
Expression of Growth Hormone-Releasing Hormone and Its Receptor Splice Variants in Primary Human Endometrial Carcinomas: Novel Therapeutic Approaches




Abstract
1. Introduction
2. Results
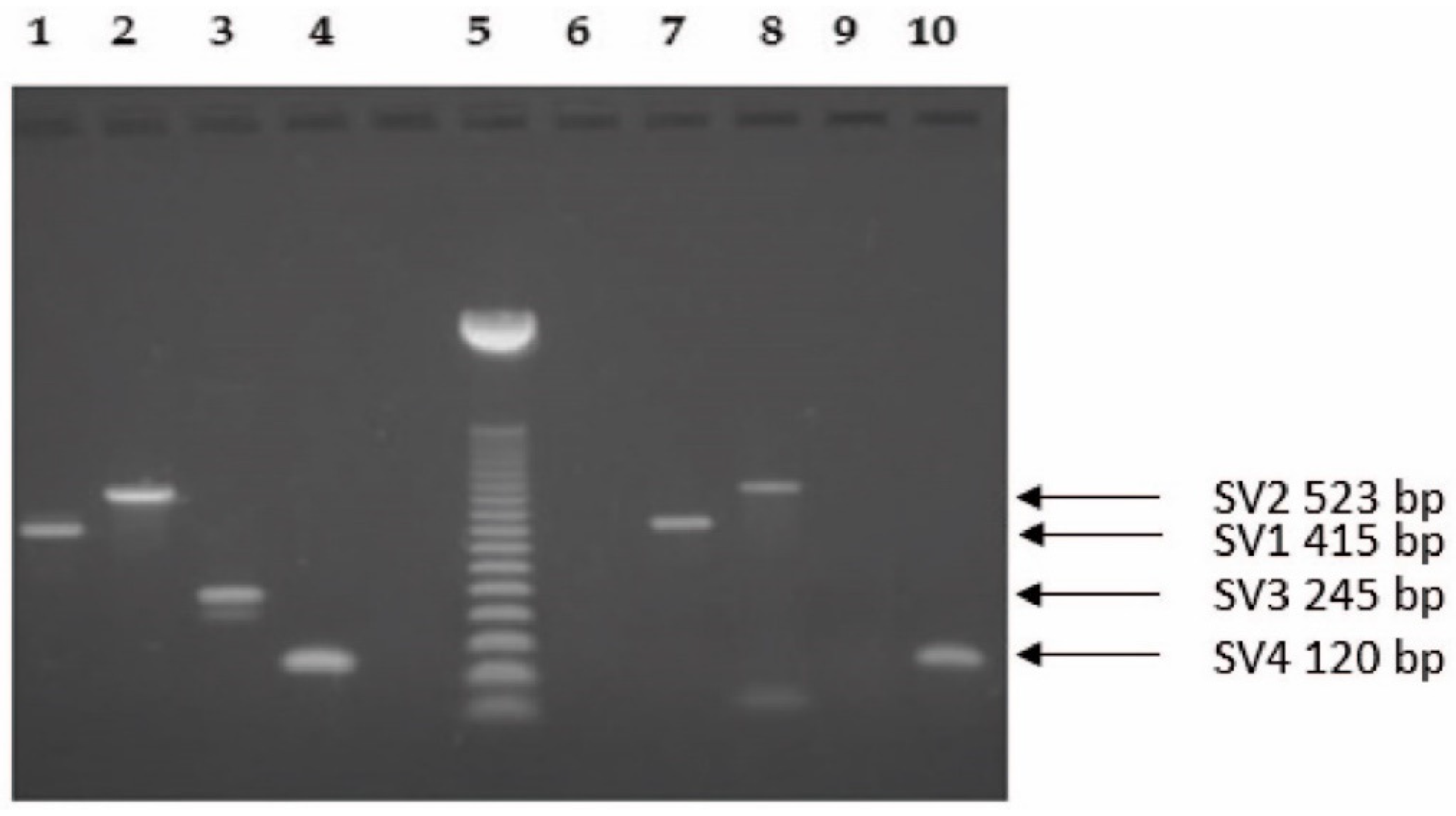
2.1. Molecular Biology Analysis
2.2. Radioligand Binding Studies
3. Discussion
4. Materials and Methods
4.1. Tissue Samples
4.2. RNA Isolation
4.3. RT-PCR
4.4. Preparation of Membranes and Radioligand Binding Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Halmos, G.; Schally, A.V.; Czompoly, T.; Krupa, M.; Varga, J.L.; Rekasi, Z. Expression of growth hormone-releasing hormone and its receptor splice variants in human prostate cancer. J. Clin. Endocrinol. Metab. 2002, 87, 4707–4714. [Google Scholar] [CrossRef] [PubMed]
- Chatzistamou, I.; Schally, A.V.; Kiaris, H.; Politi, E.; Varga, J.; Kanellis, G.; Kalofoutis, A.; Pafiti, A.; Koutselini, H. Immunohistochemical detection of GHRH and its receptor splice variant 1 in primary human breast cancers. Eur. J. Endocrinol. 2004, 151, 391–396. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Havt, A.; Schally, A.V.; Halmos, G.; Varga, J.L.; Toller, G.L.; Horvath, J.E.; Szepeshazi, K.; Köster, F.; Kovitz, K.; Groot, K.; et al. The expression of the pituitary growth hormone-releasing hormone receptor and its splice variants in normal and neoplastic human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 17424–17429. [Google Scholar] [CrossRef] [PubMed]
- Freddi, S.; Arnaldi, G.; Fazioli, F.; Scarpelli, M.; Appolloni, G.; Mancini, T.; Kola, B.; Bertagna, X.; Mantero, F.; Collu, R.; et al. Expression of growth hormone-releasing hormone receptor splicing variants in human primary adrenocortical tumours. Clin. Endocrinol. 2005, 62, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Rekasi, Z.; Czompoly, T.; Schally, A.V.; Halmos, G. Isolation and sequencing of cDNAs for splice variants of growth hormone-releasing hormone receptors from human cancers. Proc. Natl. Acad. Sci. USA 2000, 97, 10561–10566. [Google Scholar] [CrossRef]
- Barabutis, N.; Tsellou, E.; Schally, A.V.; Kouloheri, S.; Kalofoutis, A.; Kiaris, H. Stimulation of proliferation of MCF-7 breast cancer cells by a transfected splice variant of growth hormone-releasing hormone receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 5575–5579. [Google Scholar] [CrossRef]
- Kiaris, H.; Chatzistamou, I.; Schally, A.V.; Halmos, G.; Varga, J.L.; Koutselini, H.; Kalofoutis, A. Ligand-dependent and -independent effects of splice variant 1 of growth hormone-releasing hormone receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 9512–9517. [Google Scholar] [CrossRef]
- Kiaris, H.; Schally, A.V.; Busto, R.; Halmos, G.; Artavanis-Tsakonas, S.; Varga, J.L. Expression of a splice variant of the receptor for GHRH in 3T3 fibroblasts activates cell proliferation responses to GHRH analogs. Proc. Natl. Acad. Sci. USA 2002, 99, 196–200. [Google Scholar] [CrossRef]
- Csernus, V.J.; Schally, A.V.; Kiaris, H.; Armatis, P. Inhibition of growth, production of insulin-like growth factor-II (IGF-II), and expression of IGF-II mRNA of human cancer cell lines by antagonistic analogs of growth hormone-releasing hormone in vitro. Proc. Natl. Acad. Sci. USA 1999, 96, 3098–3103. [Google Scholar] [CrossRef]
- Rekasi, Z.; Varga, J.L.; Schally, A.V.; Halmos, G.; Armatis, P.; Groot, K.; Czompoly, T. Antagonists of growth hormone-releasing hormone and vasoactive intestinal peptide inhibit tumor proliferation by different mechanisms: Evidence from in vitro studies on human prostatic and pancreatic cancers. Endocrinology 2000, 141, 2120–2128. [Google Scholar] [CrossRef]
- Rekasi, Z.; Varga, J.L.; Schally, A.V.; Plonowski, A.; Halmos, G.; Csernus, B.; Armatis, P.; Groot, K. Antiproliferative actions of growth hormone-releasing hormone antagonists on MiaPaCa-2 human pancreatic cancer cells involve cAMP independent pathways. Peptides 2001, 22, 879–886. [Google Scholar] [CrossRef]
- Szepeshazi, K.; Schally, A.V.; Groot, K.; Armatis, P.; Hebert, F.; Halmos, G. Antagonists of growth hormone-releasing hormone (GH-RH) inhibit in vivo proliferation of experimental pancreatic cancers and decrease IGF-II levels in tumours. Eur. J. Cancer 2000, 36, 128–136. [Google Scholar] [CrossRef]
- Szepeshazi, K.; Schally, A.V.; Groot, K.; Armatis, P.; Halmos, G.; Herbert, F.; Szende, B.; Varga, J.L.; Zarandi, M. Antagonists of growth hormone-releasing hormone (GH-RH) inhibit IGF-II production and growth of HT-29 human colon cancers. Br. J. Cancer 2000, 82, 1724–1731. [Google Scholar] [PubMed]
- Jungwirth, A.; Schally, A.V.; Pinski, J.; Halmos, G.; Groot, K.; Armatis, P.; Vadillo-Buenfil, M. Inhibition of in vivo proliferation of androgen-independent prostate cancers by an antagonist of growth hormone-releasing hormone. Br. J. Cancer 1997, 75, 1585–1592. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lamharzi, N.; Schally, A.V.; Koppán, M.; Groot, K. Growth hormone-releasing hormone antagonist MZ-5-156 inhibits growth of DU-145 human androgen-independent prostate carcinoma in nude mice and suppresses the levels and mRNA expression of insulin-like growth factor II in tumors. Proc. Natl. Acad. Sci. USA 1998, 95, 8864–8868. [Google Scholar] [CrossRef] [PubMed]
- Letsch, M.; Schally, A.V.; Busto, R.; Bajo, A.M.; Varga, J.L. Growth hormone-releasing hormone (GHRH) antagonists inhibit the proliferation of androgen-dependent and -independent prostate cancers. Proc. Natl. Acad. Sci. USA 2003, 100, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Kahán, Z.; Varga, J.L.; Schally, A.V.; Rékási, Z.; Armatis, P.; Chatzistamou, L.; Czömpöly, T.; Halmos, G. Antagonists of growth hormone-releasing hormone arrest the growth of MDA-MB-468 estrogen-independent human breast cancers in nude mice. Breast Cancer Res. Treat. 2000, 60, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Chatzistamou, I.; Schally, A.V.; Varga, J.L.; Groot, K.; Busto, R.; Armatis, P.; Halmos, G. Inhibition of growth and metastases of MDA-MB-435 human estrogen-independent breast cancers by an antagonist of growth hormone-releasing hormone. Anti-Cancer Drugs 2001, 12, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Khanlari, M.; Schally, A.V.; Block, N.L.; Nadji, M. Expression of GHRH-R, a Potentially Targetable Biomarker, in Triple-negative Breast Cancer. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 1–5. [Google Scholar] [CrossRef]
- Chatzistamou, I.; Schally, A.V.; Varga, J.L.; Groot, K.; Armatis, P.; Bajo, A.M. Inhibition of growth and reduction in tumorigenicity of UCI-107 ovarian cancer by antagonists of growth hormone-releasing hormone and vasoactive intestinal peptide. J. Cancer Res. Clin. Oncol. 2001, 127, 645–652. [Google Scholar] [CrossRef]
- Jungwirth, A.; Schally, A.V.; Pinski, J.; Groot, K.; Armatis, P.; Halmos, G. Growth hormone-releasing hormone antagonist MZ-4-71 inhibits in vivo proliferation of Caki-I renal adenocarcinoma. Proc. Natl. Acad. Sci. USA 1997, 94, 5810–5813. [Google Scholar] [CrossRef] [PubMed]
- Pinski, J.; Schally, A.; Jungwirth, A.; Groot, K.; Halmos, G.; Armatis, P.; Zarandi, M.; Vadillobuenfil, M. Inhibition of growth of human small cell and non-small cell lung carcinomas by antagonists of growth hormone-releasing hormone (GH-RH). Int. J. Oncol. 1996, 9, 1099–1105. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kiaris, H.; Schally, A.V.; Varga, J.L.; Groot, K.; Armatis, P. Growth hormone-releasing hormone: An autocrine growth factor for small cell lung carcinoma. Proc. Natl. Acad. Sci. USA 1999, 96, 14894–14898. [Google Scholar] [CrossRef] [PubMed]
- Szereday, Z.; Schally, A.V.; Varga, J.L.; Kanashiro, C.A.; Hebert, F.; Armatis, P.; Groot, K.; Szepeshazi, K.; Halmos, G.; Busto, R. Antagonists of growth hormone-releasing hormone inhibit the proliferation of experimental non-small cell lung carcinoma. Cancer Res. 2003, 63, 7913–7919. [Google Scholar] [PubMed]
- Kiaris, H.; Schally, A.V.; Varga, J.L. Antagonists of growth hormone-releasing hormone inhibit the growth of U-87MG human glioblastoma in nude mice. Neoplasia 2000, 2, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Mezey, G.; Treszl, A.; Schally, A.V.; Block, N.L.; Vízkeleti, L.; Juhász, A.; Klekner, A.; Nagy, J.; Balázs, M.; Halmos, G.; et al. Prognosis in human glioblastoma based on expression of ligand growth hormone-releasing hormone, pituitary-type growth hormone-releasing hormone receptor, its splicing variant receptors, EGF receptor and PTEN genes. J. Cancer Res. Clin. Oncol. 2014, 140, 1641–1649. [Google Scholar] [CrossRef]
- Braczkowski, R.; Schally, A.V.; Plonowski, A.; Varga, J.L.; Groot, K.; Krupa, M.; Armatis, P. Inhibition of proliferation in human MNNG/HOS osteosarcoma and SK-ES-1 Ewing sarcoma cell lines in vitro and in vivo by antagonists of growth hormone-releasing hormone: Effects on insulin-like growth factor II. Cancer 2002, 95, 1735–1745. [Google Scholar] [CrossRef]
- Pinski, J.; Schally, A.V.; Groot, K.; Halmos, G.; Szepeshazi, K.; Zarandi, M.; Armatis, P. Inhibition of growth of human osteosarcomas by antagonists of growth hormone-releasing hormone. J. Natl. Cancer Inst. 1995, 87, 1787–1794. [Google Scholar] [CrossRef]
- Xiong, X.; Ke, X.; Wang, L.; Yao, Z.; Guo, Y.; Zhang, X.; Chen, Y.; Pang, C.P.; Schally, A.V.; Zhang, H. Splice variant of growth hormone-releasing hormone receptor drives esophageal squamous cell carcinoma conferring a therapeutic target. Proc. Natl. Acad. Sci. USA 2020, 117, 6726–6732. [Google Scholar] [CrossRef]
- Villanova, T.; Gesmundo, I.; Audrito, V.; Vitale, N.; Silvagno, F.; Musuraca, C.; Righi, L.; Libener, R.; Riganti, C.; Bironzo, P.; et al. Antagonists of growth hormone-releasing hormone (GHRH) inhibit the growth of human malignant pleural mesothelioma. Proc. Natl. Acad. Sci. USA 2019, 116, 2226–2231. [Google Scholar] [CrossRef]
- Engel, J.B.; Keller, G.; Schally, A.V.; Toller, G.L.; Groot, K.; Havt, A.; Armatis, P.; Zarandi, M.; Varga, J.L.; Halmos, G. Inhibition of growth of experimental human endometrial cancer by an antagonist of growth hormone-releasing hormone. J. Clin. Endocrinol. Metab. 2005, 90, 3614–3621. [Google Scholar] [CrossRef] [PubMed]
- Kahán, Z.; Arencibia, J.M.; Csernus, V.J.; Groot, K.; Kineman, R.D.; Robinson, W.R.; Schally, A.V. Expression of growth hormone-releasing hormone (GHRH) messenger ribonucleic acid and the presence of biologically active GHRH in human breast, endometrial, and ovarian cancers. J. Clin. Endocrinol. Metab. 1999, 84, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Chatzistamou, I.; Schally, A.V.; Pafiti, A.; Kiaris, H.; Koutselini, H. Expression of growth hormone-releasing hormone in human primary endometrial carcinomas. Eur. J. Endocrinol. 2002, 147, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Busto, R.; Schally, A.V.; Varga, J.L.; Garcia-Fernandez, M.O.; Groot, K.; Armatis, P.; Szepeshazi, K. The expression of growth hormone-releasing hormone (GHRH) and splice variants of its receptor in human gastroenteropancreatic carcinomas. Proc. Natl. Acad. Sci. USA 2002, 99, 11866–11871. [Google Scholar] [CrossRef] [PubMed]
- Plonowski, A.; Schally, A.V.; Busto, R.; Krupa, M.; Varga, J.L.; Halmos, G. Expression of growth hormone-releasing hormone (GHRH) and splice variants of GHRH receptors in human experimental prostate cancers. Peptides 2002, 23, 1127–1133. [Google Scholar] [CrossRef]
- Schally, A.V.; Wang, H.; He, J.; Cai, R.; Sha, W.; Popovics, P.; Perez, R.; Vidaurre, I.; Zhang, X. Agonists of growth hormone-releasing hormone (GHRH) inhibit human experimental cancers in vivo by down-regulating receptors for GHRH. Proc. Natl. Acad. Sci. USA 2018, 115, 12028–12033. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Zhang, X.; Cai, R.; Hare, J.M.; Granata, R.; Bartoli, M. Actions and Potential Therapeutic Applications of Growth Hormone-Releasing Hormone Agonists. Endocrinology 2019, 160, 1600–1612. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Khorram, O.; Garthwaite, M.; Grosen, E.; Golos, T. Human uterine and ovarian expression of growth hormone-releasing hormone messenger RNA in benign and malignant gynecologic conditions. Fertil. Steril. 2001, 75, 174–179. [Google Scholar] [CrossRef]
- Kővári, B.; Vranic, S.; Marchio, C.; Sapino, A.; Cserni, G. The expression of GHRH and its receptors in breast carcinomas with apocrine differentiation-further evidence of the presence of a GHRH pathway in these tumors. Hum. Pathol. 2017, 64, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Osuga, Y.; Yano, T.; Takemura, Y.; Morimoto, C.; Hirota, Y.; Schally, A.V.; Taketani, Y. Expression and possible implication of growth hormone-releasing hormone receptor splice variant 1 in endometriosis. Fertil. Steril. 2009, 92, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Wright, J.D.; Barrena Medel, N.I.; Sehouli, J.; Fujiwara, K.; Herzog, T.J. Contemporary management of endometrial cancer. Lancet 2012, 379, 1352–1360. [Google Scholar] [CrossRef]
- Arend, R.C.; Jones, B.A.; Martinez, A.; Goodfellow, P. Endometrial cancer: Molecular markers and management of advanced stage disease. Gynecol. Oncol. 2018, 150, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Dong, P.; Ihira, K.; Kudo, M.; Watari, H. Molecular-targeted therapies and precision medicine for endometrial cancer. Jpn. J. Clin. Oncol. 2019, 49, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Kanao, H.; Wang, X.; Yunokawa, M.; Omatsu, K.; Fusegi, A.; Takeshima, N. Adjuvant treatment of endometrial cancer today. Jpn. J. Clin. Oncol. 2020, 50, 753–765. [Google Scholar] [CrossRef]
- Lu, K.H.; Broaddus, R.R. Endometrial Cancer. N. Engl. J. Med. 2020, 383, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Paleari, L.; Pesce, S.; Rutigliani, M.; Greppi, M.; Obino, V.; Gorlero, F.; Vellone, V.G.; Marcenaro, E. New Insights into Endometrial Cancer. Cancers 2021, 13, 1496. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Varga, J.L.; Engel, J.B. Antagonists of growth-hormone-releasing hormone: An emerging new therapy for cancer. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 33–43. [Google Scholar] [CrossRef]
- Köster, F.; Engel, J.B.; Schally, A.V.; Hönig, A.; Schröer, A.; Seitz, S.; Hohla, F.; Ortmann, O.; Diedrich, K.; Buchholz, S. Triple-negative breast cancers express receptors for growth hormone-releasing hormone (GHRH) and respond to GHRH antagonists with growth inhibition. Breast Cancer Res. Treat. 2009, 116, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, C.; Schally, A.V.; Chatzistamou, I.; Kondi-Pafiti, A.; Lamnissou, K.; Kouloheri, S.; Kalofoutis, A.; Kiaris, H. Expression of growth hormone-releasing hormone (GHRH) and splice variant of GHRH receptors in normal mouse tissues. Regul. Pept. 2006, 136, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yano, T.; Osuga, Y.; Nakagawa, S.; Oishi, H.; Wada-Hiraike, O.; Tang, X.; Yano, N.; Kugu, K.; Schally, A.V.; et al. Cellular mechanisms of growth inhibition of human endometrial cancer cell line by an antagonist of growth hormone-releasing hormone. Int. J. Oncol. 2008, 32, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Schally, A.V.; Cheng, J.C.; Zarandi, M.; Varga, J.; Leung, P.C. Growth hormone-releasing hormone antagonist induces apoptosis of human endometrial cancer cells through PKCδ-mediated activation of p53/p21. Cancer Lett. 2010, 298, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kovács, M.; Schally, A.V.; Varga, J.L.; Zarándi, M. Endocrine and antineoplastic actions of growth hormone-releasing hormone antagonists. Curr. Med. Chem. 2008, 15, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [PubMed]
- Øvstebø, R.; Haug, K.B.; Lande, K.; Kierulf, P. PCR-based calibration curves for studies of quantitative gene expression in human monocytes: Development and evaluation. Clin. Chem. 2003, 49, 425–432. [Google Scholar] [CrossRef] [PubMed][Green Version]



{kind=link}
{kind=link}
| Gene | Positive/Total Sample Size (Tumor) | % |
|---|---|---|
| GHRH-R | 0/39 | 0 |
| GHRH | 24/39 | 61.5 |
| SV1 | 9/39 | 23.0 |
| SV2 | 3/39 | 7.7 |
| SV3 | 0/39 | 0 |
| SV4 | 8/39 | 20.5 |
| Gene | Positive/Total Sample Size (Normal) | % |
| GHRH-R | 0/7 | 0 |
| GHRH | 3/7 | 42.9 |
| SV1 | 0/7 | 0 |
| SV2 | 0/7 | 0 |
| SV3 | 0/7 | 0 |
| SV4 | 0/7 | 0 |
| Patient No | Age at Diagnosis | Histology * | Grade | Stage | GHRH-R | GHRH | SV1 | SV2 | SV3 | SV4 |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 52 | E | 2 | I/b | − | + | + | + | − | − |
| 8 | 49 | P-S | 1 | I/b | − | + | + | − | − | − |
| 9 | 65 | E | 2 | I/a | − | + | − | − | − | + |
| 12 | 67 | E | 1 | I/a | − | + | + | + | − | + |
| 13 | 76 | E | 1 | I/b | − | + | − | − | − | + |
| 14 | 66 | E | 2 | I/b | − | + | − | − | − | + |
| 16 | 67 | P-S | 1 | I/b | − | + | + | − | − | + |
| 20 | 72 | E | 2 | I/b | − | + | − | − | − | + |
| 24 | 53 | P-S | 1 | II/b | − | + | + | − | − | − |
| 29 | 48 | E | 2 | III/c | − | + | + | + | − | + |
| 30 | 43 | P-S | 3 | III/c | − | + | − | − | − | + |
| 32 | 70 | E | 2 | II/a | − | + | + | − | − | − |
| 36 | 63 | E | 2 | III/c | − | + | + | − | − | − |
| 37 | 48 | P-S | 2 | II/a | − | + | + | − | − | − |
| Patient Number | mRNA for SV1 | Kd (nM) | Bmax (fmol/mg Protein) |
|---|---|---|---|
| 6 | + | 8.81 | 509.5 |
| 7 | − | − | − |
| 8 | + | 4.02 | 297.9 |
| 12 | + | 4.78 | 276.0 |
| 16 | + | 2.17 | 474.7 |
| 24 | + | 8.74 | 486.9 |
| 29 | + | 4.77 | 415.3 |
| 32 | + | 1.63 | 482.2 |
| 34 | − | − | − |
| 36 | + | 5.77 | 249.5 |
| 37 | + | 6.86 | 273.0 |
| FIGO Stage | Grade 1 | Grade 2 | Grade 3 | Total |
|---|---|---|---|---|
| IA | 0 | 4 | 1 | 5 |
| IB | 6 | 9 | 2 | 17 |
| IC | 0 | 3 | 1 | 4 |
| Total stage I | 6 | 16 | 4 | 26 |
| IIA | 0 | 3 | 0 | 3 |
| IIB | 2 | 2 | 1 | 5 |
| Total stage II | 2 | 5 | 1 | 8 |
| IIIA | 0 | 2 | 0 | 2 |
| IIIC | 0 | 2 | 1 | 3 |
| Total stage III | 0 | 4 | 1 | 5 |
| Total | 8 | 25 | 6 | 39 |
| Endometrioid subtype | 5 | 20 | 3 | 28 |
| papillary-serous subtype | 3 | 5 | 3 | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabo, Z.; Juhasz, E.; Schally, A.V.; Dezso, B.; Huga, S.; Hernadi, Z.; Halmos, G.; Kiss, C. Expression of Growth Hormone-Releasing Hormone and Its Receptor Splice Variants in Primary Human Endometrial Carcinomas: Novel Therapeutic Approaches. Molecules 2022, 27, 2671. https://doi.org/10.3390/molecules27092671
Szabo Z, Juhasz E, Schally AV, Dezso B, Huga S, Hernadi Z, Halmos G, Kiss C. Expression of Growth Hormone-Releasing Hormone and Its Receptor Splice Variants in Primary Human Endometrial Carcinomas: Novel Therapeutic Approaches. Molecules. 2022; 27(9):2671. https://doi.org/10.3390/molecules27092671
Chicago/Turabian StyleSzabo, Zsuzsanna, Eva Juhasz, Andrew V. Schally, Balazs Dezso, Sandor Huga, Zoltan Hernadi, Gabor Halmos, and Csongor Kiss. 2022. "Expression of Growth Hormone-Releasing Hormone and Its Receptor Splice Variants in Primary Human Endometrial Carcinomas: Novel Therapeutic Approaches" Molecules 27, no. 9: 2671. https://doi.org/10.3390/molecules27092671
APA StyleSzabo, Z., Juhasz, E., Schally, A. V., Dezso, B., Huga, S., Hernadi, Z., Halmos, G., & Kiss, C. (2022). Expression of Growth Hormone-Releasing Hormone and Its Receptor Splice Variants in Primary Human Endometrial Carcinomas: Novel Therapeutic Approaches. Molecules, 27(9), 2671. https://doi.org/10.3390/molecules27092671