
Preclinical Pharmacokinetic Studies of a Novel Diuretic Inhibiting Urea Transporters



Abstract
:1. Introduction
2. Results and Discussion
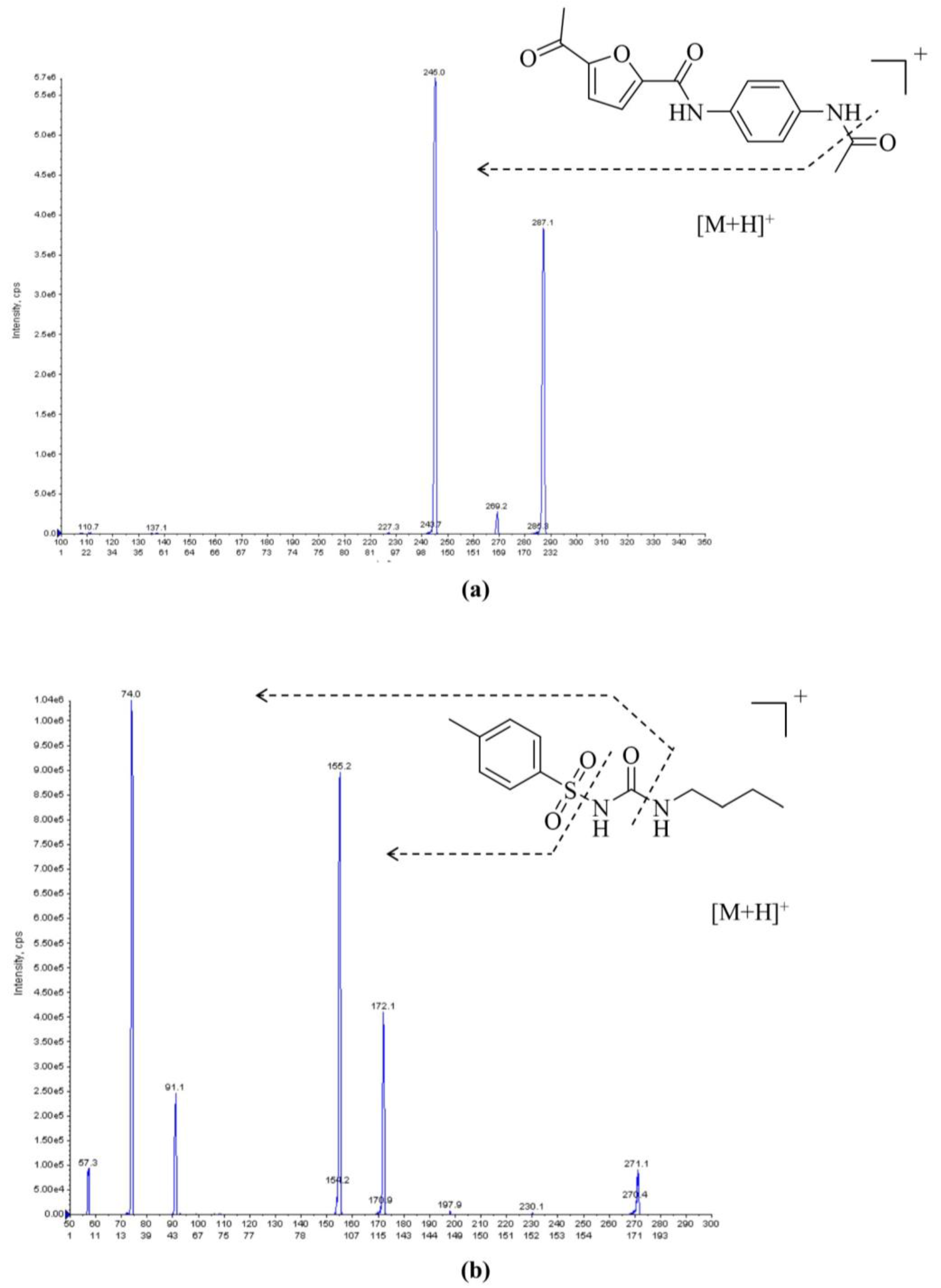
2.1. LC–MS/MS Method Development and Optimization
2.2. LC–MS/MS Validation
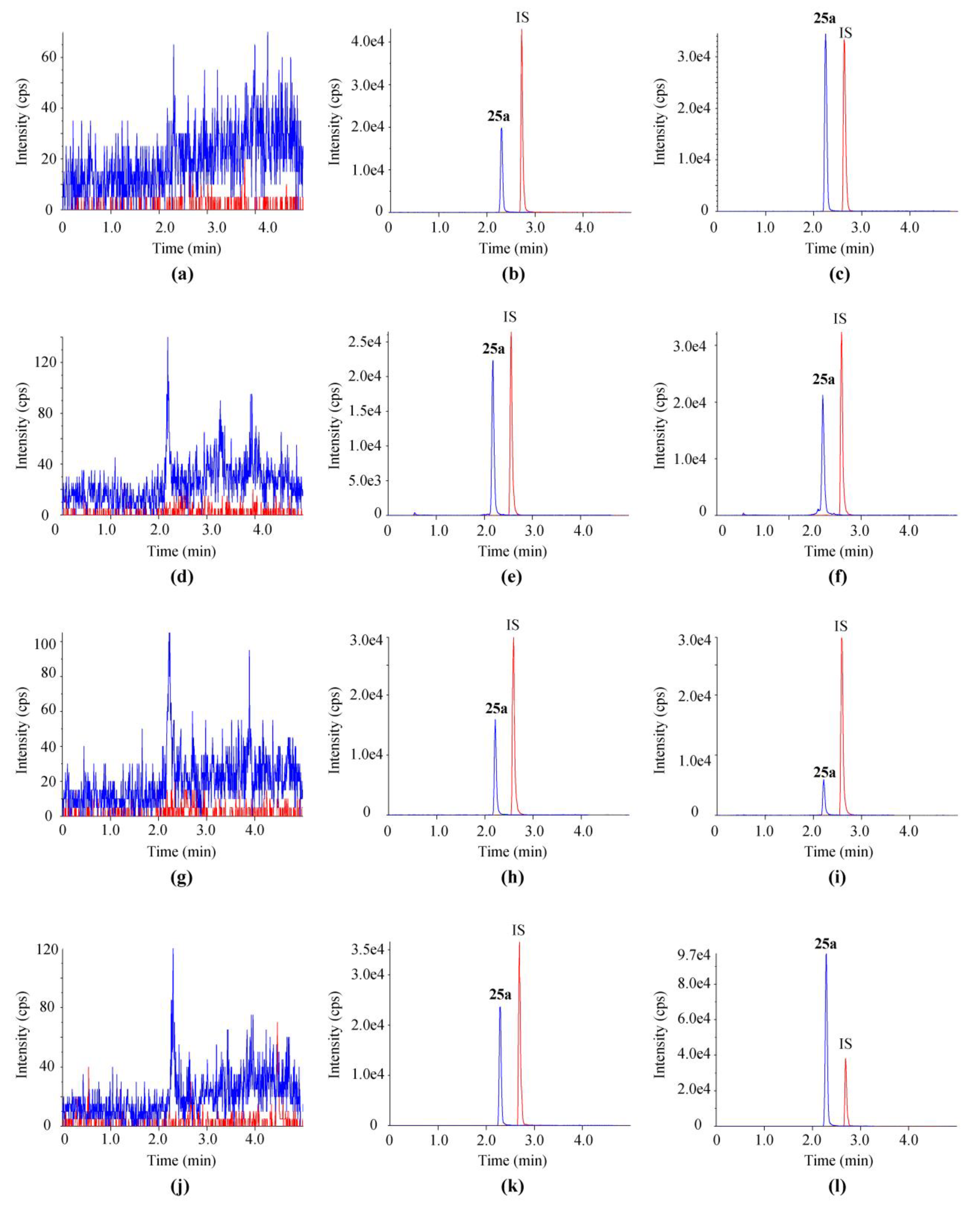
2.2.1. Selectivity
2.2.2. Linearity
2.2.3. Accuracy and Precision
2.2.4. Recovery and Matrix Effect
2.2.5. Stability Evaluation
2.2.6. Dilution Integrity
2.2.7. Carry-Over
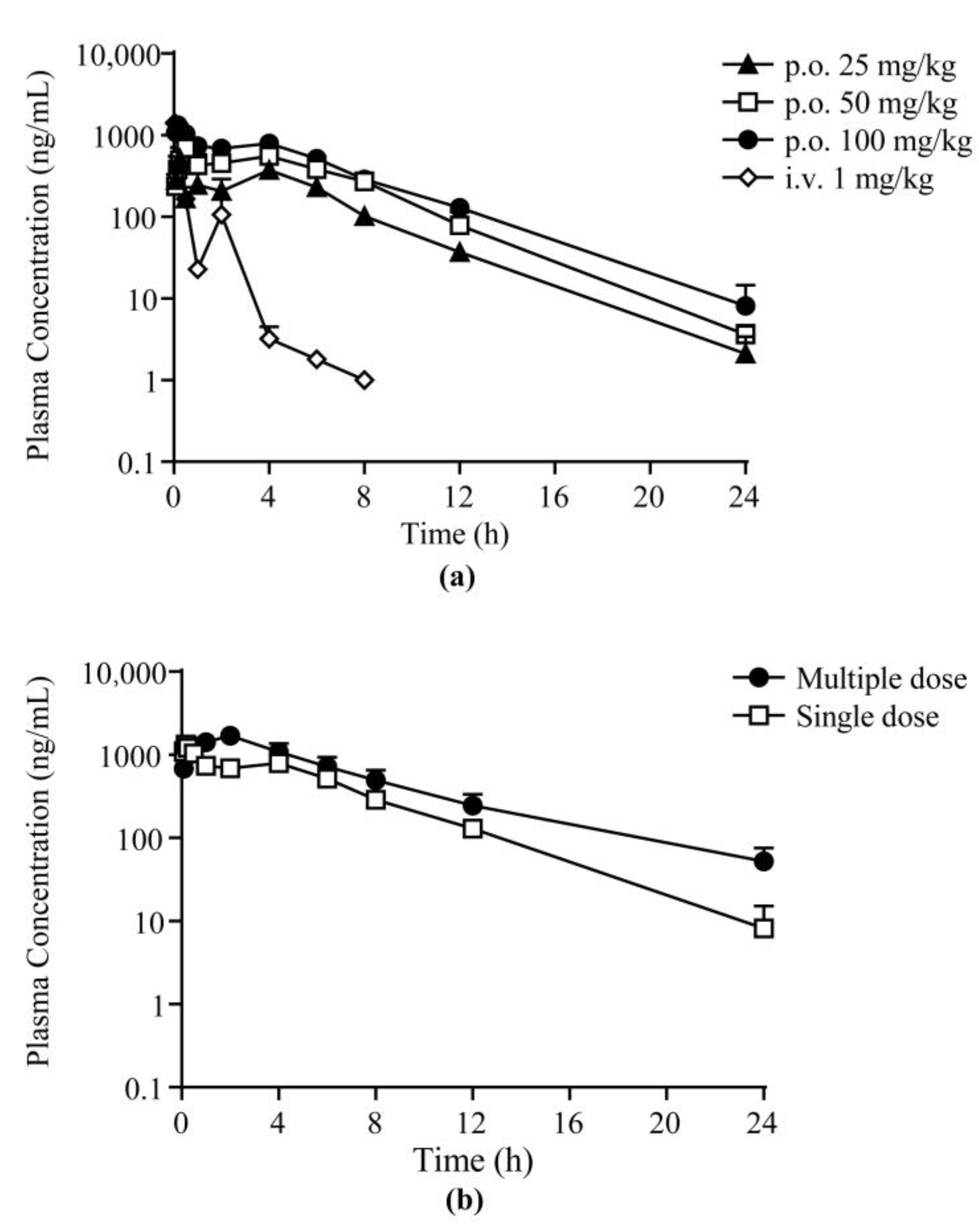
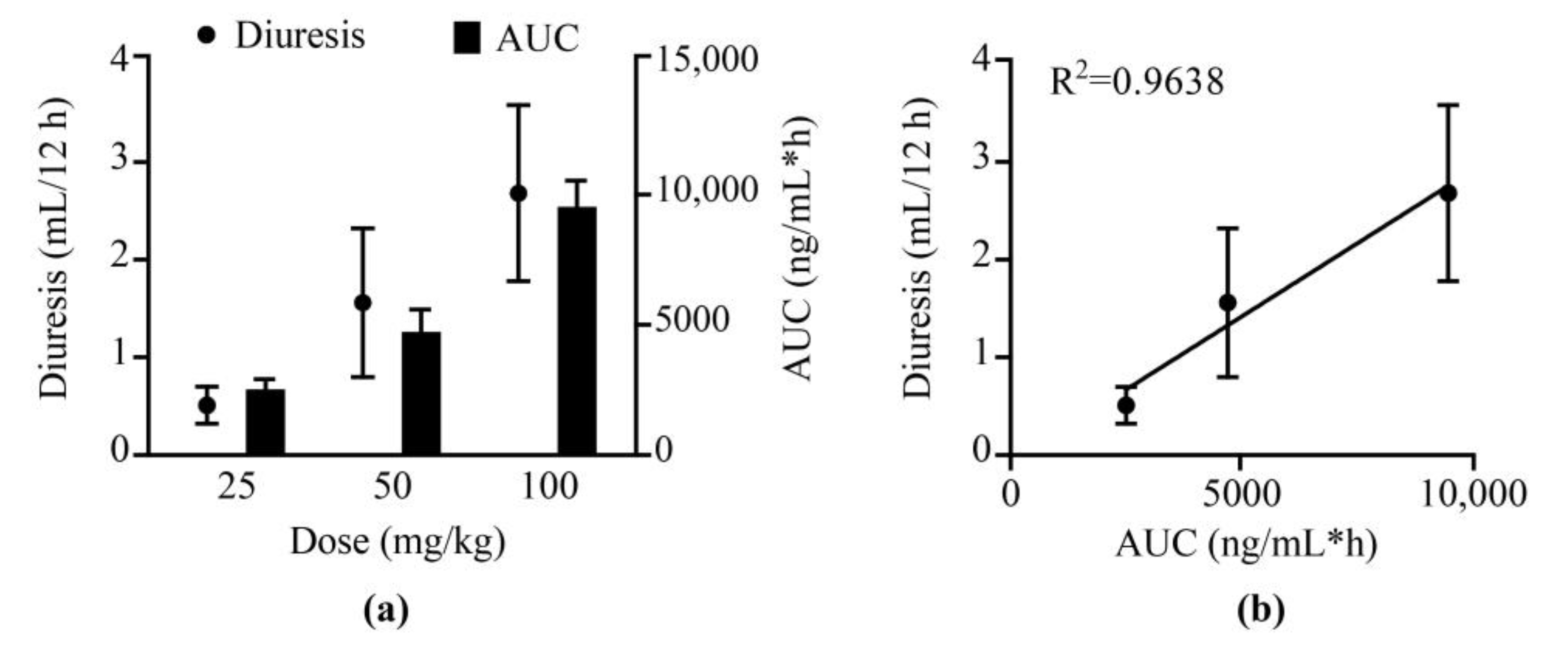
2.3. Pharmacokinetic and Pharmacodynamic Study
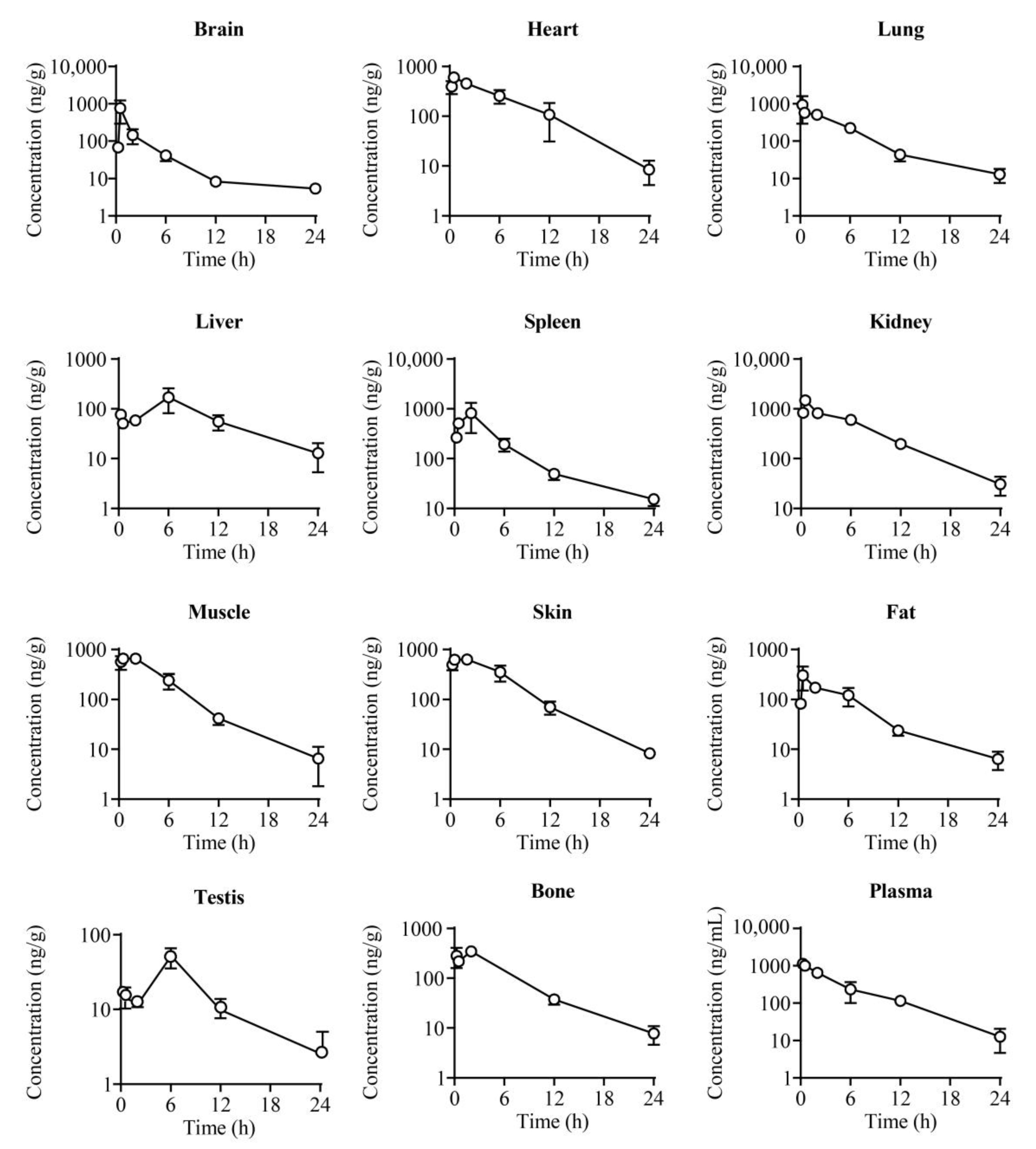
2.4. Tissue Distribution
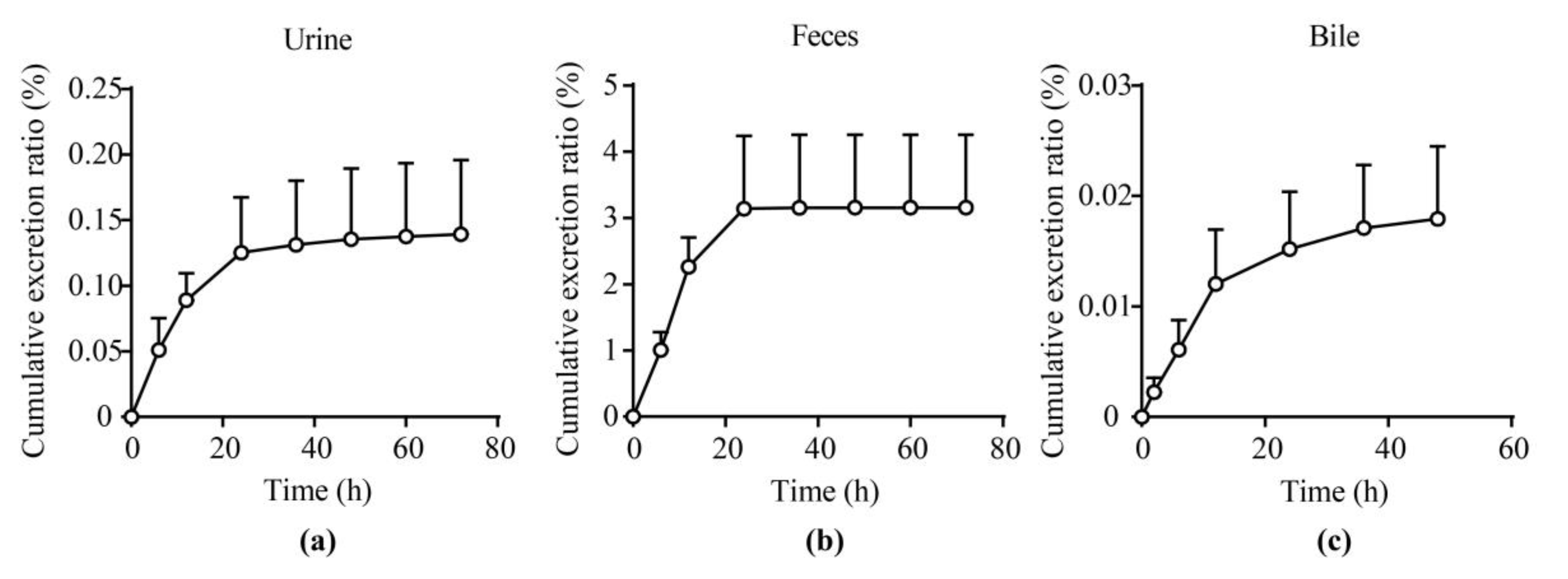
2.5. Excretion Experiments
2.6. Plasma Protein Binding and Blood Plasma Ratio
3. Materials and Methods
3.1. Chemicals and Materials
3.2. LC–MS/MS Instrument Conditions
3.3. Sample Preparation
3.4. Method Validation
3.5. Pharmacokinetic Study in Rats
3.6. Tissue Distribution Study
3.7. Excretion Experiment
3.8. Plasma Protein Binding Study
3.9. Blood Plasma Ratio Study
3.10. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Brater, D.C. Diuretic therapy. N. Engl. J. Med. 1998, 339, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Szekacs, B.; Vajo, Z.; Szepezdi, Z. Diuretics for hypertension. JAMA 1999, 282, 524. [Google Scholar] [PubMed]
- Ernst, M.E.; Moser, M. Use of diuretics in patients with hypertension. N. Engl. J. Med. 2009, 361, 2153–2164. [Google Scholar] [CrossRef] [Green Version]
- Nawar, T. Hyperkalaemia and overdose of antihypertensive agents. Lancet 1978, 1, 717. [Google Scholar] [CrossRef]
- Yang, B.; Sands, J.M. Urea Transporters; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar]
- Weiner, I.D.; Mitch, W.E.; Sands, J.M. Urea and Ammonia Metabolism and the Control of Renal Nitrogen Excretion. Clin. J. Am. Soc. Nephrol. 2015, 10, 1444–1458. [Google Scholar] [CrossRef] [Green Version]
- Fenton, R.A.; Knepper, M.A. Urea and renal function in the 21st century: Insights from knockout mice. J. Am. Soc. Nephrol. 2007, 18, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Klein, J.D.; Blount, M.A.; Sands, J.M. Urea transport in the kidney. Compr. Physiol. 2011, 1, 699–729. [Google Scholar] [CrossRef]
- Shayakul, C.; Clemencon, B.; Hediger, M.A. The urea transporter family (SLC14): Physiological, pathological and structural aspects. Mol. Asp. Med. 2013, 34, 313–322. [Google Scholar] [CrossRef]
- Geng, X.; Zhang, S.; He, J.; Ma, A.; Li, Y.; Li, M.; Zhou, H.; Chen, G.; Yang, B. The urea transporter UT-A1 plays a predominant role in a urea-dependent urine-concentrating mechanism. J. Biol. Chem. 2020, 295, 9893–9900. [Google Scholar] [CrossRef]
- Yang, B.; Bankir, L.; Gillespie, A.; Epstein, C.J.; Verkman, A.S. Urea-selective concentrating defect in transgenic mice lacking urea transporter UT-B. J. Biol. Chem. 2002, 277, 10633–10637. [Google Scholar] [CrossRef] [Green Version]
- Rianto, F.; Kuma, A.; Ellis, C.L.; Hassounah, F.; Rodriguez, E.L.; Wang, X.H.; Sands, J.M.; Klein, J.D. UT-A1/A3 knockout mice show reduced fibrosis following unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 2020, 318, F1160–F1166. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lei, T.; Zhu, J.; Wang, W.; Sun, Y.; Chen, J.; Dong, Z.; Zhou, H.; Yang, B. A novel small-molecule thienoquinolin urea transporter inhibitor acts as a potential diuretic. Kidney Int. 2013, 83, 1076–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, J.H.; Li, M.; Tou, W.I.; Lei, T.L.; Zhou, H.; Chen, C.Y.; Yang, B.X. Phenylphthalazines as small-molecule inhibitors of urea transporter UT-B and their binding model. Acta Pharmacol. Sin. 2016, 37, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, M.; Li, B.; Zhang, S.; Su, A.; Xing, Y.; Ge, Z.; Li, R.; Yang, B. Discovery and optimization of thienopyridine derivatives as novel urea transporter inhibitors. Eur. J. Med. Chem. 2019, 172, 131–142. [Google Scholar] [CrossRef]
- Li, M.; Zhao, Y.; Zhang, S.; Xu, Y.; Wang, S.Y.; Li, B.W.; Ran, J.H.; Li, R.T.; Yang, B.X. A thienopyridine, CB-20, exerts diuretic activity by inhibiting urea transporters. Acta Pharmacol. Sin. 2020, 41, 65–72. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, Y.; Wang, S.; Li, M.; Xu, Y.; Ran, J.; Geng, X.; He, J.; Meng, J.; Shao, G.; et al. Discovery of novel diarylamides as orally active diuretics targeting urea transporters. Acta Pharm. Sin. B 2021, 11, 181–202. [Google Scholar] [CrossRef]
- Yao, C.; Anderson, M.O.; Zhang, J.; Yang, B.; Phuan, P.W.; Verkman, A.S. Triazolothienopyrimidine inhibitors of urea transporter UT-B reduce urine concentration. J. Am. Soc. Nephrol. 2012, 23, 1210–1220. [Google Scholar] [CrossRef] [Green Version]
- Esteva-Font, C.; Phuan, P.W.; Anderson, M.O.; Verkman, A.S. A small molecule screen identifies selective inhibitors of urea transporter UT-A. Chem. Biol. 2013, 20, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Esteva-Font, C.; Yao, C.; Phuan, P.W.; Verkman, A.S.; Anderson, M.O. 1,1-Difluoroethyl-substituted triazolothienopyrimidines as inhibitors of a human urea transport protein (UT-B): New analogs and binding model. Bioorg. Med. Chem. Lett. 2013, 23, 3338–3341. [Google Scholar] [CrossRef] [Green Version]
- Esteva-Font, C.; Phuan, P.W.; Lee, S.; Su, T.; Anderson, M.O.; Verkman, A.S. Structure-activity analysis of thiourea analogs as inhibitors of UT-A and UT-B urea transporters. Biochim. Biophys. Acta 2015, 1848, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Cil, O.; Diez-Cecilia, E.; Anderson, M.O.; Verkman, A.S. Nanomolar-Potency 1,2,4-Triazoloquinoxaline Inhibitors of the Kidney Urea Transporter UT-A1. J. Med. Chem. 2018, 61, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, Y.; Zhao, Y.; Zhang, S.; Li, M.; Li, X.; He, J.; Zhou, H.; Ge, Z.; Li, R.; et al. N-(4-acetamidophenyl)-5-acetylfuran-2-carboxamide as a novel orally available diuretic that targets urea transporters with improved PD and PK properties. Eur J Med Chem 2021, 226, 113859. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, G.; Shen, J.X.; Aubry, A.F. Reasons for calibration standard curve slope variation in LC-MS assays and how to address it. Bioanalysis 2014, 6, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.; Yu, S.; Ronis, M.J.; Badger, T.M. Matrix effects break the LC behavior rule for analytes in LC-MS/MS analysis of biological samples. Exp. Biol. Med. 2015, 240, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parr, A.; Hidalgo, I.J.; Bode, C.; Brown, W.; Yazdanian, M.; Gonzalez, M.A.; Sagawa, K.; Miller, K.; Jiang, W.; Stippler, E.S. The Effect of Excipients on the Permeability of BCS Class III Compounds and Implications for Biowaivers. Pharm. Res. 2016, 33, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Bankir, L.; Yang, B. New insights into urea and glucose handling by the kidney, and the urine concentrating mechanism. Kidney Int. 2012, 81, 1179–1198. [Google Scholar] [CrossRef] [Green Version]
- Klein, J.D.; Wang, Y.; Mistry, A.; LaRocque, L.M.; Molina, P.A.; Rogers, R.T.; Blount, M.A.; Sands, J.M. Transgenic Restoration of Urea Transporter A1 Confers Maximal Urinary Concentration in the Absence of Urea Transporter A3. J. Am. Soc. Nephrol. 2016, 27, 1448–1455. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Xue, J.; Shao, J.; Jia, L. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov. Today 2012, 17, 475–485. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, X.; Wang, G.; Peng, Y.; Zhang, X.; Wu, H.; Yu, B.; Sun, J. Preclinical Pharmacokinetics of C118P, a Novel Prodrug of Microtubules Inhibitor and Its Metabolite C118 in Mice, Rats, and Dogs. Molecules 2018, 23, 2883. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Mobile Phase A (%) | Mobile Phase B (%) |
|---|---|---|
| 0.0 | 95 | 5 |
| 0.5 | 30 | 70 |
| 1.0 | 0 | 100 |
| 3.0 | 0 | 100 |
| 3.1 | 95 | 5 |
| 5.0 | 95 | 5 |
| Matrix | y = a × x + b * | ||
|---|---|---|---|
| a | b | r | |
| Plasma | 0.01201 | 0.00391 | 0.9983 |
| Urine | 0.00965 | 0.00204 | 0.9997 |
| Feces | 0.00967 | 0.00218 | 0.9996 |
| Bile | 0.00708 | 0.00325 | 0.9989 |
| Kidney | 0.01200 | 0.00390 | 0.9995 |
| Heart | 0.01180 | 0.00314 | 0.9997 |
| Lung | 0.01090 | 0.00400 | 0.9996 |
| Spleen | 0.01060 | 0.00646 | 0.9997 |
| Muscle | 0.01190 | 0.00332 | 0.9998 |
| Skin | 0.01030 | 0.00640 | 0.9994 |
| Bone | 0.00926 | 0.00694 | 0.9997 |
| Brain | 0.01170 | 0.00395 | 0.9999 |
| Fat | 0.01100 | 0.00259 | 0.9997 |
| Liver | 0.00961 | 0.00599 | 0.9996 |
| Testis | 0.01000 | 0.00878 | 0.9997 |
| Matrix | Sample | Nominal Conc. (ng/mL) | Intraday | Interday | ||
|---|---|---|---|---|---|---|
| RE (%) | CV (%) | RE (%) | CV (%) | |||
| Plasma | LLOQ | 1 | 1.2 | 6.0 | −4.6 | 8.4 |
| LQC | 3 | −3.9 | 2.1 | −6.8 | 5.1 | |
| MQC | 100 | 0.3 | 1.7 | −2.8 | 5.9 | |
| HQC | 800 | −3.0 | 1.6 | −3.4 | 3.8 | |
| Urine | LLOQ | 1 | −14.9 | 5.8 | −15.2 | 0.4 |
| LQC | 3 | −11.4 | 8.0 | −10.6 | 2.1 | |
| MQC | 100 | 3.3 | 1.7 | −0.7 | 6.3 | |
| HQC | 800 | −3.0 | 4.3 | −4.7 | 3.0 | |
| Feces | LLOQ | 1 | 7.0 | 2.3 | 9.1 | 2.4 |
| LQC | 3 | 2.6 | 5.4 | 7.4 | 3.9 | |
| MQC | 100 | 11.2 | 4.1 | 7.5 | 3.2 | |
| HQC | 800 | 3.9 | 4.2 | 4.5 | 1.2 | |
| Bile | LLOQ | 1 | 15.2 | 2.9 | 7.9 | 11.2 |
| LQC | 3 | −12.1 | 4.2 | −9.7 | 3.5 | |
| MQC | 100 | −7.8 | 2.8 | −8.0 | 0.6 | |
| HQC | 800 | −11.2 | 3.5 | −11.6 | 0.8 | |
| Matrix | Quality Control | Nominal Conc. (ng/mL) | Recovery | Matrix Effect | ||||
|---|---|---|---|---|---|---|---|---|
| Mean (%) | SD (%) | CV (%) | Mean (%) | SD (%) | CV (%) | |||
| Plasma | LQC | 3 | 97.6 | 6.0 | 6.1 | 105.4 | 2.8 | 2.5 |
| MQC | 100 | 98.2 | 4.5 | 4.5 | 107.5 | 2.1 | 2.1 | |
| HQC | 800 | 110.7 | 4.7 | 4.7 | 103.7 | 4.2 | 4.2 | |
| Urine | LQC | 3 | 101.8 | 7.6 | 7.4 | 98.2 | 5.3 | 4.8 |
| MQC | 100 | 94.4 | 8.4 | 8.9 | 99.3 | 2.6 | 2.4 | |
| HQC | 800 | 100.9 | 3.6 | 3.5 | 98.0 | 0.9 | 0.8 | |
| Feces | LQC | 3 | 98.5 | 8.0 | 7.5 | 102.1 | 8.0 | 6.8 |
| MQC | 100 | 105.6 | 2.8 | 5.4 | 95.0 | 2.4 | 5.0 | |
| HQC | 800 | 103.8 | 10.8 | 9.4 | 97.1 | 9.6 | 7.9 | |
| Bile | LQC | 3 | 94.1 | 10.0 | 10.6 | 88.8 | 9.0 | 8.1 |
| MQC | 100 | 88.7 | 3.1 | 3.5 | 82.8 | 9.6 | 8.6 | |
| HQC | 800 | 92.5 | 10.3 | 11.1 | 83.5 | 5.5 | 5.0 | |
| Matrix | Quality Control | Nominal Conc. (ng/mL) | 25 °C for 4 h | 4 °C for 24 h | Three Freeze–Thaw Cycles | −80 °C for 30 days | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| RE (%) | CV (%) | RE (%) | CV (%) | RE (%) | CV (%) | RE (%) | CV (%) | |||
| Plasma | LQC | 3 | −4.9 | 2.0 | 2.8 | 5.6 | −3.6 | 1.4 | 1.1 | 5.6 |
| MQC | 100 | −2.5 | 2.2 | 3.7 | 1.3 | −4.0 | 2.1 | 3.7 | 1.3 | |
| HQC | 800 | −3.1 | 0.5 | −4.5 | 1.7 | −7.6 | 2.1 | −4.5 | 1.7 | |
| Urine | LQC | 3 | 3.2 | 6.7 | −8.3 | 3.1 | 5.8 | 10.7 | 6.2 | 3.1 |
| MQC | 100 | 4.3 | 1.7 | 2.5 | 1.7 | −5.2 | 4.2 | 2.5 | 1.7 | |
| HQC | 800 | −2.3 | 4.4 | −2.3 | 3.9 | −1.5 | 3.5 | −2.3 | 3.9 | |
| Feces | LQC | 3 | −5.2 | 4.9 | 3.9 | 7.4 | −5.2 | 2.9 | 1.2 | 7.4 |
| MQC | 100 | 1.3 | 3.4 | 4.8 | 3.2 | −3.2 | 3.1 | 4.8 | 3.2 | |
| HQC | 800 | −2.4 | 0.9 | −5.3 | 1.5 | −9.1 | 4.9 | −5.3 | 1.5 | |
| Bile | LQC | 3 | −14.9 | 4.7 | −14.7 | 5.8 | −5.8 | 10.7 | −2.1 | 5.8 |
| MQC | 100 | −5.1 | 9.0 | −3.9 | 2.2 | −5.2 | 4.2 | −3.9 | 2.2 | |
| HQC | 800 | −7.8 | 3.7 | −7.1 | 3.5 | −12.2 | 3.5 | −7.1 | 3.5 | |
| Parameter | Unit | Single i.v. 1 mg/kg | Single p.o. 25 mg/kg | Single p.o. 50 mg/kg | Single p.o. 100 mg/kg | Multiple p.o. 100 mg/kg |
|---|---|---|---|---|---|---|
| t1/2 | h | 0.89 ± 0.52 | 2.86 ± 0.29 | 2.63 ± 0.20 | 3.04 ± 0.70 | 5.03 ± 0.82 |
| tmax | h | - | 0.25 ± 0.00 | 0.50 ± 0.00 | 0.17 ± 0.04 | 2.00 ± 0.00 |
| Cmax1 | ng/mL | 1711.0 ± 397.7 | 515.0 ± 106.7 | 693.2 ± 260.4 | 1324.0 ± 227.5 | 1711.7 ± 185.0 |
| AUC0–t | μg/L·h | 500.9 ± 124.3 | 2517.9 ± 395.2 | 4729.6 ± 854.3 | 6921.7 ± 377.4 | 11,753.9 ± 1145.6 |
| AUC0–∞ | μg/L·h | 502.7 ± 124.4 | 2526.5 ± 362.4 | 4743.3 ± 854.4 | 6957.4 ± 345.1 | 12,132.7 ± 1187.2 |
| MRT | h | 0.6 ± 0.1 | 5.2 ± 0.4 | 5.4 ± 0.8 | 5.3 ± 0.3 | 6.5 ± 0.6 |
| V/F 2 | L/kg | 2.6 ± 1.4 | 40.9 ± 7.4 | 40.1 ± 10.8 | 63.0 ± 17.6 | 59.8 ± 11.8 |
| CL/F 3 | L/kg/h | 2.1 ± 0.6 | 9.9 ± 1.6 | 10.5 ± 2.2 | 14.4 ± 0.7 | 8.2 ± 0.8 |
| F | % | - | 20.1 ± 0.8 | 18.9 ± 1.7 | 13.8 ± 0.8 | 23.5 ± 2.3 |
| Species | Plasma Protein Binding (%) | ||
|---|---|---|---|
| 50 ng/mL | 500 ng/mL | 2500 ng/mL | |
| Human | 44.1 ± 2.5 | 38.8 ± 12.0 | 40.5 ± 2.5 |
| Rat | 63.2 ± 3.3 | 61.4 ± 1.2 | 64.5 ± 2.5 |
| Species | Blood Plasma Ratio | ||
|---|---|---|---|
| 50 ng/mL | 500 ng/mL | 2500 ng/mL | |
| Human | 1.39 ± 0.14 | 1.28 ± 0.05 | 1.22 ± 0.32 |
| Rat | 0.84 ± 0.26 | 0.74 ± 0.06 | 0.79 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Zhang, H.; Li, N.; Ma, W.; Wang, S.; Sun, J.; Yang, B. Preclinical Pharmacokinetic Studies of a Novel Diuretic Inhibiting Urea Transporters. Molecules 2022, 27, 2451. https://doi.org/10.3390/molecules27082451
Xu Y, Zhang H, Li N, Ma W, Wang S, Sun J, Yang B. Preclinical Pharmacokinetic Studies of a Novel Diuretic Inhibiting Urea Transporters. Molecules. 2022; 27(8):2451. https://doi.org/10.3390/molecules27082451
Chicago/Turabian StyleXu, Yue, Hang Zhang, Nannan Li, Wen Ma, Shuyuan Wang, Jianguo Sun, and Baoxue Yang. 2022. "Preclinical Pharmacokinetic Studies of a Novel Diuretic Inhibiting Urea Transporters" Molecules 27, no. 8: 2451. https://doi.org/10.3390/molecules27082451
APA StyleXu, Y., Zhang, H., Li, N., Ma, W., Wang, S., Sun, J., & Yang, B. (2022). Preclinical Pharmacokinetic Studies of a Novel Diuretic Inhibiting Urea Transporters. Molecules, 27(8), 2451. https://doi.org/10.3390/molecules27082451