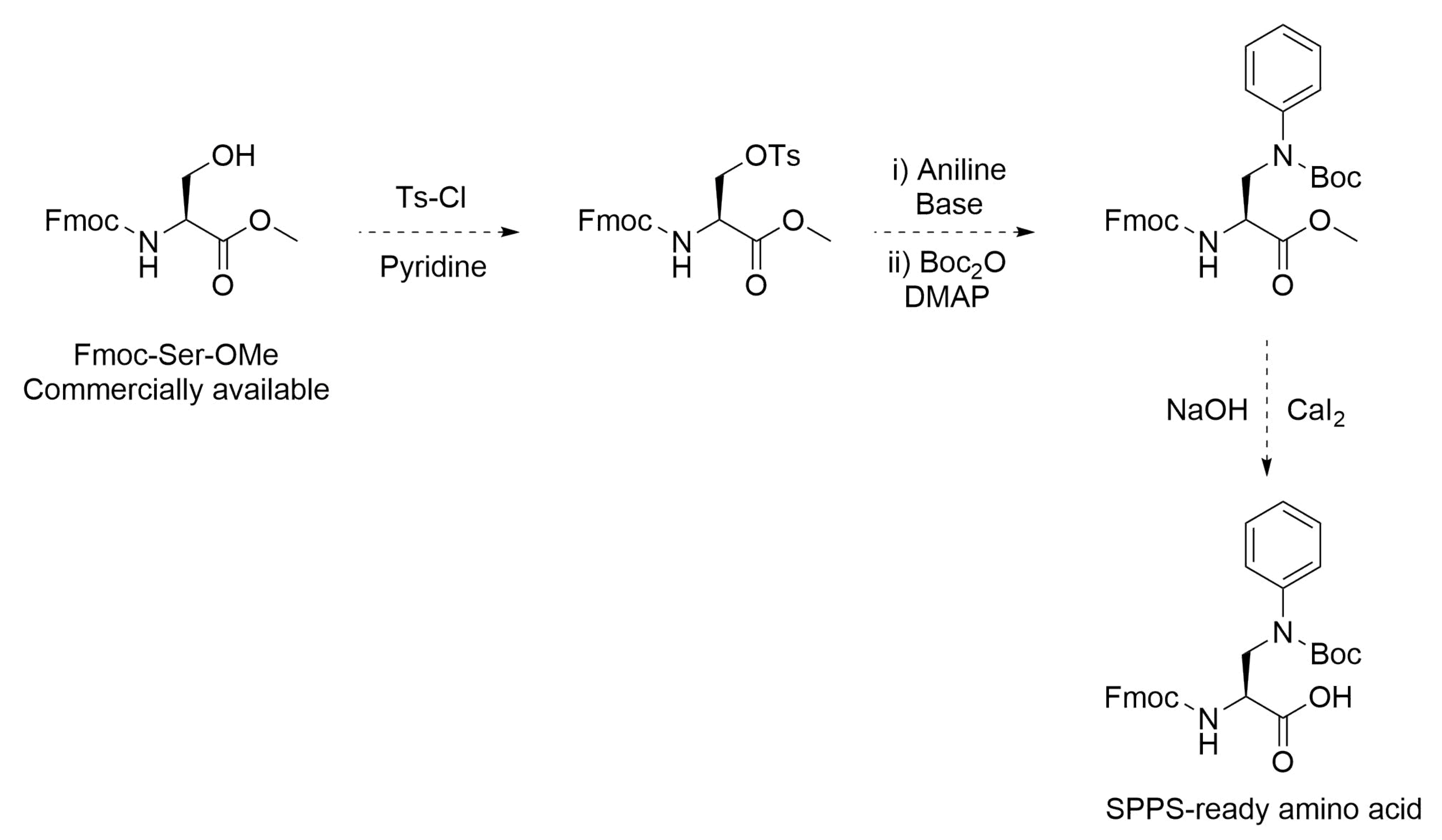
3.7. Example of Newly Accessible Amino Acid Chemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Isidro-Llobet, A.; Álvarez, M.; Albericio, F. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef] [PubMed]
- Merker, R.L.; Scott, M.J. The Reaction of Alkyl Halides with Carboxylic Acids and Phenols in the Presence of Tertiary Amines. J. Org. Chem. 1961, 26, 5180–5182. [Google Scholar] [CrossRef]
- Pascal, R.; Sola, R. Preservation of the protective group under alkaline conditions by using CaCl Applications in peptide synthesis. Tetrahedron Lett. 1998, 39, 5031–5034. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Estrada, A.A.; Zak, M.; Lee, S.H.; Safina, B.S. A Mild and Selective Method for the Hydrolysis of Esters with Trimethyltin Hydroxide. Angew. Chem. Int. Ed. 2005, 44, 1378–1382. [Google Scholar] [CrossRef]
- Hikal, A.H.; Lipe, G.W.; Slikker, W.; Scallet, A.C.; Ali, S.F.; Newport, G.D. Determination of amino acids in different regions of the rat brain application to the acute effects of tetrahydrocannabinol (THC) and trimethyltin (TMT). Life Sci. 1988, 42, 2029–2035. [Google Scholar] [CrossRef]
- Besser, R.; Kramer, G.; Thümler, R.; Bohl, J.; Gutmann, L.; Hopf, H.C. Acute trimethyltin limbic-cerebellar syndrome. Neurology 1987, 37, 945. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, M.; Dawson, A.P.; Selwyn, M.J. Effects of Trialkyltin and Triphenyltin Copounds on Mitochondrial Respiration. JBIC J. Biol. Inorg. Chem. 1970, 15, 342–351. [Google Scholar] [CrossRef]
- Henschler, D. Toxicity of Chlorinated Organic Compounds: Effects of the Introduction of Chlorine in Organic Molecules. Angew. Chem. Int. Ed. 1994, 33, 1920–1935. [Google Scholar] [CrossRef]
- Martínez, A.G.; Barcinaa, J.O.; del Veccio, G.H.; Hanack, M.; Subramanian, L. Non-hydrolytic cleavage of esters with magnesium iodide in aprotic non-polar solvents. Tetrahedron Lett. 1991, 32, 5931–5934. [Google Scholar] [CrossRef]
- Berthet, M.; Davanier, F.; Dujardin, G.; Martinez, J.; Parrot, I. MgI2-Mediated Chemoselective Cleavage of Protecting Groups: An Alternative to Conventional Deprotection Methodologies. Chem.–A Eur. J. 2015, 21, 11014–11016. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Kemp, R.; Keegan, S.E. Calcium Chloride. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA a04_547: Weinheim, Germany, 2000. [Google Scholar] [CrossRef]
- Otera, J. Transesterification. Chem. Rev. 1993, 93, 1449–1470. [Google Scholar] [CrossRef]
- Prat, D.; Hayler, J.; Wells, A. A survey of solvent selection guides. Green Chem. 2014, 16, 4546–4551. [Google Scholar] [CrossRef]
- Capello, C.; Fischer, U.; Hungerbühler, K. What is a green solvent? A comprehensive framework for the environmental assessment of solvents. Green Chem. 2007, 9, 927–934. [Google Scholar] [CrossRef]
- Hayashi, K.; Ichimaru, Y.; Sugiura, K.; Maeda, A.; Harada, Y.; Kojima, Y.; Nakayama, K.; Imai, M. Efficiency of Lithium Cations in Hydrolysis Reactions of Esters in Aqueous Tetrahydrofuran. Chem. Pharm. Bull. 2021, 69, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Huffman, J.W.; Harris, P.R. A Convenient Procedure for the Hydrolysis of Hindered Esters. Synth. Commun. 1976, 6, 481–484. [Google Scholar] [CrossRef]
- Meyer, K.; Bloch, H. Naphthoresorcinol. Org. Synth. 1945, 25, 73. [Google Scholar] [CrossRef]
- Moir, M.E.; Crawford, R.J. Model studies of competing hydrolysis and epimerization of some tetrapeptides of interest in amino acid racemization studies in geochronology. Can. J. Chem. 1988, 66, 2903–2913. [Google Scholar] [CrossRef]
- Stroud, E.D.; Fife, D.J.; Smith, G.G. A method for the determination of the pKa of the .alpha.-hydrogen in amino acids using racemization and exchange studies. J. Org. Chem. 1983, 48, 5368–5369. [Google Scholar] [CrossRef]
- Li, Y.H.; Baek, C.; Jo, B.; Lee, W. Direct Chiral Separation of N-Fluorenylmethoxycarbonyl α-Amino Acids by HPLC for Determination of Enantiomeric Purity. Bull. Korean Chem. Soc. 2005, 26, 998–1000. [Google Scholar] [CrossRef][Green Version]
- Shinbara, K.; Liu, W.; Van Neer, R.H.P.; Katoh, T.; Suga, H. Methodologies for Backbone Macrocyclic Peptide Synthesis Compatible with Screening Technologies. Front. Chem. 2020, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.P.; Gordon, J.J.; Watson, H.B. The influence of alkyl groups upon reaction velocities in solution. Part III. The alkaline hydrolysis of saturated aliphatic esters. J. Chem. Soc. 1938, 1439–1444. [Google Scholar] [CrossRef]
- Goodman, M.; McGahren, W. Mechanistic studies of peptide oxazolone racemization. Tetrahedron 1967, 23, 2031–2050. [Google Scholar] [CrossRef]
- Santagada, V.; Fiorino, F.; Perissutti, E.; Severino, B.; De Filippis, V.; Vivenzio, B.; Caliendo, G. Microwave-enhanced solution coupling of the α,α-dialkyl amino acid, Aib. Tetrahedron Lett. 2001, 42, 5171–5173. [Google Scholar] [CrossRef]
- Khan, M.N. Suggested Improvement in the Ing−Manske Procedure and Gabriel Synthesis of Primary Amines: Kinetic Study on Alkaline Hydrolysis of N-Phthaloylglycine and Acid Hydrolysis of N-(o-Carboxybenzoyl)glycine in Aqueous Organic Solvents. J. Org. Chem. 1996, 61, 8063–8068. [Google Scholar] [CrossRef]
- Osby, J.O.; Martin, M.G.; Ganem, B. An exceptionally mild deprotection of phthalimides. Tetrahedron Lett. 1984, 25, 2093–2096. [Google Scholar] [CrossRef]
- Clausen, J.D.; Linderoth, L.; Nielsen, H.M.; Franzyk, H. Solid-phase route to Fmoc-protected cationic amino acid building blocks. Amino Acids 2012, 43, 1633–1641. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Salt (eq.) | % Acid | % Ester | % Dibenzofulvene |
|---|---|---|---|
| CaCl2 (30) | 65.5 | 17.7 | 13.4 |
| LiCl (30) | 65.9 | 10.2 | 22.8 |
| MgCl2 (30) | 22.1 | 64.0 | 13.9 |
| BaCl2 (6) | 74.4 | 2.8 | 21.9 |
| LaCl3 (15) | 28.1 | 41.9 | 23.2 |
| KCl (30) | 49.8 | 26.2 | 23.9 |
| ZnCl2 (30) | 5.0 | 88.6 | 6.2 |
| FeCl2 (30) | 2.3 | 92.0 | 5.7 |
| FeCl3 (20) | 3.6 | 87.2 | 9.1 |
| Amino Acid | Crude Yield (%) * | Dibenzofulvene Formation (%) * | Category | Average Yield Per Category (%) * |
|---|---|---|---|---|
| Fmoc-Gly-OMe | 88.8 | 9.2 | 1: Standard amino methyl esters | 83.9 |
| Fmoc-Ala-OMe | 80.4 | 13.3 | ||
| Fmoc-Arg(Pbf)-OMe | 76.3 | 5.3 | ||
| Fmoc-Asn(Trt)-OMe | 95.6 | Not observed | ||
| Fmoc-Asp(tBu)-OMe | 77.6 | 11.8 | ||
| Fmoc-Cys(Trt)-OMe | 83.1 | 12.2 | ||
| Fmoc-Gln(Trt)-OMe | 94.1 | Not observed | ||
| Fmoc-Glu(tBu)-OMe | 73.6 | 9.2 | ||
| Fmoc-His(Trt)-OMe | 84.4 | 8.4 | ||
| Fmoc-Leu-OMe | 86.5 | 13.1 | ||
| Fmoc-Lys(Boc)-OMe | 79.9 | 13.0 | ||
| Fmoc-Lys(Alloc)-OMe | 84.9 | 12.4 | ||
| Fmoc-Met-OMe | 80.6 | 9.3 | ||
| Fmoc-Phe-OMe | 84.3 | 12.2 | ||
| Fmoc-Pro-OMe | 87.4 | 9.7 | ||
| Fmoc-Ser(tBu)-OMe | 81.4 | 13.7 | ||
| Fmoc-Trp(Boc)-OMe | 83.1 | 7.7 | ||
| Fmoc-Tyr(tBu)-OMe | 88.9 | 7.8 | ||
| Fmoc-Thr(tBu)-OMe | 51.1 | 11.0 | 2: β-hindered amino methyl esters | 52.3 |
| Fmoc-Val-OMe | 59.4 | 11.1 | ||
| Fmoc-Ile-OMe | 46.5 | Not observed | ||
| Fmoc-Gly-OEt | 79.3 | 7.3 | 3: Other hindered amino esters | 62.4 |
| Fmoc-Gly-OiPr | 83.8 | 11.8 | ||
| Fmoc-Gly-OBn | 85.3 | 9.1 | ||
| Fmoc-Gly-OtBu | 30.6 | 10.7 | ||
| Fmoc-Aib-OMe | 32.9 | 9.2 | ||
| Phth-Gly-OMe | 0 | N/A | 4: Non-compatible amino methyl esters | Non-compatible (high degradation) |
| Fmoc-Asp(Allyl)-OMe | 36.1 | 4.2 | ||
| Fmoc-Gly-OMe (Scale-up, 3g) | 95.6 ** | 2.1 | Scale-up | 95.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binette, R.; Desgagné, M.; Theaud, C.; Boudreault, P.-L. Efficient Fmoc-Protected Amino Ester Hydrolysis Using Green Calcium(II) Iodide as a Protective Agent. Molecules 2022, 27, 2788. https://doi.org/10.3390/molecules27092788
Binette R, Desgagné M, Theaud C, Boudreault P-L. Efficient Fmoc-Protected Amino Ester Hydrolysis Using Green Calcium(II) Iodide as a Protective Agent. Molecules. 2022; 27(9):2788. https://doi.org/10.3390/molecules27092788
Chicago/Turabian StyleBinette, Renaud, Michael Desgagné, Camille Theaud, and Pierre-Luc Boudreault. 2022. "Efficient Fmoc-Protected Amino Ester Hydrolysis Using Green Calcium(II) Iodide as a Protective Agent" Molecules 27, no. 9: 2788. https://doi.org/10.3390/molecules27092788
APA StyleBinette, R., Desgagné, M., Theaud, C., & Boudreault, P.-L. (2022). Efficient Fmoc-Protected Amino Ester Hydrolysis Using Green Calcium(II) Iodide as a Protective Agent. Molecules, 27(9), 2788. https://doi.org/10.3390/molecules27092788