
Surface Modification Strategy for Enhanced NO2 Capture in Metal–Organic Frameworks

 ,
,
Abstract
:1. Introduction
2. Computational Methods
2.1. Density Functional Theory
2.2. Grand Canonical Monte Carlo
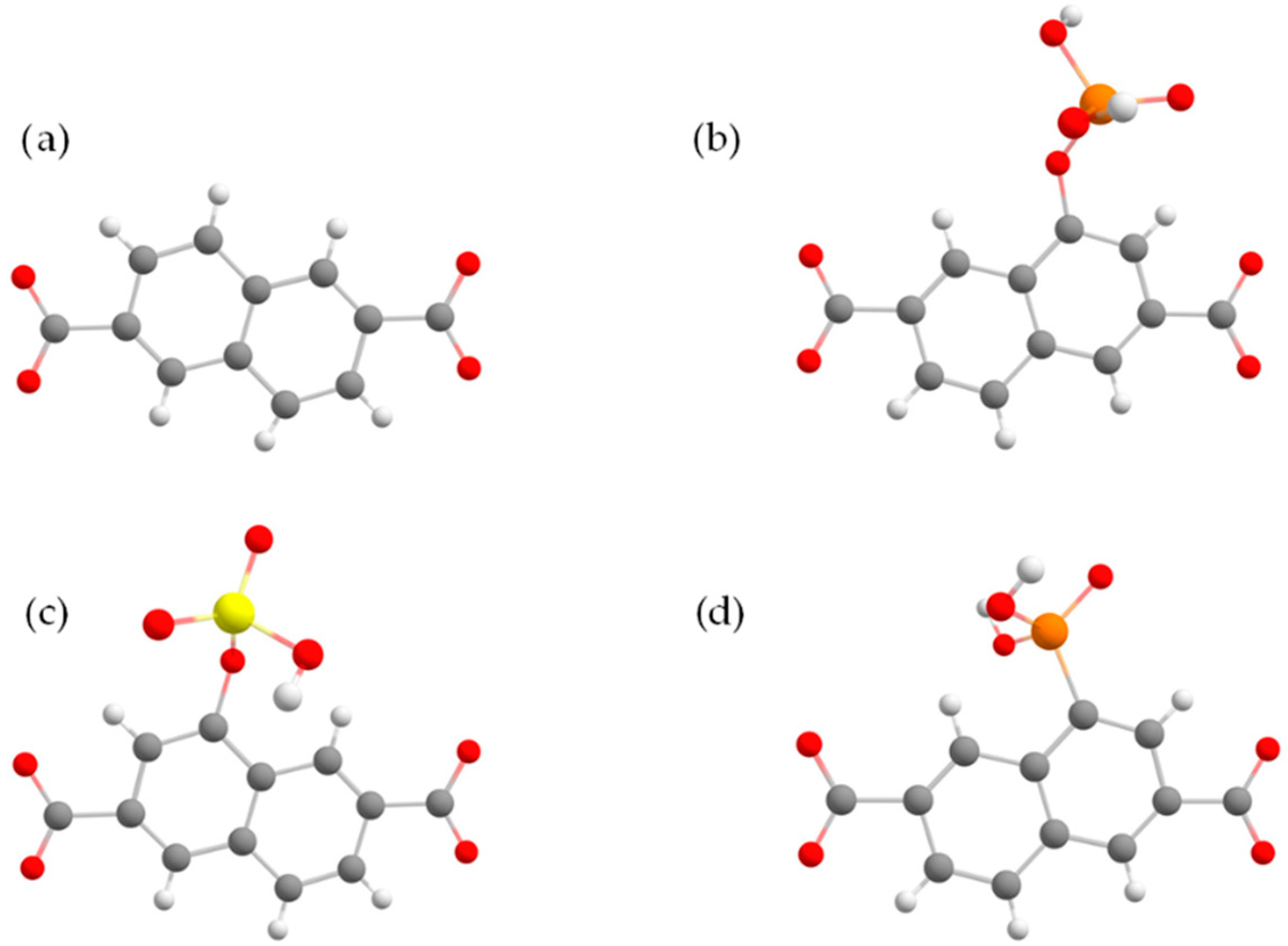
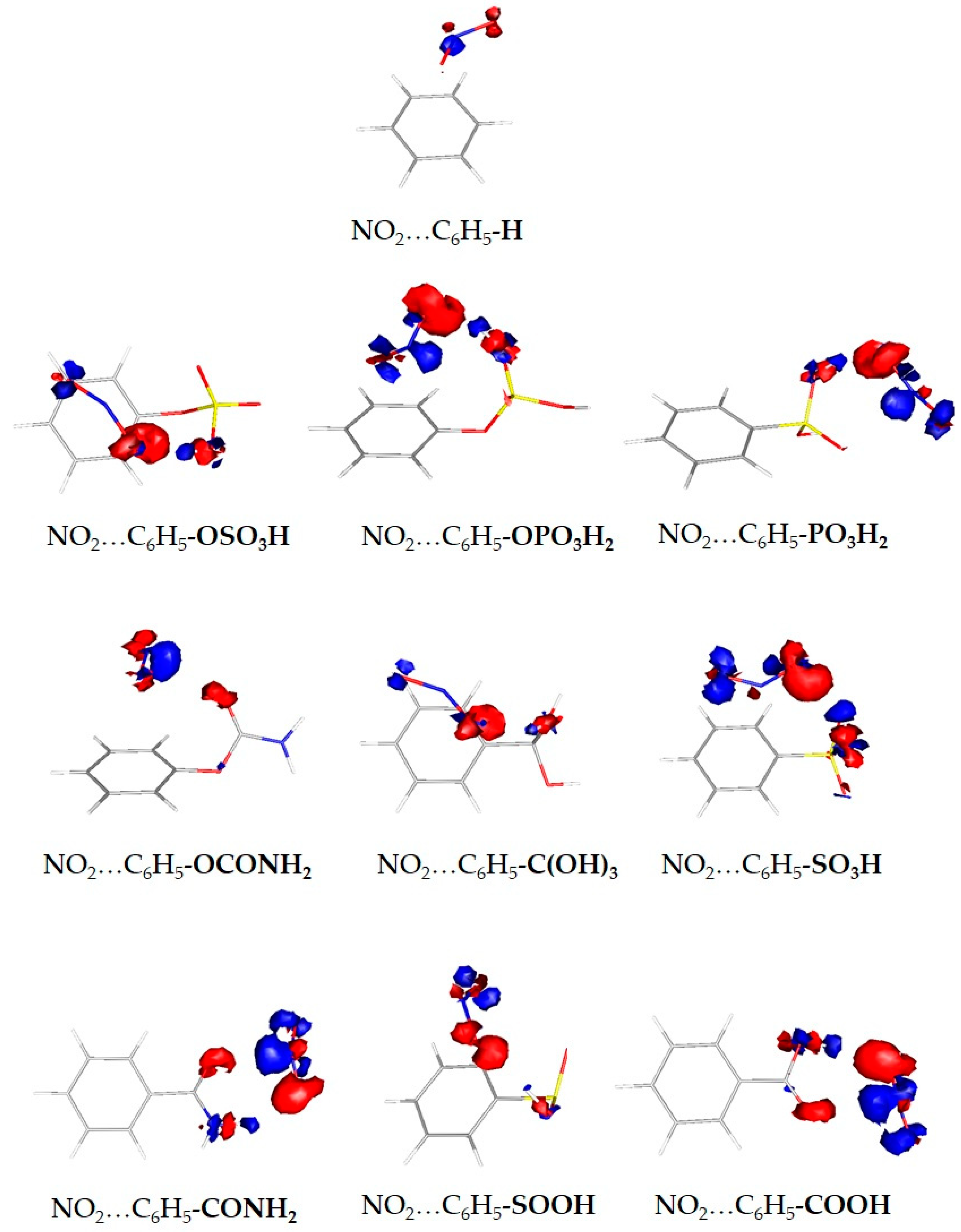
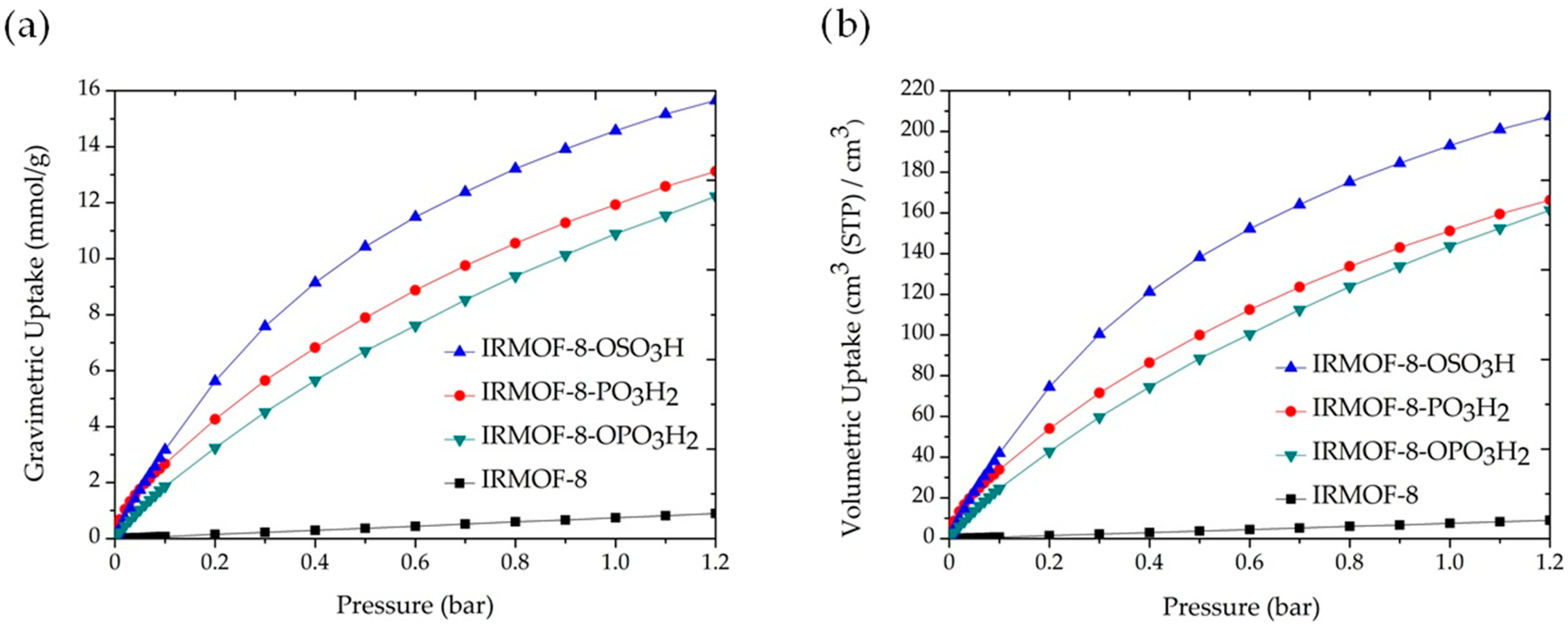
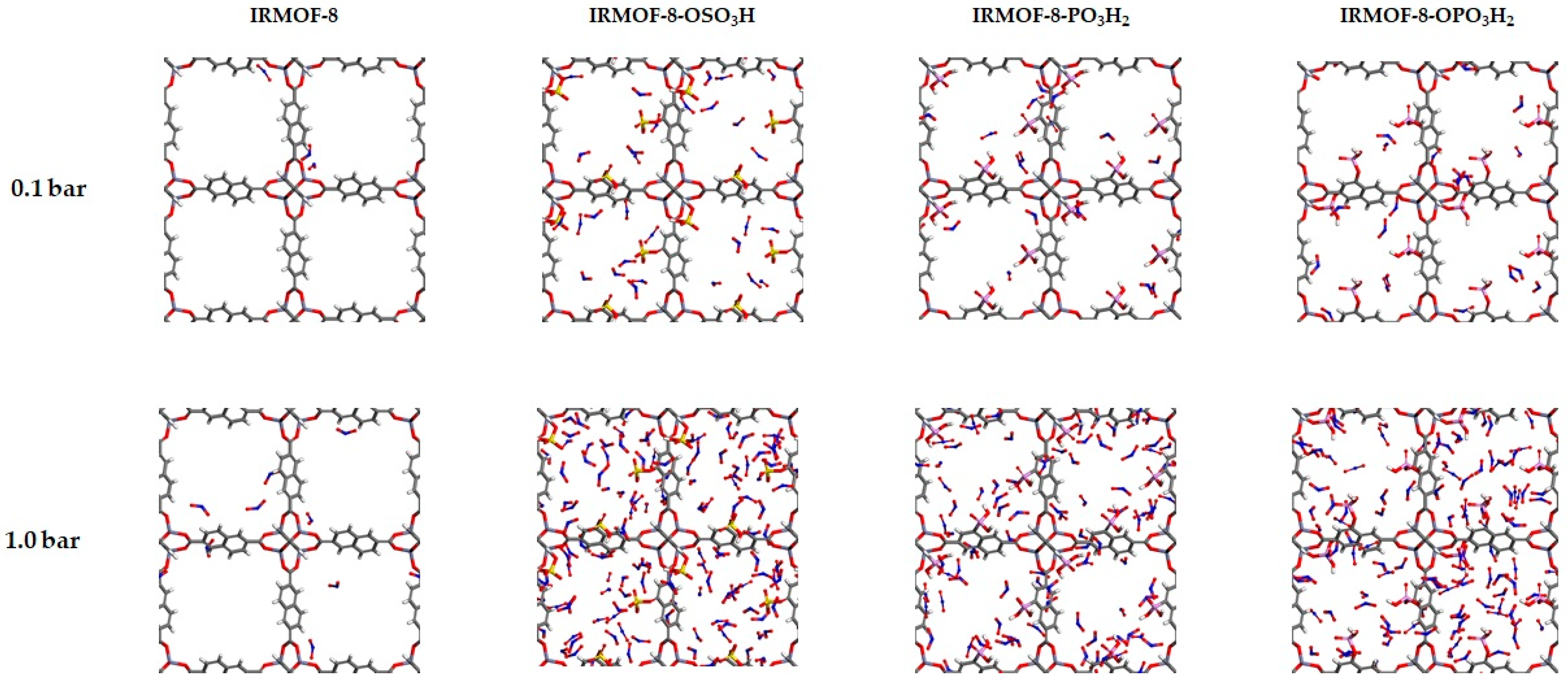
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hill, S.C.; Douglas Smoot, L. Modeling of Nitrogen Oxides Formation and Destruction in Combustion Systems. Prog. Energy Combust. Sci. 2000, 26, 417–458. [Google Scholar] [CrossRef]
- Camargo, J.A.; Alonso, Á. Ecological and Toxicological Effects of Inorganic Nitrogen Pollution in Aquatic Ecosystems: A Global Assessment. Environ. Int. 2006, 32, 831–849. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Bian, H.; Feng, Y.; Liu, A.; Li, X.; Zeng, F.; Zhang, X. Analysis of the Relationship between O3, NO and NO2 in Tianjin, China. Aerosol Air Qual. Res. 2011, 11, 128–139. [Google Scholar] [CrossRef] [Green Version]
- EPA. U.S. Integrated Science Assessment for Oxides of Nitrogen–Health Criteria; US Environmental Protection Agency: Washington, DC, USA, 2016.
- Samet, J.M.; Utell, M.J. The Risk of Nitrogen Dioxide: What Have We Learned from Epidemiological and Clinical Studies? Toxicol. Ind. Health 1990, 6, 247–262. [Google Scholar] [CrossRef]
- Hesterberg, T.W.; Bunn, W.B.; McClellan, R.O.; Hamade, A.K.; Long, C.M.; Valberg, P.A. Critical Review of the Human Data on Short-Term Nitrogen Dioxide (NO2) Exposures: Evidence for NO2 No-Effect Levels. Crit. Rev. Toxicol. 2009, 39, 743–781. [Google Scholar] [CrossRef]
- Chokbunpiam, T.; Chanajaree, R.; Caro, J.; Janke, W.; Remsungnen, T.; Hannongbua, S.; Fritzsche, S. Separation of Nitrogen Dioxide from the Gas Mixture with Nitrogen by Use of ZIF Materials; Computer Simulation Studies. Comput. Mater. Sci. 2019, 168, 246–252. [Google Scholar] [CrossRef]
- Gal, A.; Kurahashi, M.; Kuzumoto, M. Effect of O3 on NO2 Sorption from Gas over H-Y Zeolite: Supposition on the Nitrate Anion Formation with NO2 and O3 as Coreactants. J. Phys. Chem. A 2000, 104, 10821–10824. [Google Scholar] [CrossRef]
- Muckenhuber, H.; Grothe, H. A DRIFTS Study of the Heterogeneous Reaction of NO2 with Carbonaceous Materials at Elevated Temperature. Carbon N. Y. 2007, 45, 321–329. [Google Scholar] [CrossRef]
- Pietrzak, R.; Bandosz, T.J. Reactive Adsorption of NO2 at Dry Conditions on Sewage Sludge-Derived Materials. Environ. Sci. Technol. 2007, 41, 7516–7522. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, L.; Zhang, M.; Ma, C. Effect of N-Doping on NO2 Adsorption and Reduction over Activated Carbon: An Experimental and Computational Study. Fuel 2019, 258, 116109. [Google Scholar] [CrossRef]
- Iranimanesh, A.; Yousefi, M.; Mirzaei, M. DFT Approach on SiC Nanotube for NO2 Gas Pollutant Removal. Lab-in-Silico 2021, 2, 38–43. [Google Scholar] [CrossRef]
- Li, J.R.; Kuppler, R.J.; Zhou, H.C. Selective Gas Adsorption and Separation in Metal-Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, A.M.; Levasseur, B.; Bandosz, T.J. Interactions of NO2 with Zr-Based MOF: Effects of the Size of Organic Linkers on NO2 Adsorption at Ambient Conditions. Langmuir 2013, 29, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Hasan, Z.; Jhung, S.H. Adsorptive Removal of Hazardous Materials Using Metal-Organic Frameworks (MOFs): A Review. J. Hazard. Mater. 2013, 244–245, 444–456. [Google Scholar] [CrossRef]
- Wang, W.; Xu, X.; Zhou, W.; Shao, Z. Recent Progress in Metal-Organic Frameworks for Applications in Electrocatalytic and Photocatalytic Water Splitting. Adv. Sci. 2017, 4, 1600371. [Google Scholar] [CrossRef]
- Della Rocca, J.; Liu, D.; Lin, W. Nanoscale Metal-Organic Frameworks for Biomedical Imaging and Drug Delivery. Acc. Chem. Res. 2011, 44, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.C.; Long, J.R.; Yaghi, O.M. Introduction to Metal-Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- Zhou, H.C.J.; Kitagawa, S. Metal-Organic Frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. [Google Scholar] [CrossRef] [Green Version]
- Bobbitt, N.S.; Mendonca, M.L.; Howarth, A.J.; Islamoglu, T.; Hupp, J.T.; Farha, O.K.; Snurr, R.Q. Metal-Organic Frameworks for the Removal of Toxic Industrial Chemicals and Chemical Warfare Agents. Chem. Soc. Rev. 2017, 46, 3357–3385. [Google Scholar] [CrossRef]
- Frysali, M.G.; Klontzas, E.; Tylianakis, E.; Froudakis, G.E. Tuning the Interaction Strength and the Adsorption of CO2 in Metal Organic Frameworks by Functionalization of the Organic Linkers. Microporous Mesoporous Mater. 2016, 227, 144–151. [Google Scholar] [CrossRef]
- Klontzas, E.; Mavrandonakis, A.; Tylianakis, E.; Froudakis, G.E. Improving Hydrogen Storage Capacity of MOF by Functionalization of the Organic Linker with Lithium Atoms. Nano Lett. 2008, 8, 1572–1576. [Google Scholar] [CrossRef] [PubMed]
- Fioretos, K.A.; Psofogiannakis, G.M.; Froudakis, G.E. Ab-Initio Study of the Adsorption and Separation of NO x and SO x Gases in Functionalized IRMOF Ligands. J. Phys. Chem. C 2011, 115, 24906–24914. [Google Scholar] [CrossRef]
- Stergiannakos, T.; Tylianakis, E.; Klontzas, E.; Froudakis, G.E. Enhancement of Hydrogen Adsorption in Metal-Organic Frameworks by Mg 2+ Functionalization: A Multiscale Computational Study. J. Phys. Chem. C 2010, 114, 16855–16858. [Google Scholar] [CrossRef]
- Frysali, M.G.; Klontzas, E.; Froudakis, G.E. Ab Initio Study of the Adsorption of CO(2) on Functionalized Benzenes. Chemphyschem 2014, 15, 905–911. [Google Scholar] [CrossRef]
- Giappa, R.M.; Tylianakis, E.; Di Gennaro, M.; Gkagkas, K.; Froudakis, G.E. A Combination of Multi-Scale Calculations with Machine Learning for Investigating Hydrogen Storage in Metal Organic Frameworks. Int. J. Hydrogen Energy 2021, 46, 27612–27621. [Google Scholar] [CrossRef]
- Giappa, R.M.; Papadopoulos, A.G.; Klontzas, E.; Tylianakis, E.; Froudakis, G.E. Linker Functionalization Strategy for Water Adsorption in Metal–Organic Frameworks. Molecules 2022, 27, 2614. [Google Scholar] [CrossRef]
- Skylaris, C.-K.; Gagliardi, L.; Handy, N.C.; Ioannou, A.G.; Spencer, S.; Willetts, A. On the Resolution of Identity Coulomb Energy Approximation in Density Functional Theory. J. Mol. Struct. THEOCHEM 2000, 501–502, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Kozuch, S.; Gruzman, D.; Martin, J.M.L. DSD-BLYP: A General Purpose Double Hybrid Density Functional Including Spin Component Scaling and Dispersion Correction. J. Phys. Chem. C 2010, 114, 20801–20808. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 Dispersion Coefficient Model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Laaksonen, L. A Graphics Program for the Analysis and Display of Molecular Dynamics Trajectories. J. Mol. Graph. 1992, 10, 33–34. [Google Scholar] [CrossRef]
- Bergman, D.L.; Laaksonen, L.; Laaksonen, A. Visualization of Solvation Structures in Liquid Mixtures. J. Mol. Graph. Model. 1997, 15, 301–306. [Google Scholar] [CrossRef]
- Peng, D.-Y.; Robinson, D.B. A New Two-Constant Equation of State. Ind. Eng. Chem. Fundam. 1976, 15, 59–64. [Google Scholar] [CrossRef]
- Mohamed, E.; Jaheon, K.; Nathaniel, R.; David, V.; Joseph, W.; Michael, O.; Yaghi, O.M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.E.; Chapman, S. On the Determination of Molecular Fields.—II. From the Equation of State of a Gas. Proc. R. Soc. London. Ser. A Contain. Pap. A Math. Phys. Character 1924, 106, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Breneman, C.M.; Wiberg, K.B. Determining Atom-centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Yang, J.; Ren, Y.; Tian, A.; Sun, H. COMPASS Force Field for 14 Inorganic Molecules, He, Ne, Ar, Kr, Xe, H2, O2, N2, NO, CO, CO2, NO2, CS2, and SO2, in Liquid Phases. J. Phys. Chem. B 2000, 104, 4951–4957. [Google Scholar] [CrossRef]
- Matito-Martos, I.; Rahbari, A.; Martin-Calvo, A.; Dubbeldam, D.; Vlugt, T.J.H.; Calero, S. Adsorption Equilibrium of Nitrogen Dioxide in Porous Materials. Phys. Chem. Chem. Phys. 2018, 20, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Dubbeldam, D.; Calero, S.; Ellis, D.E.; Snurr, R.Q. RASPA: Molecular Simulation Software for Adsorption and Diffusion in Flexible Nanoporous Materials. Mol. Simul. 2016, 42, 81–101. [Google Scholar] [CrossRef] [Green Version]
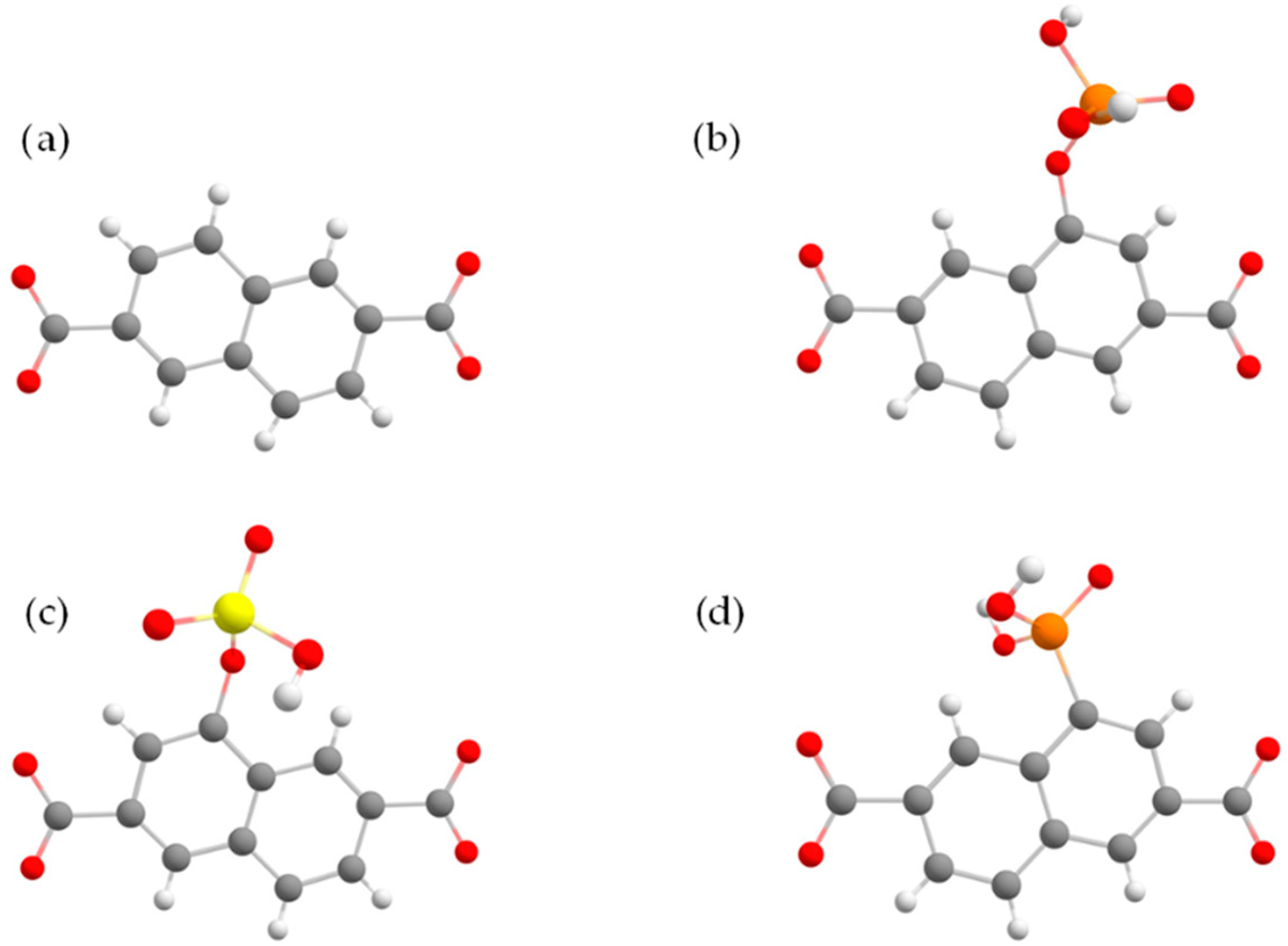

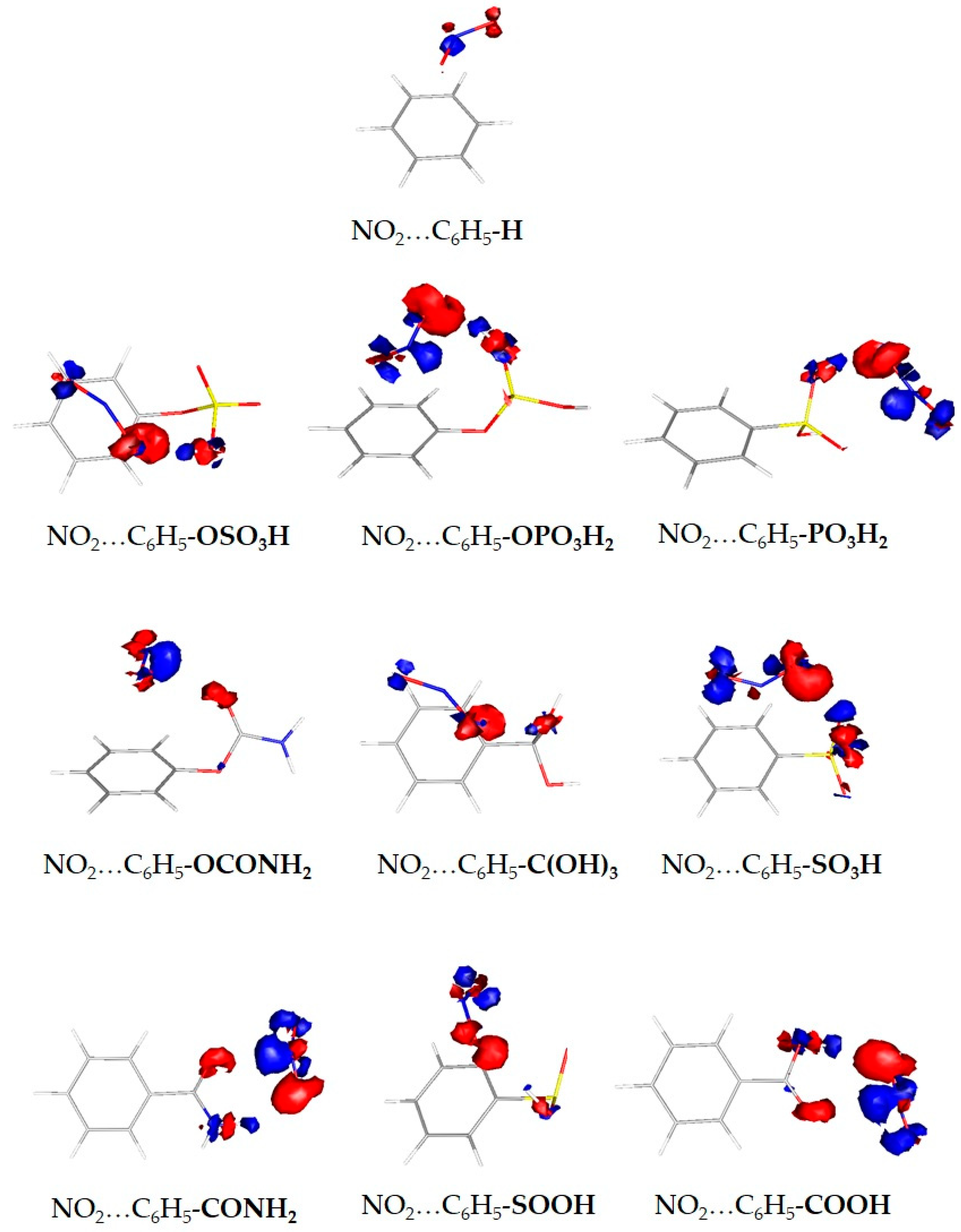
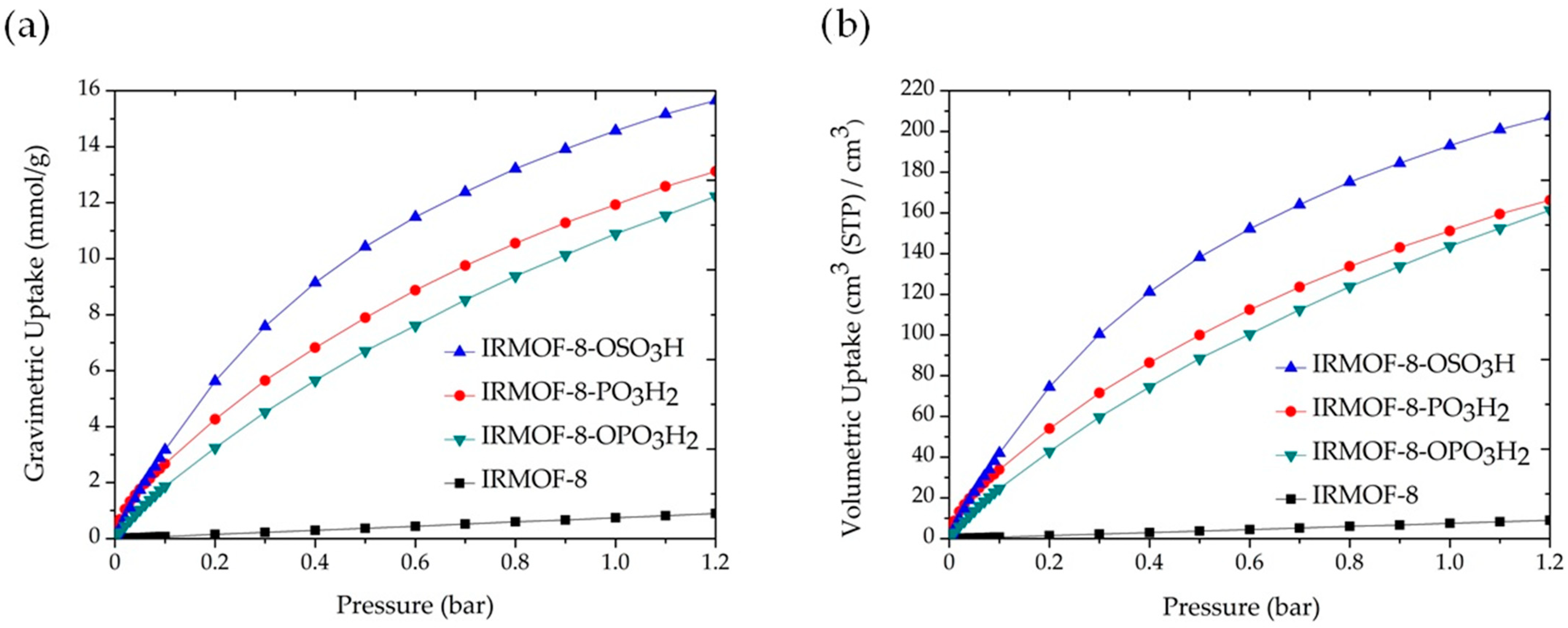
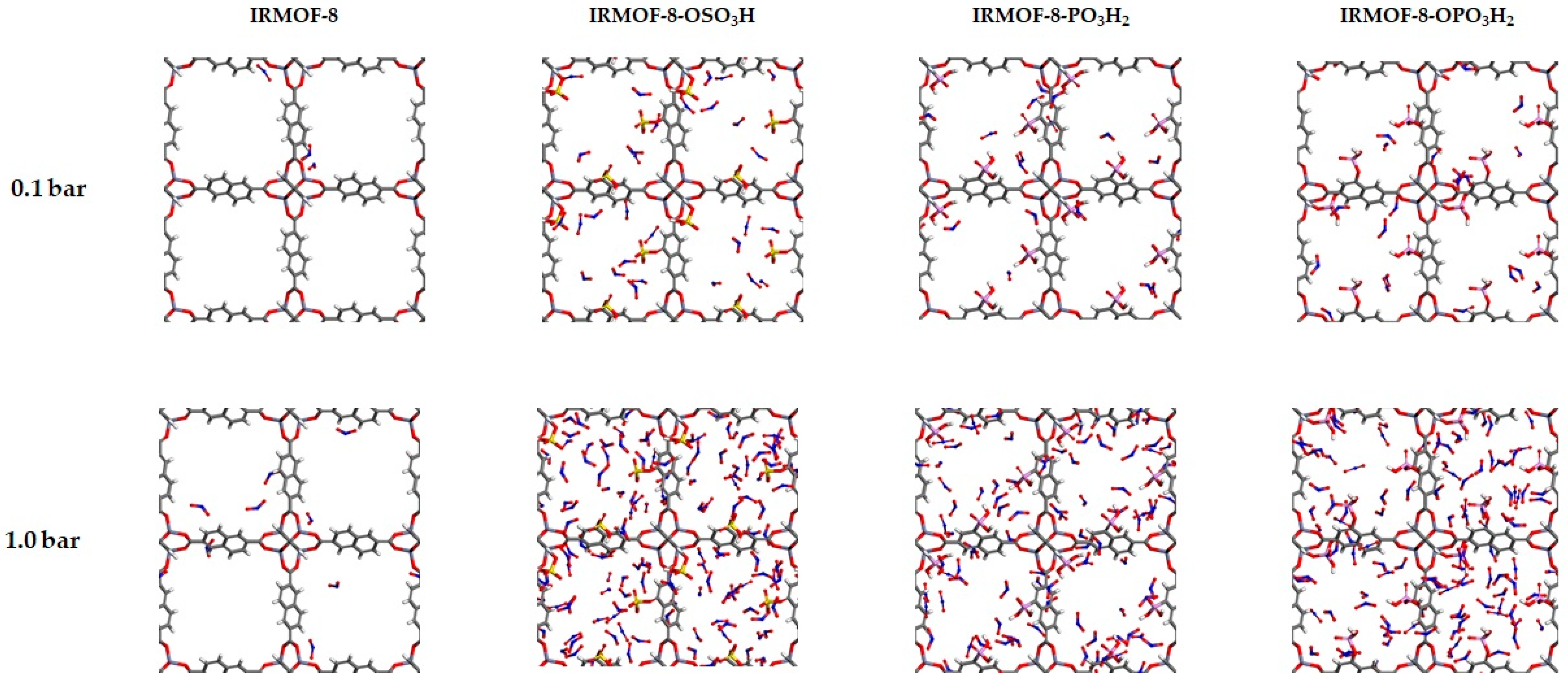

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
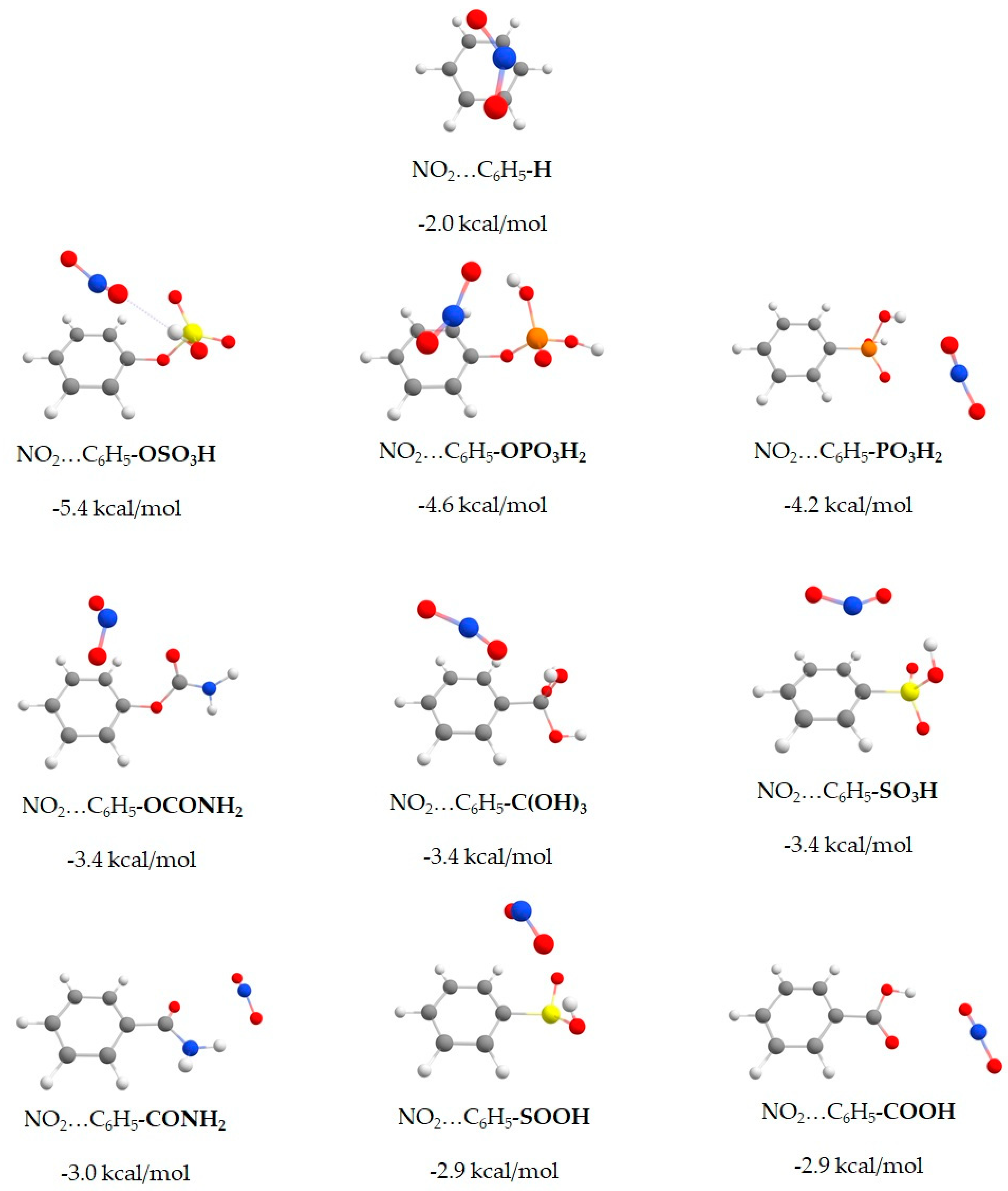
| System | Binding Energy (kcal/mol) | Binding Energy Enhancement (%) |
|---|---|---|
| NO2…C6H5-OSO3H | −5.4 | 170% |
| NO2…C6H5-OPO3H2 | −4.6 | 131% |
| NO2…C6H5-PO3H2 | −4.2 | 110% |
| NO2…C6H5-OCONH2 | −3.4 | 70% |
| NO2…C6H5-C(OH)3 | −3.4 | 70% |
| NO2…C6H5-SO3H | −3.4 | 70% |
| NO2…C6H5-CONH2 | −3.0 | 50% |
| NO2…C6H5-SOOH | −2.9 | 44% |
| NO2…C6H5-COOH | −2.9 | 44% |
| NO2…C6H5-H | −2.0 | 0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raptis, D.; Livas, C.; Stavroglou, G.; Giappa, R.M.; Tylianakis, E.; Stergiannakos, T.; Froudakis, G.E. Surface Modification Strategy for Enhanced NO2 Capture in Metal–Organic Frameworks. Molecules 2022, 27, 3448. https://doi.org/10.3390/molecules27113448
Raptis D, Livas C, Stavroglou G, Giappa RM, Tylianakis E, Stergiannakos T, Froudakis GE. Surface Modification Strategy for Enhanced NO2 Capture in Metal–Organic Frameworks. Molecules. 2022; 27(11):3448. https://doi.org/10.3390/molecules27113448
Chicago/Turabian StyleRaptis, Dionysios, Charalampos Livas, George Stavroglou, Rafaela Maria Giappa, Emmanuel Tylianakis, Taxiarchis Stergiannakos, and George E. Froudakis. 2022. "Surface Modification Strategy for Enhanced NO2 Capture in Metal–Organic Frameworks" Molecules 27, no. 11: 3448. https://doi.org/10.3390/molecules27113448
APA StyleRaptis, D., Livas, C., Stavroglou, G., Giappa, R. M., Tylianakis, E., Stergiannakos, T., & Froudakis, G. E. (2022). Surface Modification Strategy for Enhanced NO2 Capture in Metal–Organic Frameworks. Molecules, 27(11), 3448. https://doi.org/10.3390/molecules27113448