
Linker Functionalization Strategy for Water Adsorption in Metal–Organic Frameworks

 , and
, and
Abstract
:1. Introduction
2. Methodology
2.1. Quantum Chemical Calculations
2.2. Monte Carlo Simulations
3. Results and Discussion
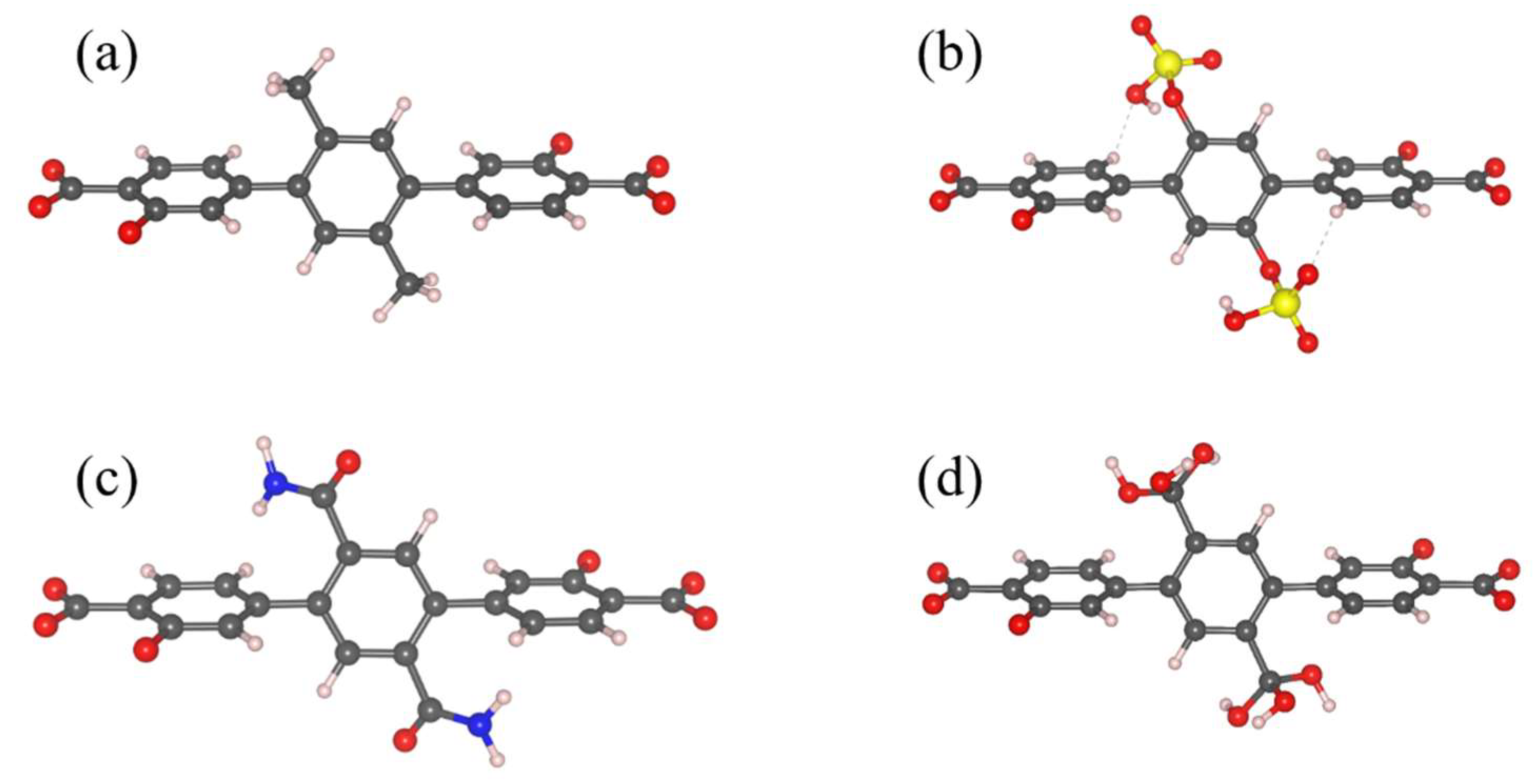
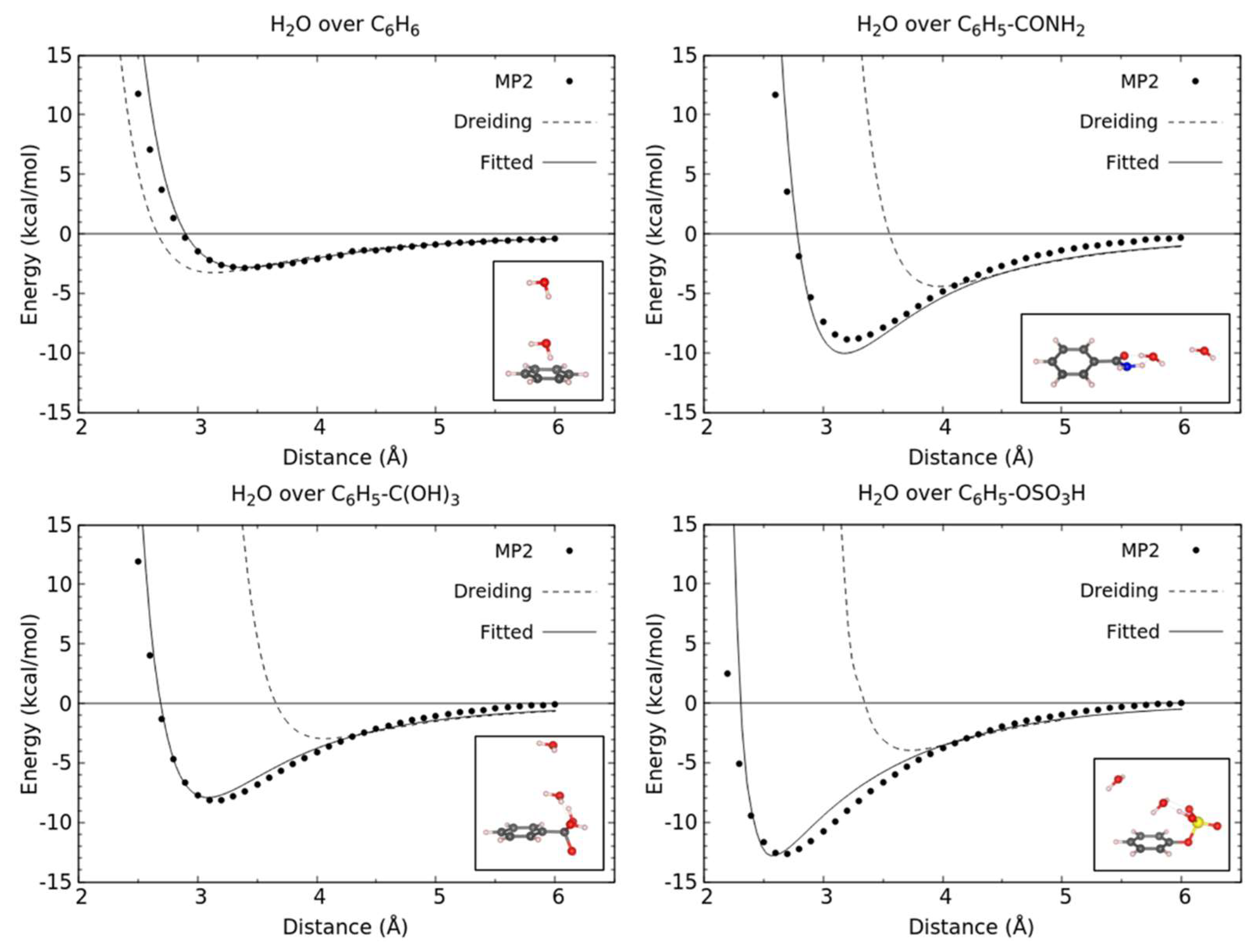
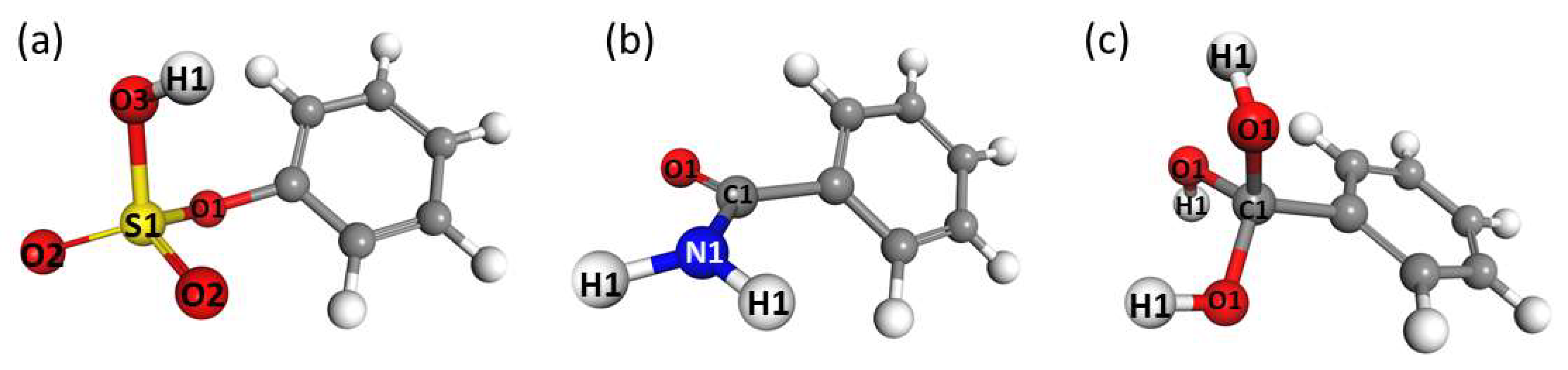
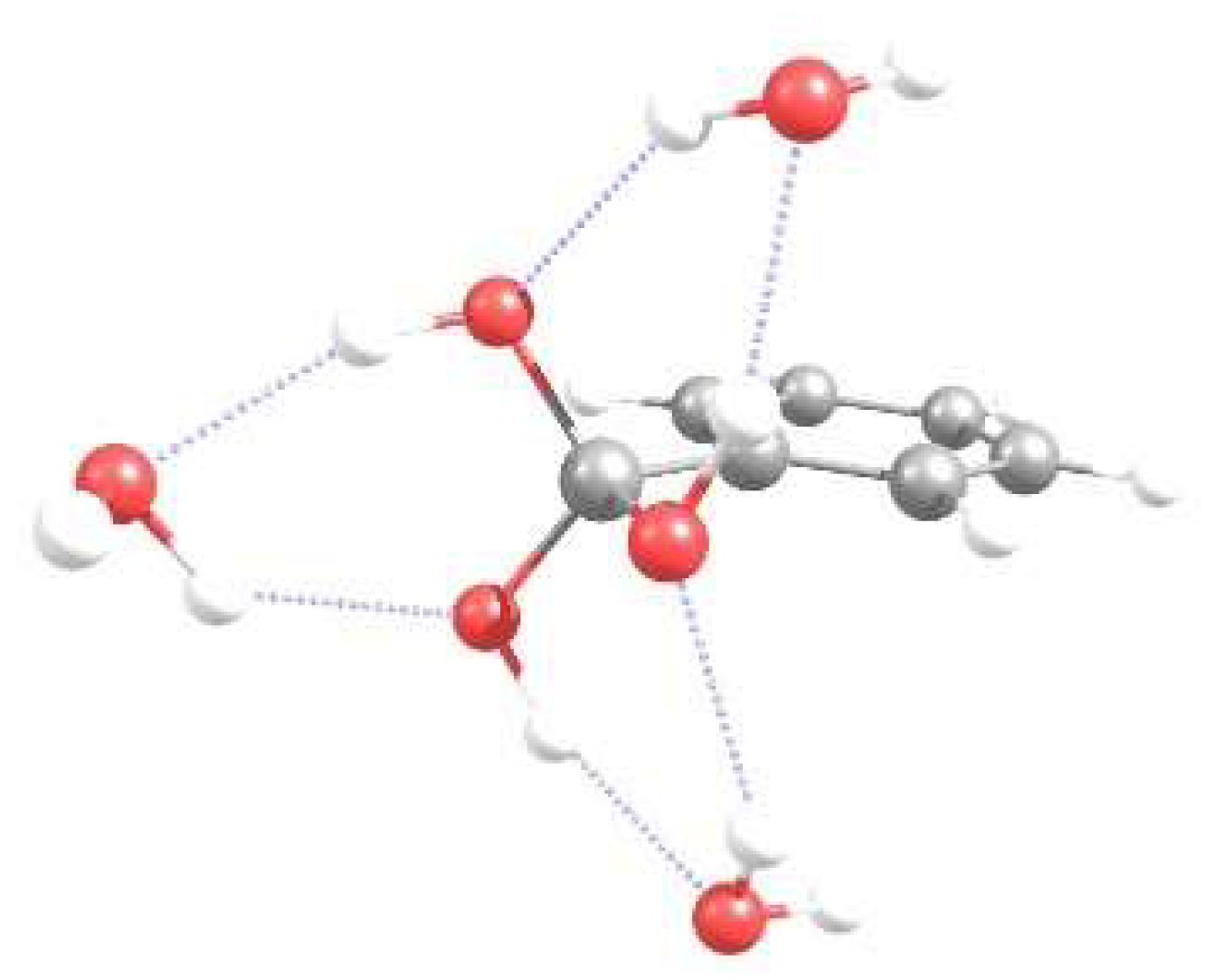
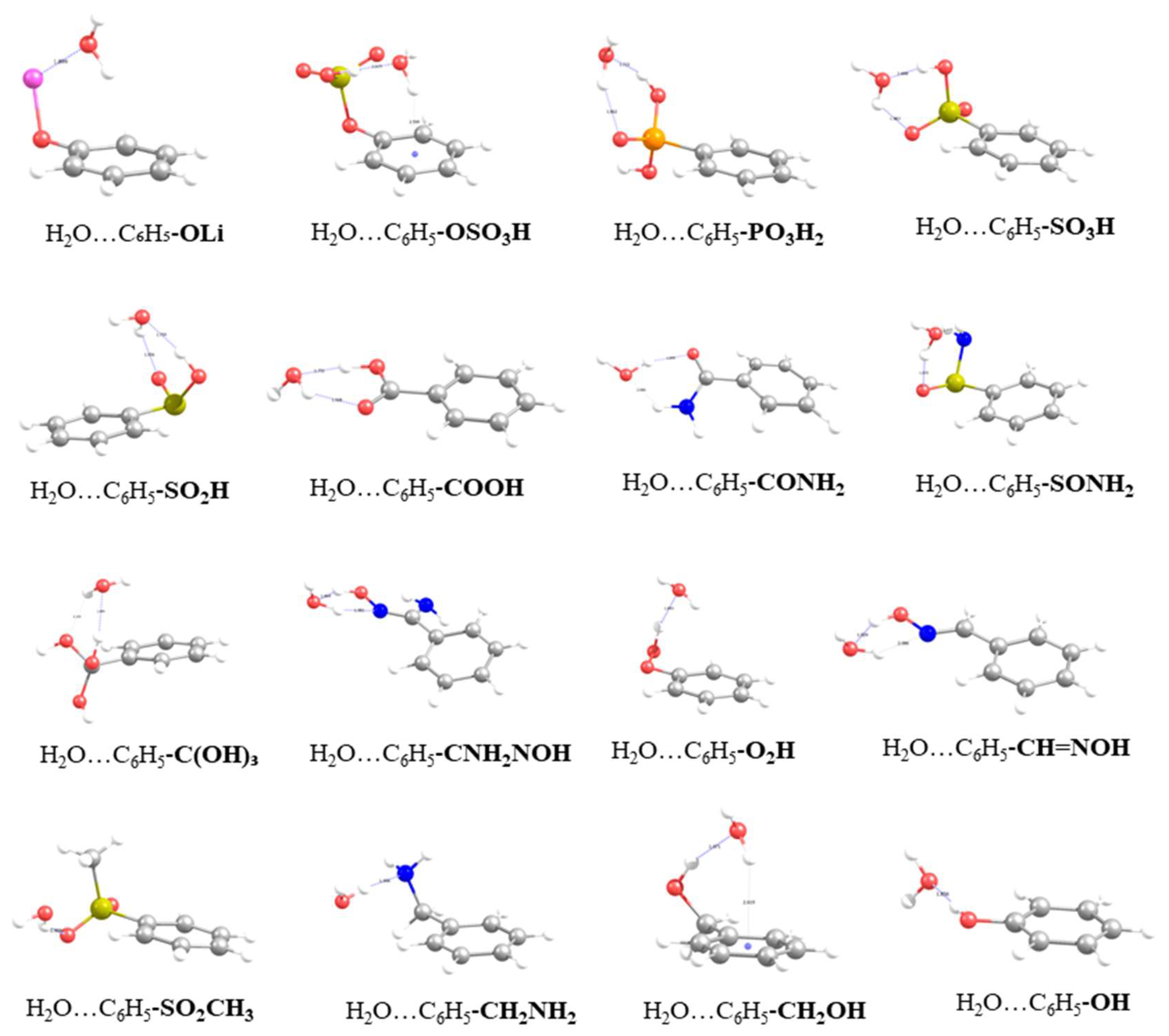
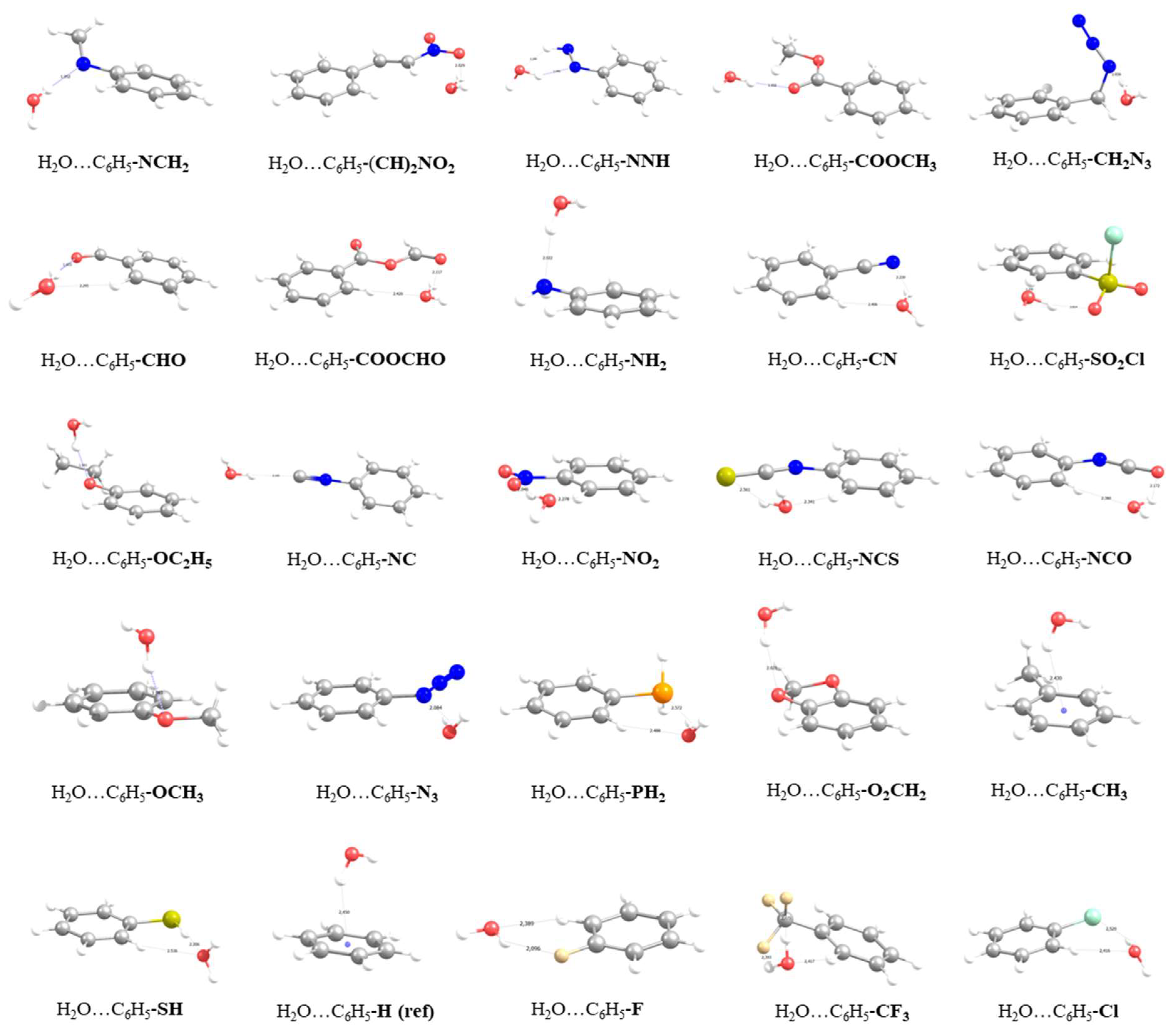
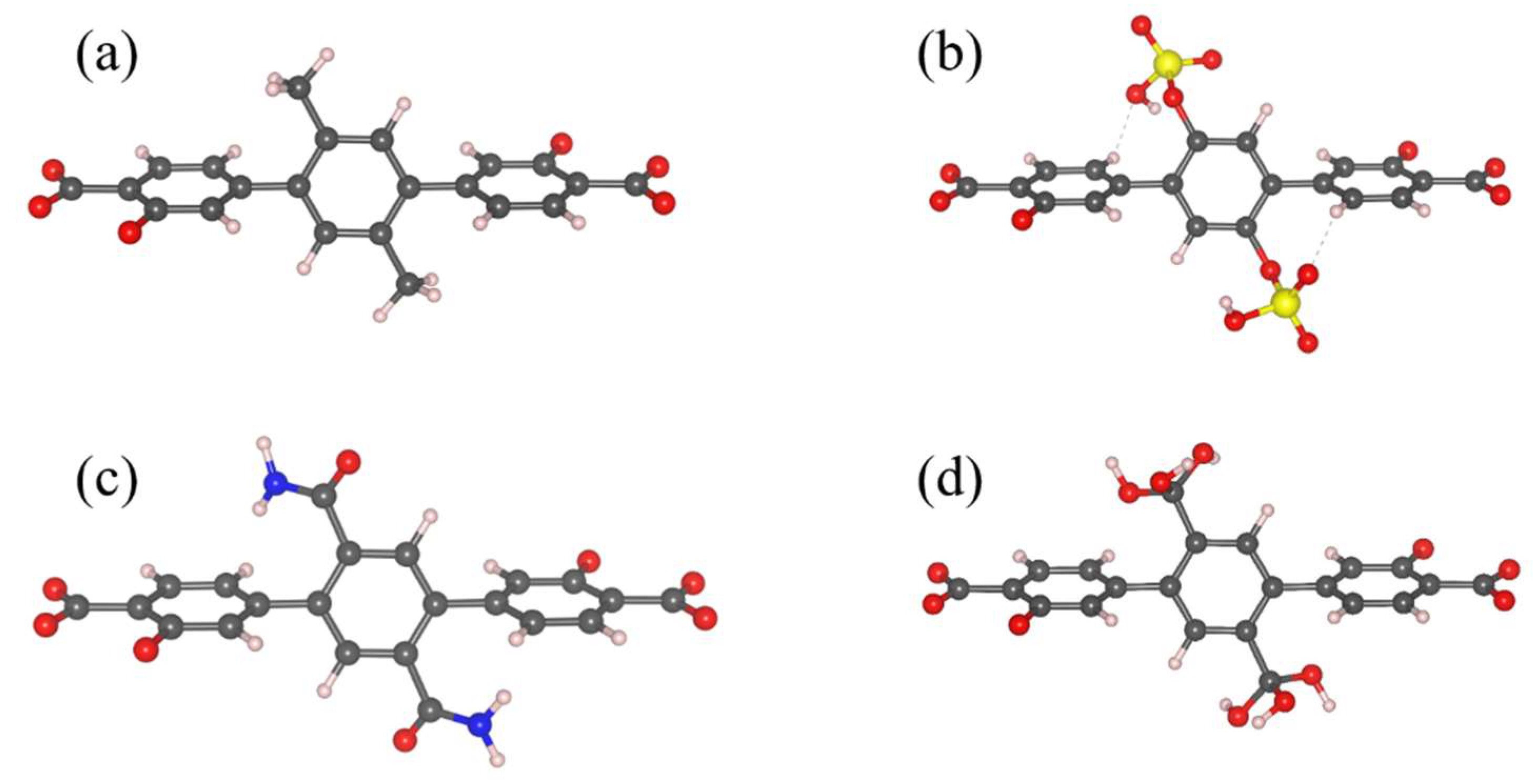
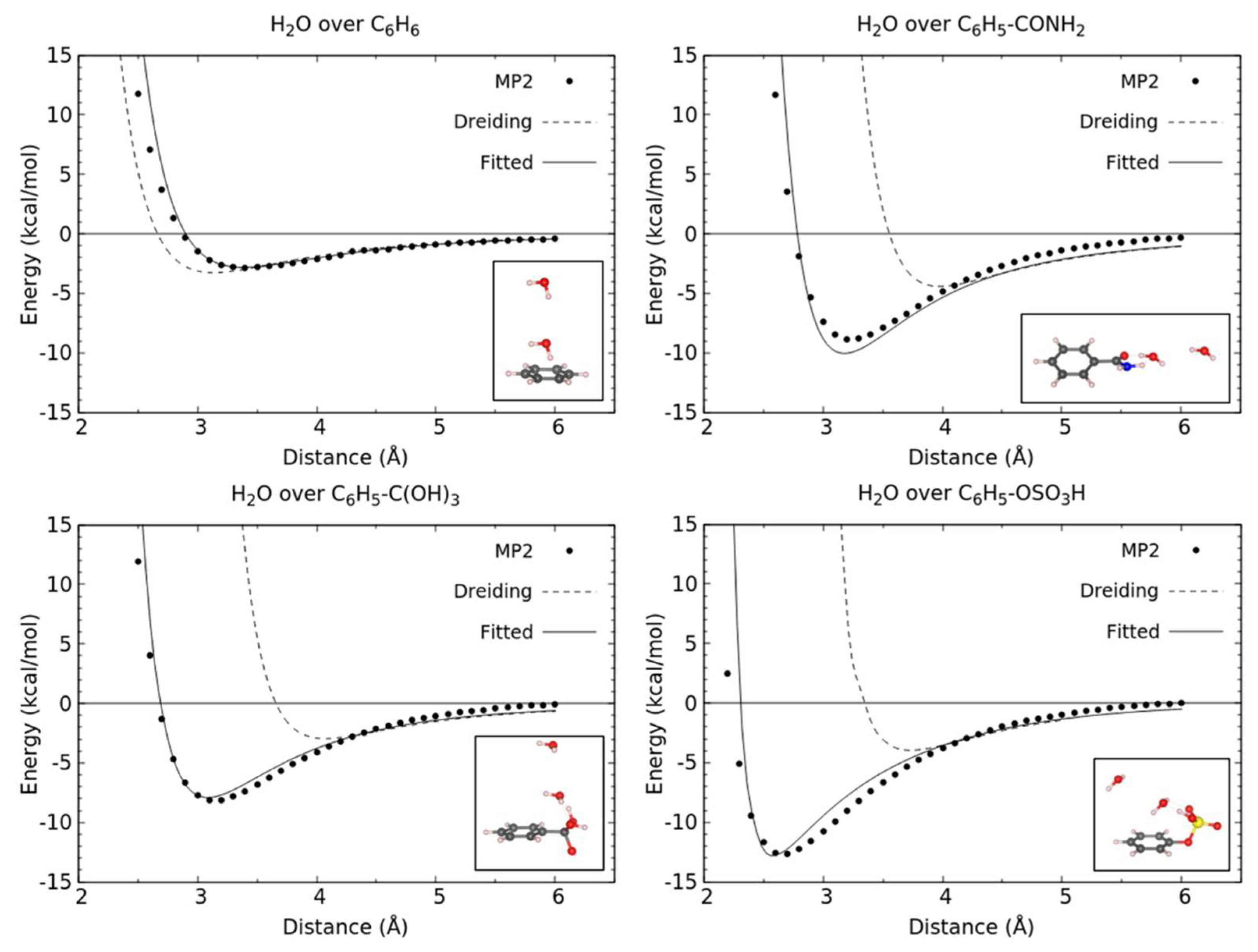
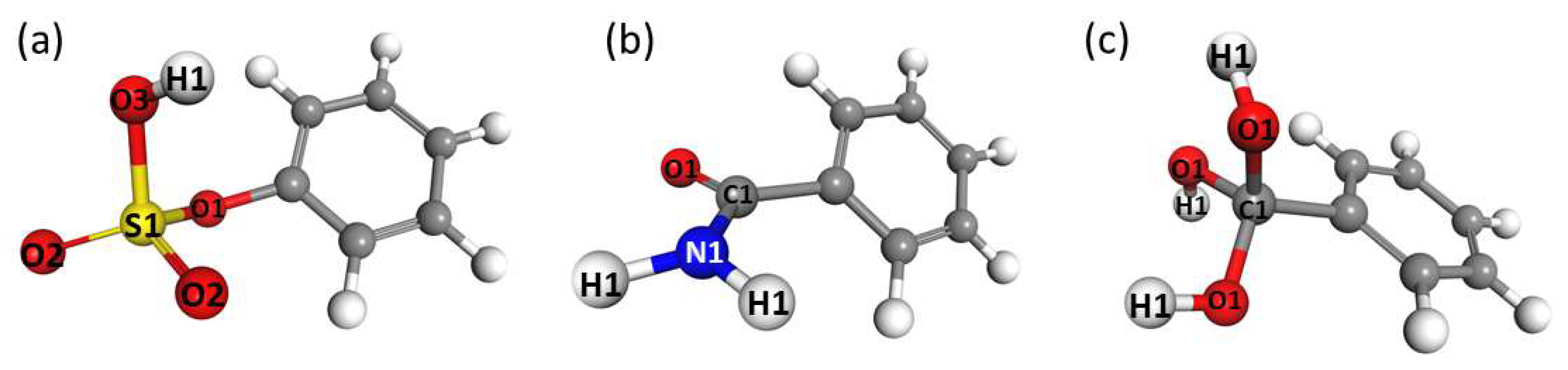
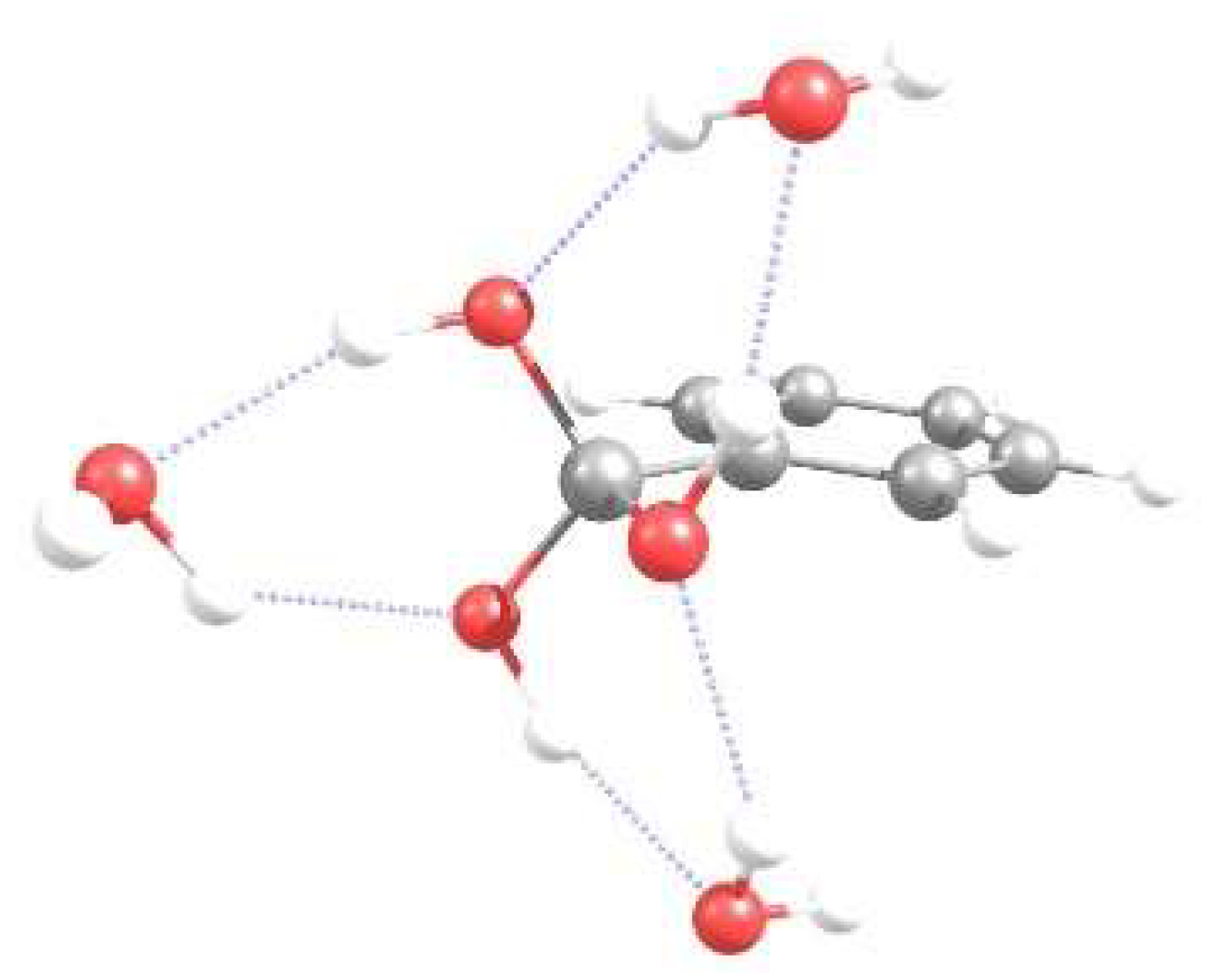
3.1. Energetics and Optimized Geometries of Monohydrated Functionalized Benzenes from ab initio Calculations
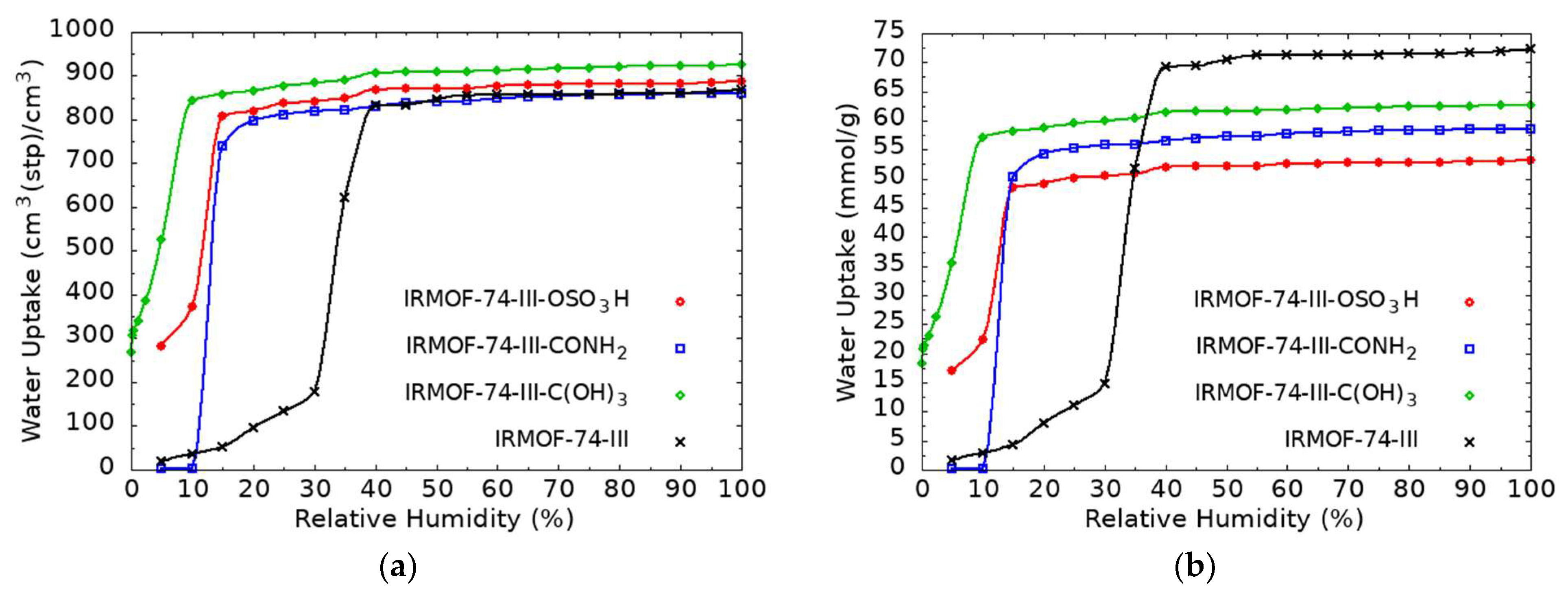
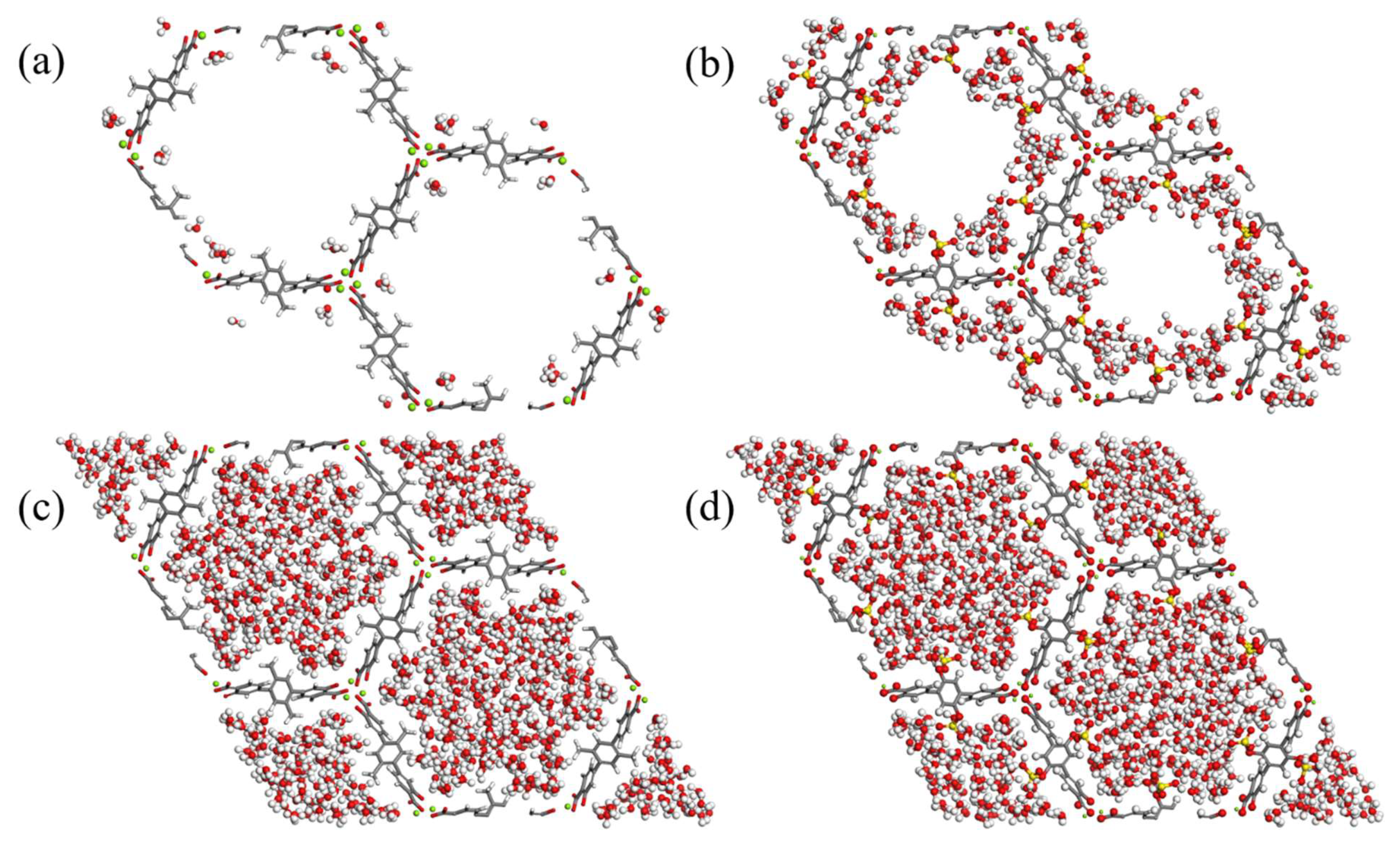
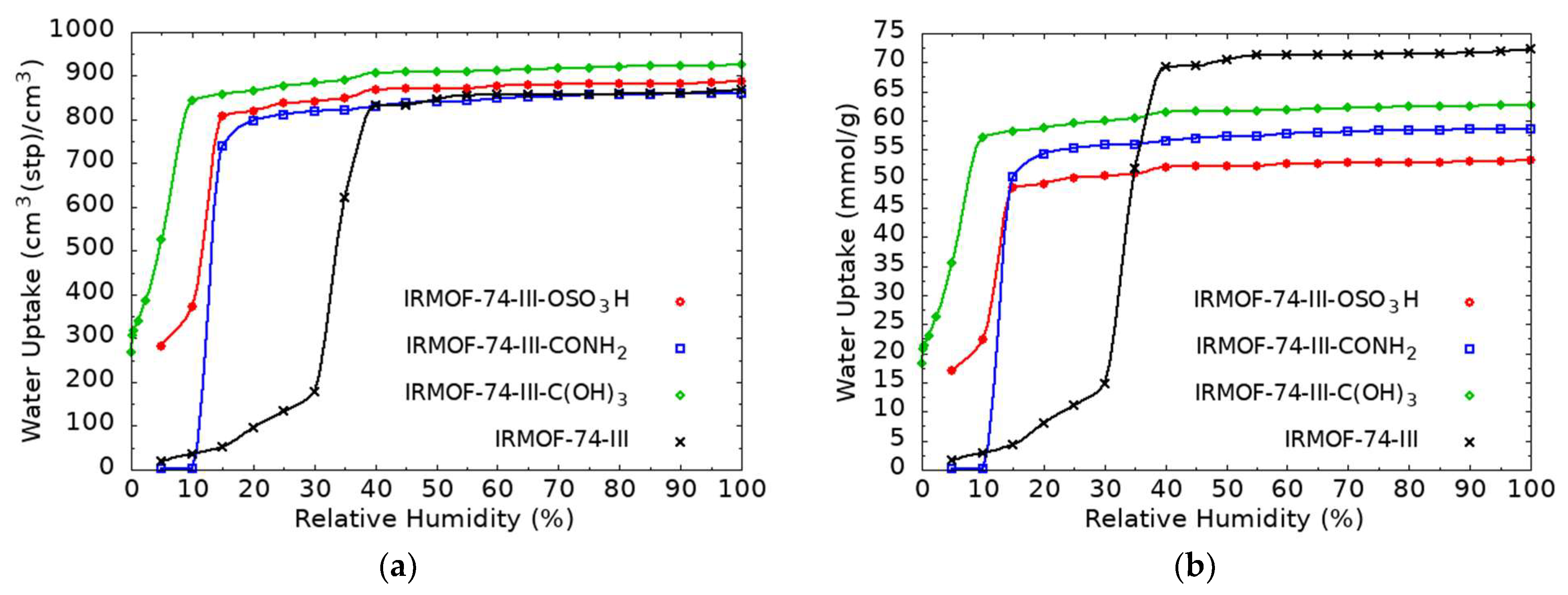
3.2. GCMC Simulations for H2O Uptake in Functionalized Mg-MOF-74-III
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Boretti, A.; Rosa, L. Reassessing the projections of the world water development report. NPJ Clean Water 2019, 2, 15. [Google Scholar] [CrossRef]
- United Nations World Water Development Report 2018: Nature-Based Solutions for Water, UN. 2018. Available online: https://www.unwater.org/world-water-development-report-2018-nature-based-solutions-for-water/ (accessed on 14 April 2022).
- Yang, K.; Pan, T.; Lei, Q.; Dong, X.; Cheng, Q.; Han, Y. A Roadmap to sorption-based atmospheric water harvesting: From molecular sorption mechanism to sorbent design and system optimization. Environ. Sci. Technol. 2021, 55, 6542–6560. [Google Scholar] [CrossRef] [PubMed]
- Rattner, A.S.; Garimella, S. Energy harvesting, reuse and upgrade to reduce primary energy usage in the USA. Energy 2011, 36, 6172–6183. [Google Scholar] [CrossRef]
- Wang, Y.; Levan, M.D. Adsorption equilibrium of carbon dioxide and water vapor on zeolites 5A and 13X and silica gel: Pure components. J. Chem. Eng. Data 2009, 54, 2839–2844. [Google Scholar] [CrossRef]
- Burtch, N.C.; Jasuja, H.; Walton, K.S. Water stability and adsorption in Metal-organic frameworks. Chem. Rev. 2014, 114, 10575–10612. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.S.; Shim, W.G.; Lee, J.W.; Kim, J.H.; Moon, H.; Seo, G. Adsorption equilibrium of water vapor on mesoporous materials. J. Chem. Eng. Data 2003, 48, 1458–1462. [Google Scholar] [CrossRef]
- Tu, Y.D.; Wang, R.Z.; Zhang, Y.N.; Wang, J.Y. Progress and expectation of atmospheric water harvesting. Joule 2018, 2, 1452–1475. [Google Scholar] [CrossRef] [Green Version]
- Yaghi, O.M.; Kalmutzki, M.J.; Diercks, C.S. Introduction to Reticular Chemistry: Metal-Organic Frameworks and Covalent Organic Frameworks; Wiley VCH: Weinheim, Germany, 2019. [Google Scholar]
- Yaghi, O.M. Reticular chemistry: Molecular precision in infinite 2D and 3D. Mol. Front. J. 2019, 03, 66–83. [Google Scholar] [CrossRef]
- Freund, R.; Zaremba, O.; Arnauts, G.; Ameloot, R.; Skorupskii, G.; Dincă, M.; Bavykina, A.; Gascon, J.; Ejsmont, A.; Gościańska, J.; et al. The current status of MOF and COF applications. Angew. Chem. Int. Ed. 2021, 60, 23975–24001. [Google Scholar] [CrossRef]
- Wang, C.; Liu, X.; Keser Demir, N.; Chen, J.P.; Li, K. Applications of water stable metal−organic frameworks. Chem. Soc. Rev. 2016, 45, 5107–5134. [Google Scholar] [CrossRef]
- Kalmutzki, M.J.; Diercks, C.S.; Yaghi, O.M. Metal-Organic Frameworks for water harvesting from air. Adv. Mater. 2018, 30, 1704304. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, K.; Sun, Y.; Lollar, C.T.; Li, J.; Zhou, H.C. Recent advances in gas storage and separation using metal–organic frameworks. Mater. Today 2018, 21, 108–121. [Google Scholar] [CrossRef]
- Qian, Q.; Asinger, P.A.; Lee, M.J.; Han, G.; Mizrahi Rodriguez, K.; Lin, S.; Benedetti, F.M.; Wu, A.X.; Chi, W.S.; Smith, Z.P. MOF-based membranes for gas separations. Chem. Rev. 2020, 120, 8161–8266. [Google Scholar] [CrossRef]
- Zhang, L.-T.; Zhou, Y.; Han, S.-T. The role of metal–organic frameworks in electronic sensors. Angew. Chem. Int. Ed. 2020, 60, 15192–15212. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, A.; Li, Z.; Garcia, H. Catalysis and photocatalysis by metal organic frameworks. Chem. Soc. Rev. 2018, 47, 8134–8172. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.V.; Dong, H.C.; Nguyen-Manh, D.; Vu, N.H.; Trinh, T.T.; Phan, T.B. Effect of hydrogen-bonding networks in water on the proton conductivity properties of metal–organic frameworks. J. Sci. Adv. Mater. Devices 2021, 6, 509–515. [Google Scholar] [CrossRef]
- Gordeeva, L.G.; Solovyeva, M.V.; Sapienza, A.; Aristov, Y.I. Potable water extraction from the atmosphere: Potential of MOFs. Renew. Energy 2020, 148, 72–80. [Google Scholar] [CrossRef]
- de Lange, M.F.; Karlijn, V.J.F.M.; Vlugt, T.J.H.; Gascon, J.; Kapteijn, F. Adsorption- driven heat pumps: The potential of metal–organic frameworks. Chem. Rev. 2015, 115, 12205–12250. [Google Scholar] [CrossRef]
- Borges, D.D.; Maurin, G.; Galvão, D.S. Design of porous metal- organic frameworks for adsorption driven thermal batteries. MRS Adv. 2017, 2, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Thu, K.; Ng, K.C.; Saha, B.B.; Chakraborty, A.; Koyama, S. Operational strategy of adsorption desalination systems. Int. J. Heat Mass Transf. 2009, 52, 1811–1816. [Google Scholar] [CrossRef]
- Zu, K.; Qin, M.; Cui, S. Progress and potential of metal-organic frameworks (MOFs) as novel desiccants for built environment control: A review. Renew. Sustain. Energ. Rev. 2020, 133, 110246. [Google Scholar] [CrossRef]
- Liu, X.; Wang, X.; Kapteijn, F. Water and metal-organic frameworks: From interaction towards utilization. Chem. Rev. 2020, 120, 8303–8377. [Google Scholar] [CrossRef] [PubMed]
- Reinsch, H.; van der Veen, M.A.; Gil, B.; Marszalek, B.; Verbiest, T.; de Vos, D.; Stock, N. Structures, sorption characteristics, and nonlinear optical properties of a new series of highly stable aluminum MOFs. Chem. Mater. 2013, 25, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Solovyeva, M.V.; Shkatulov, A.I.; Gordeeva, L.G.; Fedorova, E.A.; Krieger, T.A.; Aristov, Y.I. Water vapor adsorption on CAU-10-X: Effect of functional groups on adsorption equilibrium and mechanisms. Langmuir 2021, 37, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, H.; Wang, X.; Li, F.; Gong, Y.; Zhong, C.; Li, J.-R. Proton conductivities in functionalized UiO-66: Tuned properties, thermogravimetry mass, and molecular simulation analyses. Cryst. Growth Des. 2015, 15, 5827–5833. [Google Scholar] [CrossRef]
- Ko, N.; Choi, P.G.; Hong, J.; Yeo, M.; Sung, S.; Cordova, K.E.; Park, H.J.; Yang, J.K.; Kim, J. Tailoring the water adsorption properties of MIL-101 metal-organic frameworks by partial functionalization. J. Mater. Chem. A 2015, 3, 2057–2064. [Google Scholar] [CrossRef]
- Shi, W.; Zhu, Y.; Shen, C.; Shi, J.; Xu, G.; Xiao, X.; Cao, R. water sorption properties of functionalized MIL-101 (Cr)-X (X=-NH2, -SO3H, H, -CH3, -F) based composites as thermochemical heat storage materials. Micropor. Mesopor. Mater. 2019, 285, 129–136. [Google Scholar] [CrossRef]
- Akiyama, G.; Matsuda, R.; Sato, H.; Hori, A.; Takata, M.; Kitagawa, S. Effect of functional groups in MIL-101 on water sorption behavior. Micropor. Mesopor. Mater. 2012, 157, 89–93. [Google Scholar] [CrossRef]
- Fan, W.; Zhang, X.; Kang, Z.; Liu, X.; Sun, D. Isoreticular chemistry within metal-organic frameworks for gas storage and separation. Coord. Chem. Rev. 2021, 440, 213968. [Google Scholar] [CrossRef]
- Giappa, R.M.; Tylianakis, E.; Di Gennaro, M.; Gkagkas, K.; Froudakis, G.E. A combination of multi-scale calculations with machine learning for investigating hydrogen storage in metal organic frameworks. Int. J. Hydrogen Energy 2021, 46, 27612–27621. [Google Scholar] [CrossRef]
- Frysali, M.G.; Klontzas, E.; Tylianakis, E.; Froudakis, G.E. Tuning the interaction strength and the adsorption of CO2 in metal organic frameworks by functionalization of the organic linkers. Micropor. Mesopor. Mater. 2016, 227, 144–151. [Google Scholar] [CrossRef]
- Frysali, M.G.; Klontzas, E.; Froudakis, G.E. Ab initio study of the adsorption of CO2 on functionalized benzenes. Chem. Phys. Chem. 2014, 15, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.G.H.; Cordova, K.E.; Valente, C.; Furukawa, H.; Hmadeh, M.; Gándara, F.; Whalley, A.C.; Liu, Z.; Asahina, S.; Kazumori, H.; et al. Large-pore apertures in a series of metal-organic frameworks. Science 2012, 336, 1018–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fracaroli, A.M.; Furukawa, H.; Suzuki, M.; Dodd, M.; Okajima, S.; Gándara, F.; Reimer, J.A.; Yaghi, O.M. Metal–organic frameworks with precisely designed interior for carbon dioxide capture in the presence of water. J. Am. Chem. Soc. 2014, 136, 8863–8866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fracaroli, A.M.; Siman, P.; Nagib, D.A.; Suzuki, M.; Furukawa, H.; Toste, F.D.; Yaghi, O.M. Seven post-synthetic covalent reactions in tandem leading to enzyme-like complexity within metal–organic framework crystals. J. Am. Chem. Soc. 2016, 138, 8352–8355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geary, J.; Wong, A.H.; Xiao, D.J. Thermolabile cross-linkers for templating precise multicomponent metal-organic framework pores. J. Am. Chem. Soc. 2021, 143, 10317–10323. [Google Scholar] [CrossRef]
- Rieth, A.J.; Yang, S.; Wang, E.N.; Dinca, M. Record atmospheric fresh water capture and heat transfer with a material operating at the water uptake reversibility limit. ACS Cent. Sci. 2017, 3, 668–672. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmer, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- TURBOMOLE V7.3 2018, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 14 April 2022).
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 18 April 2022).
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Adams, D.J. On the use of the Ewald summation in computer simulation. J. Chem. Phys. 1983, 78, 2585–2590. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dubbeldam, D.; Calero, S.; Ellis, D.E.; Snurr, R.Q. RASPA: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simulat. 2016, 42, 81–101. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Group | B.E. (Kcal/mol) | % Enhancement | |
|---|---|---|---|
| 1 | -OLi | −18.7 | 545% |
| 2 | -OSO3H | −12.7 | 338% |
| 3 | -PO3H2 | −11.8 | 307% |
| 4 | -SO3H | −11.0 | 279% |
| 5 | -SO2H | −10.1 | 248% |
| 6 | -COOH | −9.4 | 224% |
| 7 | -CONH2 | −8.9 | 207% |
| 8 | -SONH2 | −8.3 | 186% |
| 9 | -C(OH)3 | −8.2 | 182% |
| 10 | -CNH2NOH | −8.1 | 179% |
| 11 | -O2H | −7.4 | 155% |
| 12 | -CHNOH | −7.1 | 145% |
| 13 | -SO2CH3 | −6.9 | 138% |
| 14 | -CH2NH2 | −6.9 | 138% |
| 15 | -CH2OH | −6.5 | 124% |
| 16 | -OH | −6.4 | 121% |
| 17 | -NCH2 | −5.9 | 103% |
| 18 | -(CH)2NO2 | −5.7 | 97% |
| 19 | -N=NH | −5.6 | 93% |
| 20 | -COOCH3 | −5.5 | 90% |
| 21 | -CH2N3 | −5.4 | 86% |
| 22 | -CHO | −5.4 | 86% |
| 23 | -COOCHO | −5.4 | 86% |
| 24 | -NH2 | −5.1 | 76% |
| 25 | -C≡N | −4.9 | 69% |
| 26 | -SO2Cl | −4.9 | 69% |
| 27 | -OC2H5 | −4.4 | 52% |
| 28 | -N≡C | −4.4 | 52% |
| 29 | -NO2 | −4.1 | 41% |
| 30 | -N=C=S | −4.1 | 41% |
| 31 | -N=C=O | −4.0 | 38% |
| 32 | -OCH3 | −3.9 | 53% |
| 33 | -N3 | −3.7 | 28% |
| 34 | -PH2 | −3.5 | 21% |
| 35 | -O2CH2 | −3.5 | 21% |
| 36 | -CH3 | −3.4 | 17% |
| 37 | -SH | −3.4 | 17% |
| 38 | -H | −2.9 | 0% (ref) |
| 39 | -F | −2.9 | 0% |
| 40 | -CF3 | −2.7 | −7% |
| 41 | -Cl | −2.6 | −10% |
| Functional Group | B.E., Kcal/mol | OH···Ow, Å | OHw···O(N), Å | Ow-H-O(N), ° | Ow-Hw-O(N), ° | Δr OH, Å | H-Ow (1), Å | H-Ow (2), Å | Hw-COM ring, Å | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | -OLi | −18.7 | 0.957 | 0.978 | 2.689 | |||||
| 2 | -OSO3H | −12.7 | 1.619 | 2.295 | 169.453 | 122.289 | 0.03849 | 0.965 | 0.964 | 2.509 |
| 3 | -PO3H2 | −11.8 | 1.733 | 1.862 | 156.068 | 146.685 | 0.02433 | 0.976 | 0.958 | |
| 4 | -SO3H | −11.0 | 1.688 | 1.983 | 162.974 | 133.704 | 0.02661 | 0.969 | 0.959 | |
| 5 | -SO2H | −10.1 | 1.759 | 1.956 | 153.22 | 142.999 | 0.02132 | 0.973 | 0.96 | |
| 6 | -COOH | −9.4 | 1.751 | 1.928 | 158.802 | 140.045 | 0.01921 | 0.972 | 0.959 | |
| 7 | -CONH2 | −8.9 | 2.006 | 1.849 | 140.504 | 153.63 | 0.00605 | 0.974 | 0.958 | |
| 8 | -SONH2 | −8.3 | 2.075 | 1.87 | 140.482 | 151.84 | 0.00421 | 0.973 | 0.958 | |
| 9 | -C(OH)3 | −8.2 | 1.899 | 2.104 | 151.809 | 104.017 | 0.012 | 0.966 | 0.96 | |
| 10 | -CNH2NOH | −8.1 | 1.964 | 1.981 | 140.17 | 140.17 | 0.00933 | 0.971 | 0.958 | |
| 11 | -O2H | −7.4 | 1.843 | 157.52 | 0.01273 | 0.963 | 0.96 | |||
| 12 | -CH=NOH | −7.1 | 1.924 | 2.086 | 143.1 | 128.61 | 0.01028 | 0.967 | 0.958 | |
| 13 | -SO2CH3 | −6.9 | 1.906 | 153.78 | 0.968 | 0.958 | ||||
| 14 | -CH2NH2 | −6.9 | 1.906 | 164.72 | 0.976 | 0.958 | ||||
| 15 | -CH2OH | −6.5 | 1.971 | 158.707 | 0.00639 | 0.963 | 0.959 | 2.619 | ||
| 16 | -OH | −6.4 | 1.856 | 175.49 | 0.00917 | 0.959 | 0.959 | |||
| 17 | -NCH2 | −5.9 | 1.952 | 167.84 | 0.971 | 0.958 | ||||
| 18 | -(CH)2NO2 | −5.7 | 2.029 | 151.56 | 0.965 | 0.958 | ||||
| 19 | -N=NH | −5.6 | 2.299 | 1.995 | 127.693 | 141.95 | 0.0005 | 0.969 | 0.958 | |
| 20 | -COOCH3 | −5.5 | 1.932 | 169.59 | 0.966 | 0.957 | ||||
| 21 | -CH2N3 | −5.4 | 2.036 | 147.784 | 0.966 | 0.958 | ||||
| 22 | -CHO | −5.4 | 1.933 | 166.16 | 0.966 | 0.957 | ||||
| 23 | -COOCHO | −5.4 | 2.117 | 167.73 | 0.964 | 0.958 | ||||
| 24 | -NH2 | −5.1 | 2.022 | 165.24 | 0.967 | 0.958 | ||||
| 25 | -C≡N | −4.9 | 2.23 | 148.7 | 0.963 | 0.958 | ||||
| 26 | -SO2Cl | −4.9 | 2.014 | 0.963 | 0.958 | |||||
| 27 | -OC2H5 | −4.4 | 1.941 | 162.81 | 0.965 | 0.958 | ||||
| 28 | -N≡C | −4.4 | 2.165 | 178.45 | 0.965 | 0.957 | ||||
| 29 | -NO2 | −4.1 | 2.046 | 156.07 | 0.962 | 0.958 | ||||
| 30 | -N=C=S | −4.1 | 2.561 | 159.05 | 0.963 | 0.959 | ||||
| 31 | -N=C=O | −4.0 | 138.39 | 0.962 | 0.958 | |||||
| 32 | -OCH3 | −3.9 | 1.983 | 162.152 | 0.964 | 0.958 | ||||
| 33 | -N3 | −3.7 | 2.084 | 158.62 | 0.964 | 0.958 | ||||
| 34 | -PH2 | −3.5 | 2.572 | 151.67 | 0.964 | 0.958 | ||||
| 35 | -O2CH2 | −3.5 | 2.021 | 153.58 | 0.963 | 0.958 | ||||
| 36 | -CH3 | −3.4 | 0.962 | 0.959 | 2.43 | |||||
| 37 | -SH | −3.4 | 2.206 | 172.98 | 0.0014 | 0.959 | 0.959 | |||
| 38 | -H | −2.9 | 0.961 | 0.958 | 2.45 | |||||
| 39 | -F | −2.9 | 2.096 | 148.47 | 0.96 | 0.958 | ||||
| 40 | -CF3 | −2.7 | 0.96 | 0.959 | ||||||
| 41 | -Cl | −2.6 | 2.416 | 2.529 | 0.961 | 0.958 |
| -CH3 (Parent Mg-MOF-74-III) | -OSO3H | -CONH2 | -C(OH)3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ε (Κ) | σ (Å) | ε (Κ) | σ (Å) | ε (Κ) | σ (Å) | ε (Κ) | σ (Å) | ||||
| C-Ow | 61.09 | 3.51 | O1-Ow | 61.29 | 3.09 | C1-Ow | 61.09 | 3.51 | C1-Ow | 47.85 | 3.87 |
| O-Ow | 61.29 | 3.09 | O2-Ow | 61.29 | 2.69 | O1-Ow | 61.29 | 3.09 | O1-Ow | 48.16 | 2.22 |
| H-Ow | 66.01 | 3.13 | O3-Ow | 61.29 | 2.69 | N1-Ow | 55.12 | 3.21 | H1-Ow | 7.65 | 0.65 |
| Mg-Ow | 24.43 | 3.20 | S1-Ow | 116.2 | 3.27 | H1-Ow | 24.43 | 2.40 | |||
| H1-Ow | 24.43 | 1.92 | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giappa, R.M.; Papadopoulos, A.G.; Klontzas, E.; Tylianakis, E.; Froudakis, G.E. Linker Functionalization Strategy for Water Adsorption in Metal–Organic Frameworks. Molecules 2022, 27, 2614. https://doi.org/10.3390/molecules27092614
Giappa RM, Papadopoulos AG, Klontzas E, Tylianakis E, Froudakis GE. Linker Functionalization Strategy for Water Adsorption in Metal–Organic Frameworks. Molecules. 2022; 27(9):2614. https://doi.org/10.3390/molecules27092614
Chicago/Turabian StyleGiappa, Rafaela Maria, Anastasios G. Papadopoulos, Emmanuel Klontzas, Emmanuel Tylianakis, and George E. Froudakis. 2022. "Linker Functionalization Strategy for Water Adsorption in Metal–Organic Frameworks" Molecules 27, no. 9: 2614. https://doi.org/10.3390/molecules27092614
APA StyleGiappa, R. M., Papadopoulos, A. G., Klontzas, E., Tylianakis, E., & Froudakis, G. E. (2022). Linker Functionalization Strategy for Water Adsorption in Metal–Organic Frameworks. Molecules, 27(9), 2614. https://doi.org/10.3390/molecules27092614