
A2A Adenosine Receptor Antagonists: Are Triazolotriazine and Purine Scaffolds Interchangeable?

 ,
,  , ,
, ,  ,
,  , ,
, ,  ,
,  ,
,  ,
,  and
and
Abstract
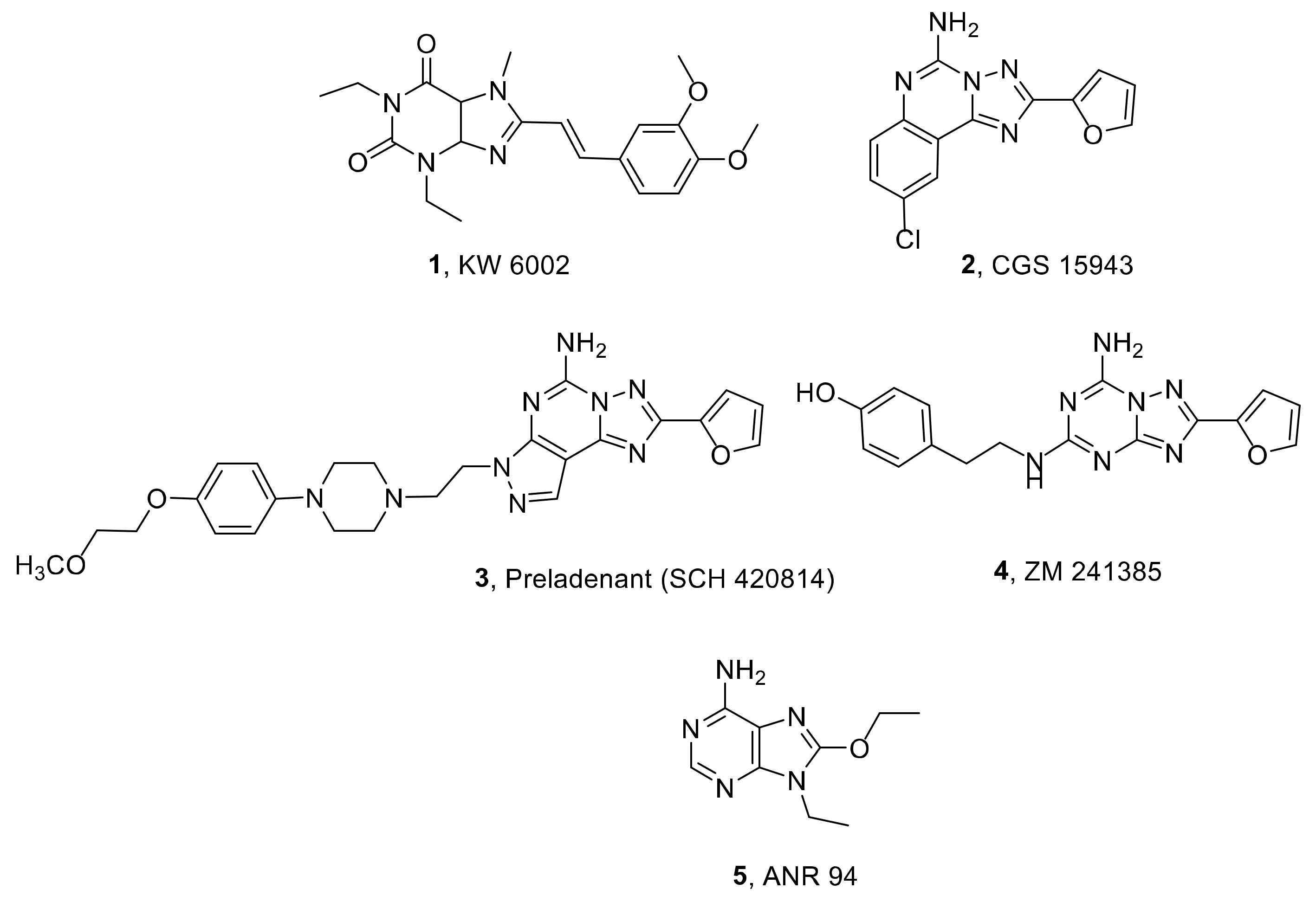
:1. Introduction
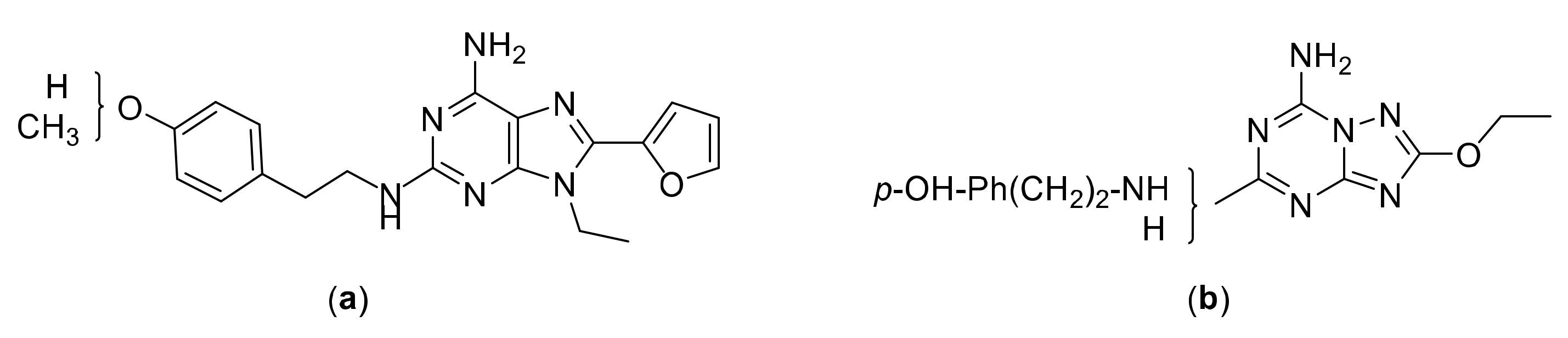
2. Results and Discussion
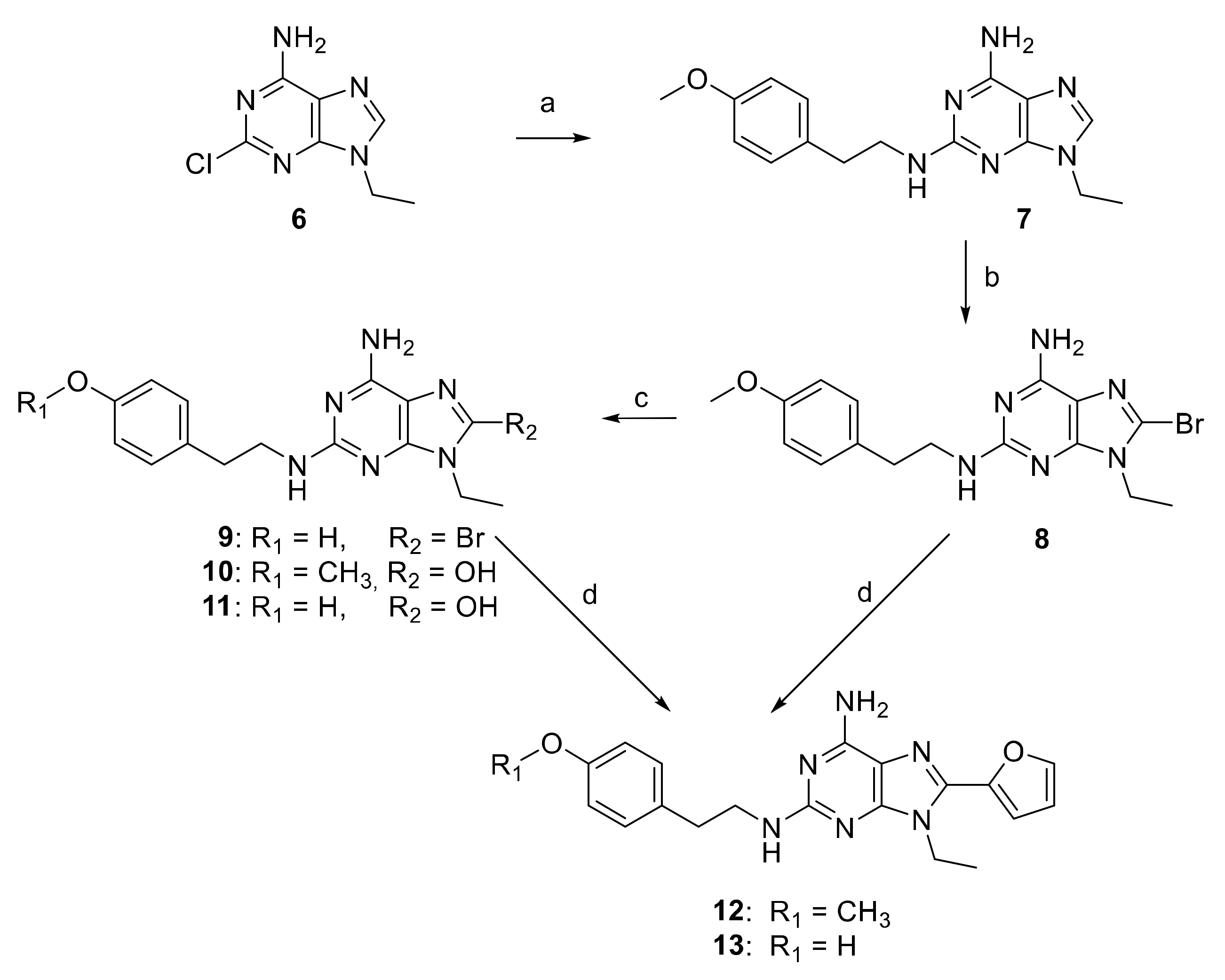
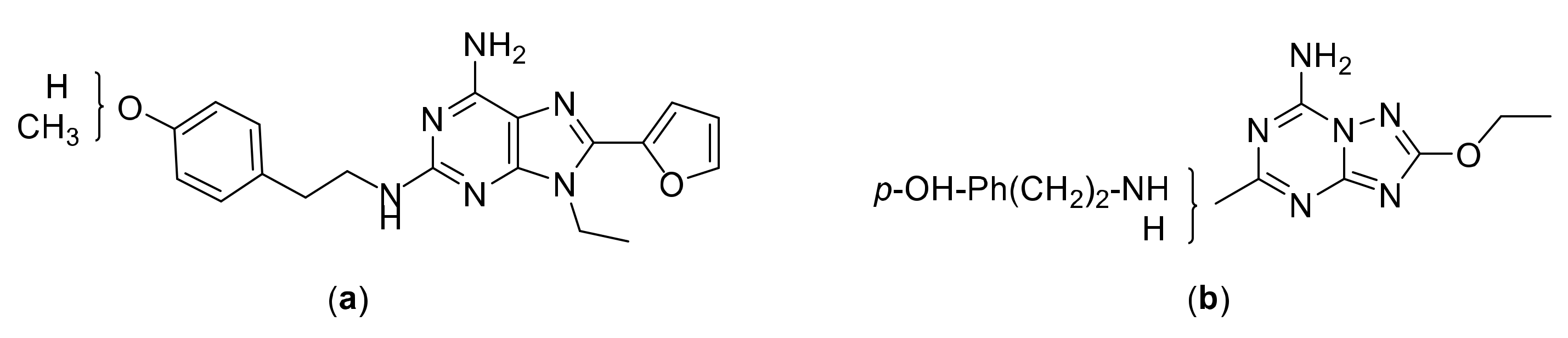
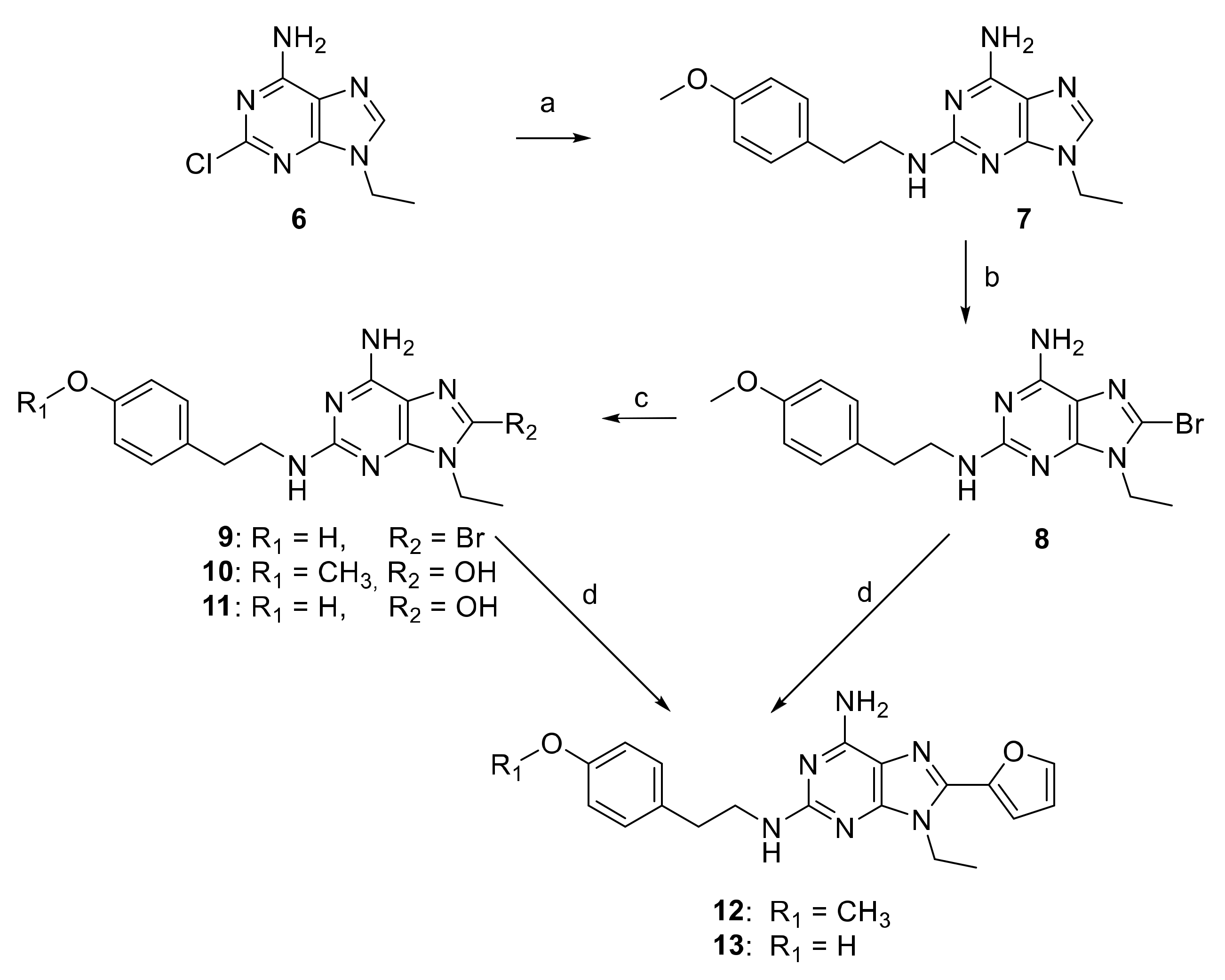
2.1. Chemical Synthesis
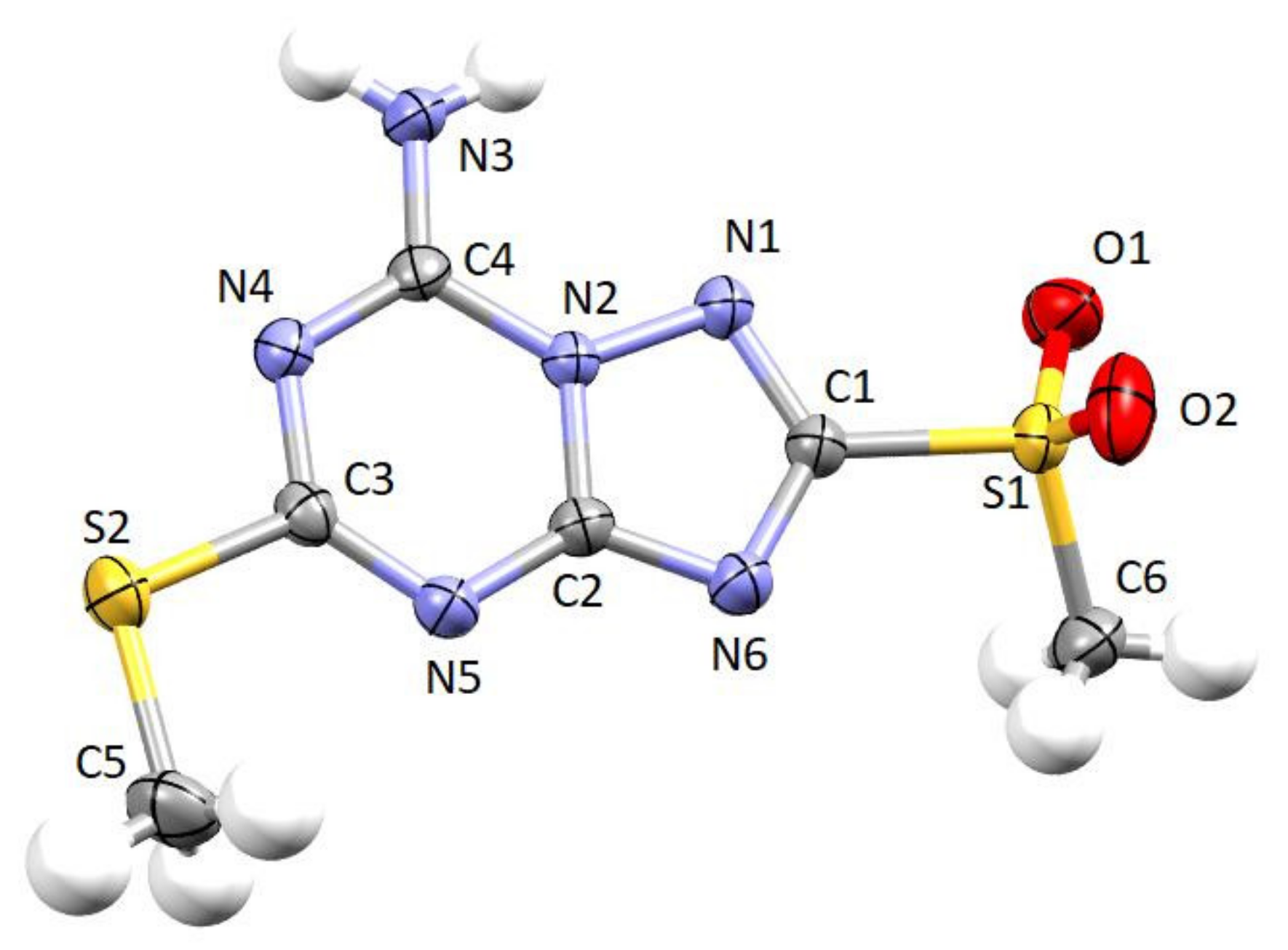
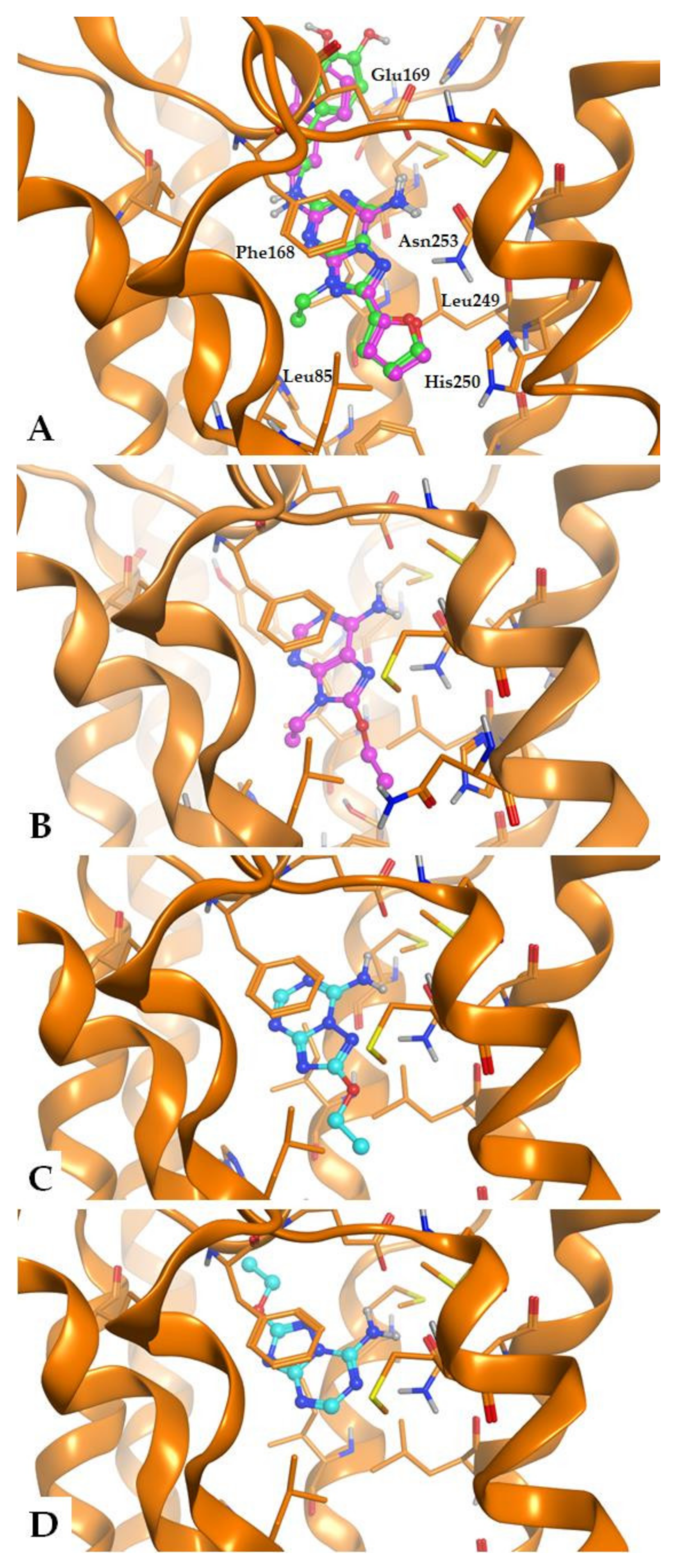
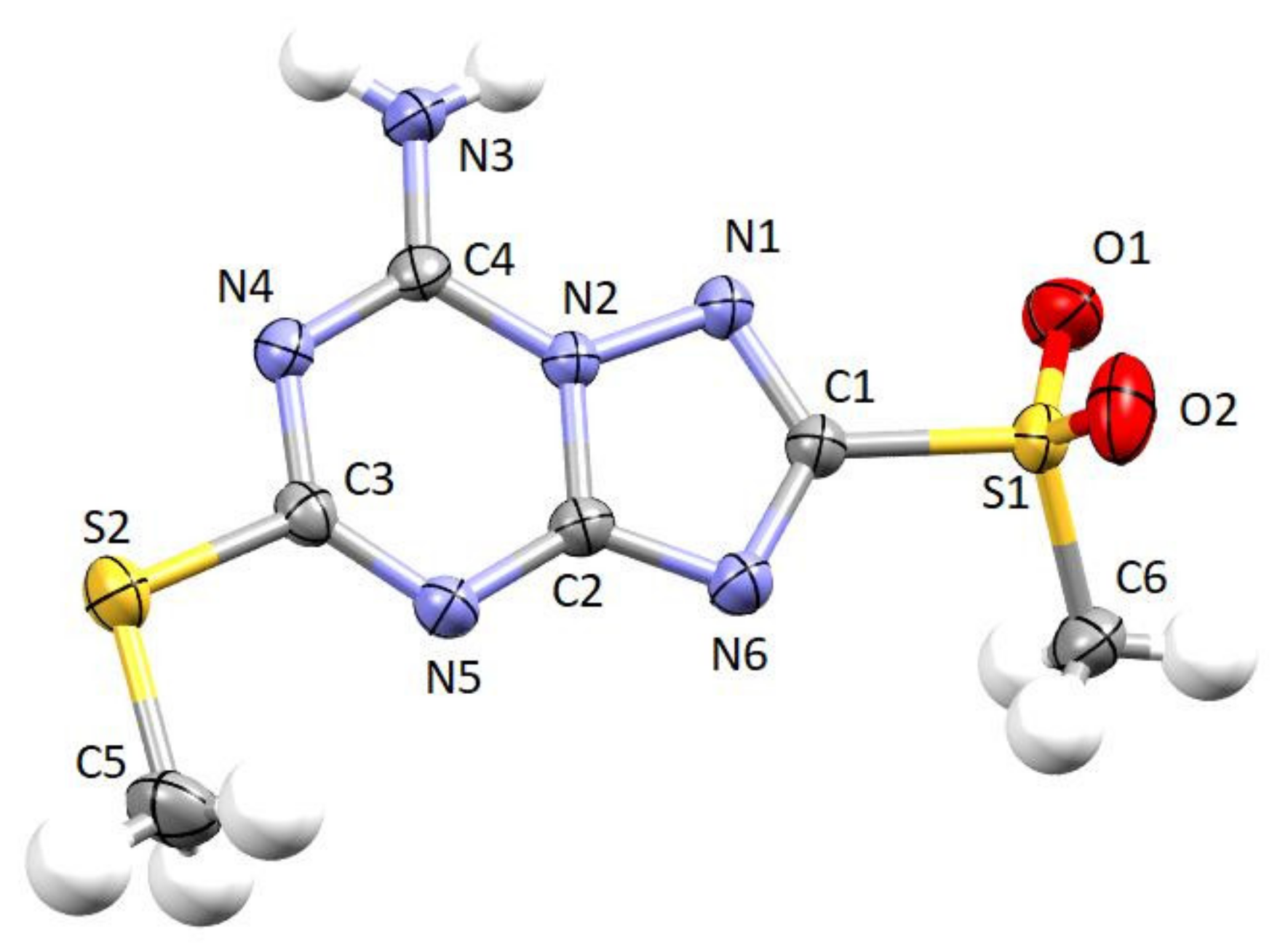
2.2. Crystallographic X-ray Analysis
2.3. Biological Evaluation
2.3.1. Binding Evaluation at ARs
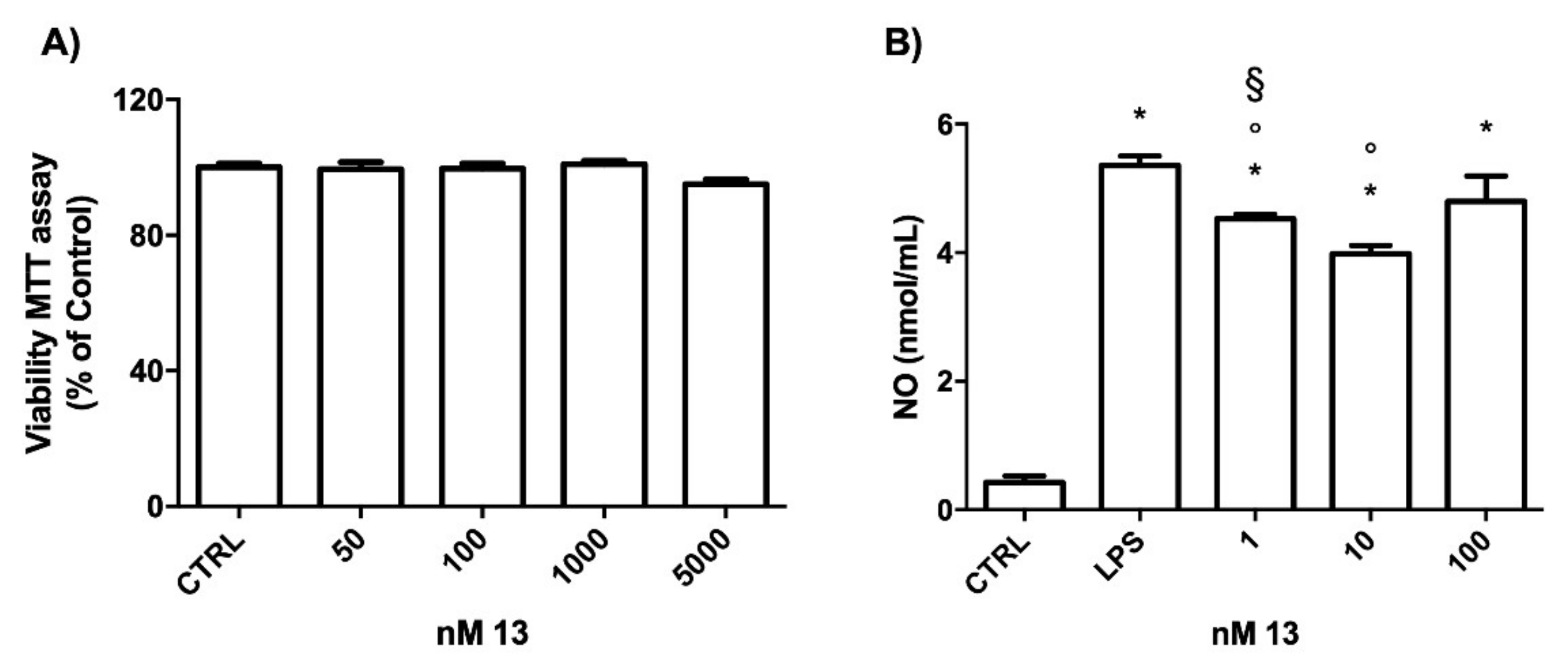
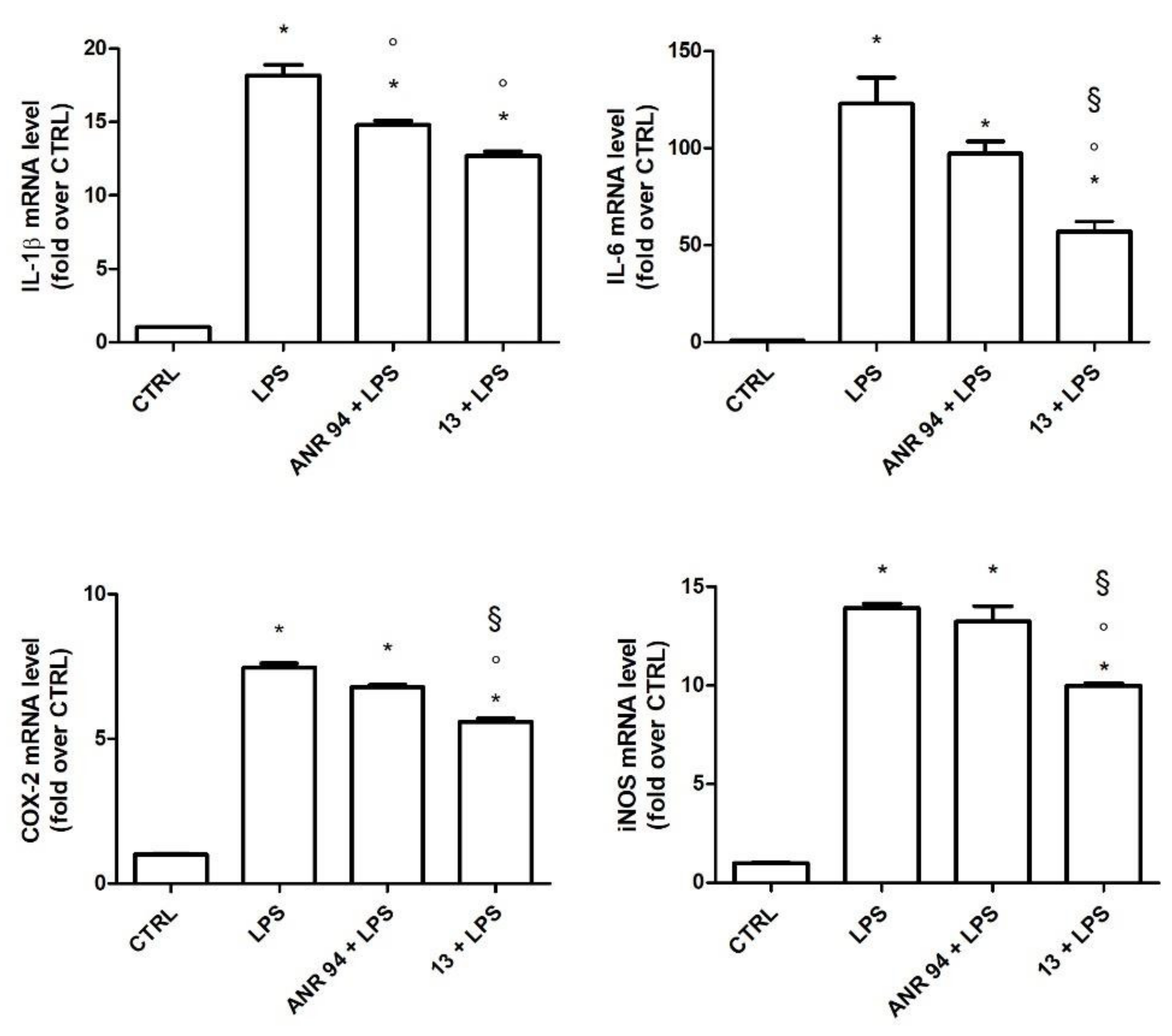
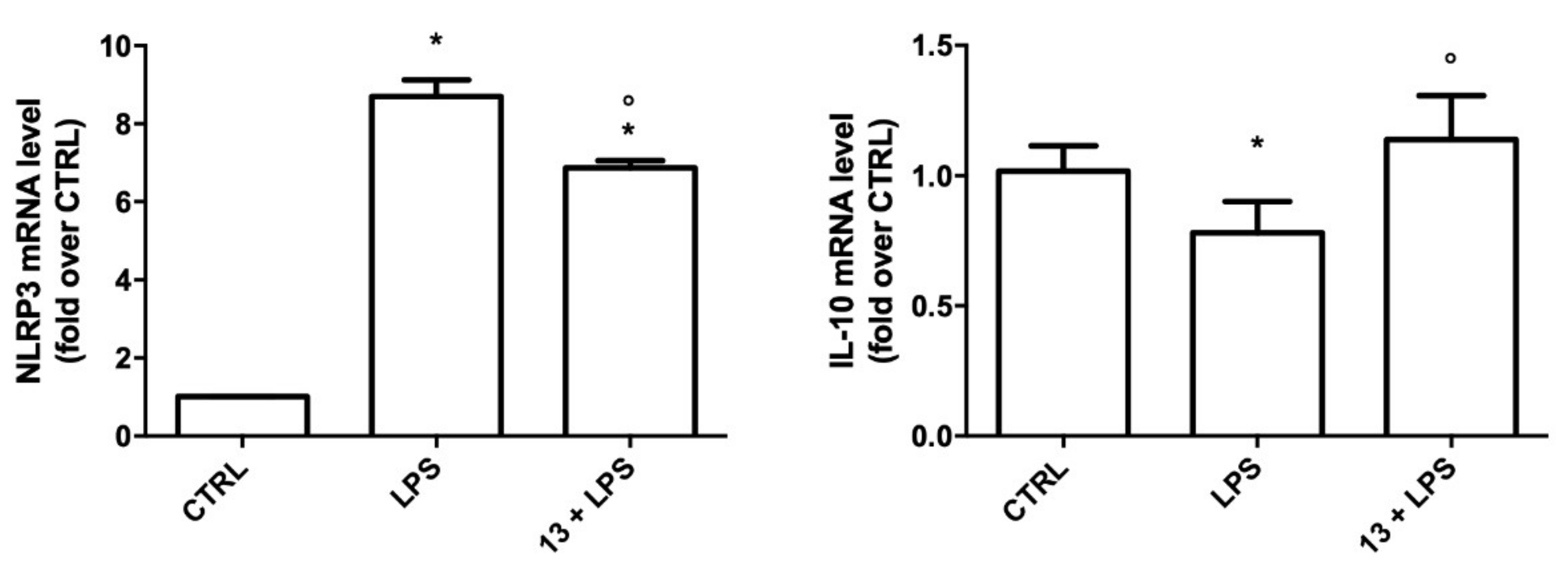
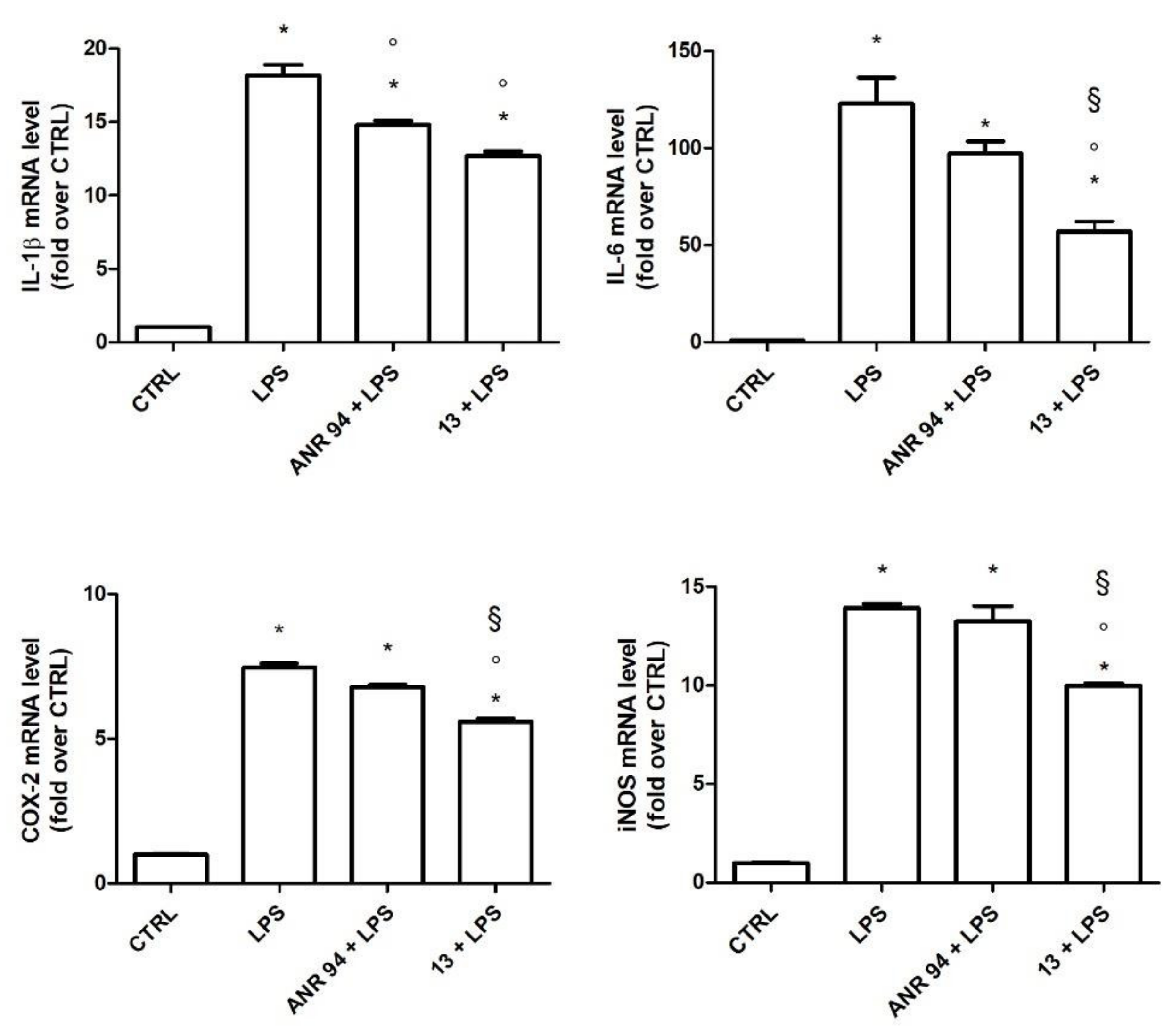
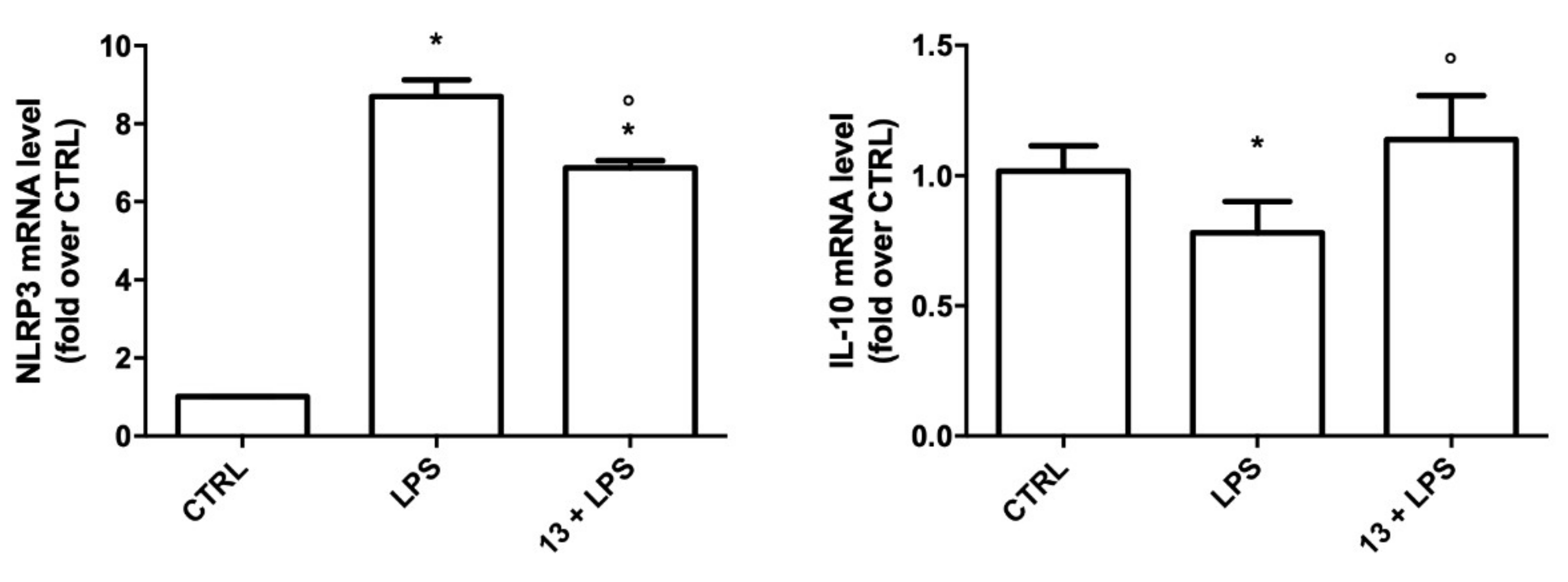
2.3.2. Study of the Anti-Inflammatory Activity of 13
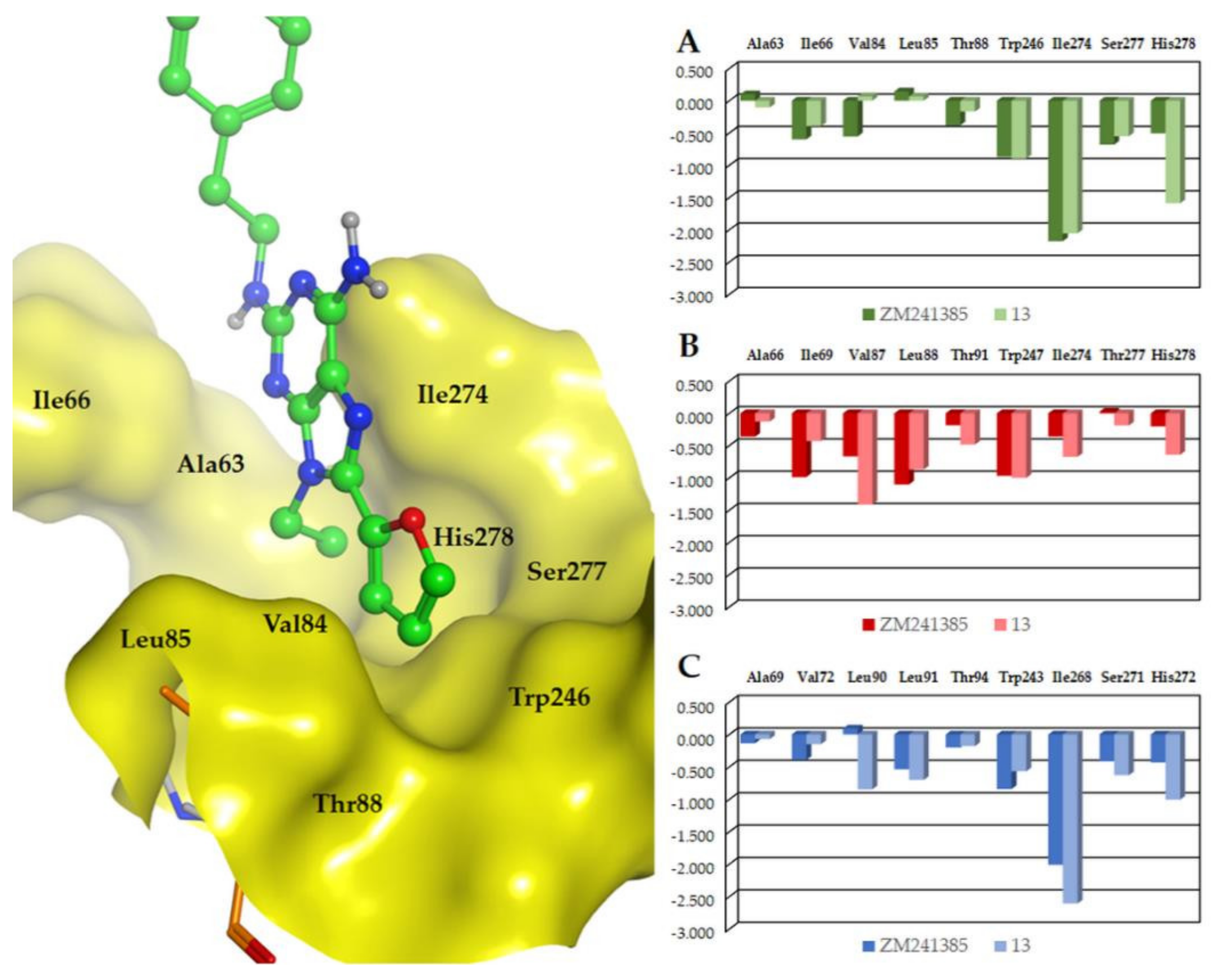
2.4. Molecular Modeling Studies
3. Materials and Methods
3.1. Chemical Synthesis
General Methods
3.2. Crystallographic Study
3.3. Biological Assays
3.3.1. Binding Evaluation at ARs
3.3.2. Anti-Inflammatory Activity Assays
3.4. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors-An update. Pharm. Rev 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.N.; Mustafa, S.J. Adenosine Receptors in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2009; Volume 193. [Google Scholar]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Cronstein, B. Regulation of inflammation by adenosine. Front. Immunol 2013, 4, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; Pacher, P.; Hasko, G. Adenosine and inflammation: It’s time to (re)solve the problem. Trends Pharmacol. Sci. 2022, 43, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M. Impact of Adenosine A2 Receptor Ligands on BCL2 Expression in Skeletal Muscle Cells. Appl. Sci. 2021, 11, 2272. [Google Scholar] [CrossRef]
- Rebola, N.; Simoes, A.P.; Canas, P.M.; Tome, A.R.; Andrade, G.M.; Barry, C.E.; Agostinho, P.M.; Lynch, M.A.; Cunha, R.A. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J. Neurochem. 2011, 117, 100–111. [Google Scholar] [CrossRef]
- Pinna, A.; Tronci, E.; Schintu, N.; Simola, N.; Volpini, R.; Pontis, S.; Cristalli, G.; Morelli, M. A new ethyladenine antagonist of adenosine A2A receptors: Behavioral and biochemical characterization as an antiparkinsonian drug. Neuropharmacology 2010, 58, 613–623. [Google Scholar] [CrossRef]
- Marti Navia, A.; Dal Ben, D.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Marques-Morgado, I.; Coelho, J.E.; Lopes, L.V.; Marucci, G.; Buccioni, M. Adenosine Receptors as Neuroinflammation Modulators: Role of A1 Agonists and A2A Antagonists. Cells 2020, 9, 1739. [Google Scholar] [CrossRef]
- Allan, S.M.; Tyrrell, P.J.; Rothwell, N.J. Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 2005, 5, 629–640. [Google Scholar] [CrossRef]
- Lai, T.T.; Kim, Y.J.; Ma, H.I.; Kim, Y.E. Evidence of Inflammation in Parkinson’s Disease and Its Contribution to Synucleinopathy. J. Mov. Disord. 2022, 15, 1–14. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Hauser, R.; Morelli, M.; Fredholm, B.B.; Chen, J.F. Adenosine, adenosine A2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A.; Serra, M.; Morelli, M.; Simola, N. Role of adenosine A2A receptors in motor control: Relevance to Parkinson’s disease and dyskinesia. J. Neural Transm. 2018, 125, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Shang, P.; Baker, M.; Banks, S.; Hong, S.I.; Chol, D.S. Emerging Nondopaminergic Medications for Parkinson’s Disease: Focusing on A2A Receptor Antagonists and GLP1 Receptor Agonists. J. Mov. Disord. 2021, 14, 193–203. [Google Scholar] [CrossRef]
- Hauser, R.A.; Hattori, N.; Fernandez, H.; Isaacson, S.H.; Mochizuki, H.; Rascol, O.; Stocchi, F.; Li, J.; Mori, A.; Nakajima, Y.; et al. Efficacy of Istradefylline, an Adenosine A2A Receptor Antagonist, as Adjunctive Therapy to Levodopa in Parkinson’s Disease: A Pooled Analysis of 8 Phase 2b/3 Trials. J. Parkinson’s Dis. 2021, 11, 1663–1675. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Mori, A.; Aradi, S.D.; Hauser, R.A. Istradefylline-A first generation adenosine A2A antagonist for the treatment of Parkinson’s disease. Expert Rev. Neurother. 2021, 21, 317–333. [Google Scholar] [CrossRef]
- Chen, J.F.; Cunha, R.A. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef]
- FDA Approves New Add-On Drug to Treat off Episodes in Adults with Parkinson’s Disease. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-add-drug-treat-episodes-adults-parkinsons-disease (accessed on 22 February 2022).
- Muller, C.E.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharm. 2011, 200, 151–199. [Google Scholar]
- Jacobson, K.A.; IJzerman, A.P.; Muller, C.E. Medicinal chemistry of P2 and adenosine receptors: Common scaffolds adapted for multiple targets. Biochem. Pharmacol. 2021, 187, 114311. [Google Scholar] [CrossRef]
- Williams, M.; Francis, J.; Ghai, G.; Braunwalder, A.; Psychoyos, S.; Stone, G.A.; Cash, W.D. Biochemical characterization of the triazoloquinazoline, CGS 15943, a novel, non-xanthine adenosine antagonist. J. Pharmacol. Exp. Ther. 1987, 241, 415–420. [Google Scholar]
- LeWitt, P.A.; Aradi, S.D.; Hauser, R.A.; Rascol, O. The challenge of developing adenosine A2A antagonists for Parkinson disease: Istradefylline, preladenant, and tozadenant. Parkinsonism Relat. Disord. 2020, 80, S54–S63. [Google Scholar] [CrossRef] [PubMed]
- Merck Provides Update on Phase III Clinical Program for Preladenant, the Company’s Investigational Parkinson’s Disease Medicine. Available online: www.merck.com/news/merck-provides-update-on-phase-iii-clinical-program-for-preladenant-the-companys-investigational-parkinsons-disease-medicine (accessed on 24 February 2022).
- Hauser, R.A.; Stocchi, F.; Rascol, O.; Huyck, S.B.; Capece, R.; Ho, T.W.; Sklar, P.; Lines, C.; Michelson, D.; Hewitt, D. Preladenant as an Adjunctive Therapy with Levodopa in Parkinson Disease Two Randomized Clinical Trials and Lessons Learned. JAMA Neurol. 2015, 72, 1491–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, T.; Olieric, N.; Cheng, R.; Brunle, S.; James, D.; Ozerov, D.; Gashi, D.; Vera, L.; Marsh, M.; Jaeger, K.; et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 2017, 8, 542. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Lebon, G. Human Adenosine A2A Receptor: Molecular Mechanism of Ligand Binding and Activation. Front. Pharm. 2017, 8, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Lambertucci, C.; Spinaci, A.; Buccioni, M.; Dal Ben, D.; Ngouadjeu Ngnintedem, M.A.; Kachler, S.; Marucci, G.; Klotz, K.N.; Volpini, R. New A2A adenosine receptor antagonists: A structure-based upside-down interaction in the receptor cavity. Bioorg. Chem. 2019, 92, 103183. [Google Scholar] [CrossRef]
- Lambertucci, C.; Marucci, G.; Catarzi, D.; Colotta, V.; Francucci, B.; Spinaci, A.; Varano, F.; Volpini, R. A2A Adenosine Receptor Antagonists and their Potential in Neurological Disorders. Curr. Med. Chem. 2022. accepted. [Google Scholar] [CrossRef]
- Lambertucci, C.; Antonini, I.; Buccioni, M.; Dal Ben, D.; Kachare, D.D.; Volpini, R.; Klotz, K.N.; Cristalli, G. 8-Bromo-9-alkyl adenine derivatives as tools for developing new adenosine A2A and A2B receptors ligands. Bioorg. Med. Chem. 2009, 17, 2812–2822. [Google Scholar] [CrossRef]
- Camaioni, E.; Costanzi, S.; Vittori, S.; Volpini, R.; Klotz, K.N.; Cristalli, G. New substituted 9-alkylpurines as adenosine receptor ligands. Bioorg. Med. Chem. 1998, 6, 523–533. [Google Scholar] [CrossRef]
- Jorg, M.; Agostino, M.; Yuriev, E.; Mak, F.S.; Miller, N.D.; White, J.M.; Scammells, P.J.; Capuano, B. Synthesis, molecular structure, NMR spectroscopic and computational analysis of a selective adenosine A2A antagonist, ZM241385. Struct. Chem. 2013, 24, 1241–1251. [Google Scholar] [CrossRef]
- Caulkett, P.W.R.; Jones, G.; McPartlin, M.; Renshaw, N.D.; Stewart, S.K.; Wright, B. Adenine isosteres with bridgehead nitrogen. Part 1. Two independent syntheses of the [1,2,4]triazolo[1,5-a][1,3,5]triazine ring system leading to a range of substituents in the 2, 5 and 7 positions. J. Chem. Soc. Perkin Trans. 1995, 1, 801–808. [Google Scholar] [CrossRef]
- Klotz, K.N.; Falgner, N.; Kachler, S.; Lambertucci, C.; Vittori, S.; Volpini, R.; Cristalli, G. [3H]HEMADO-a novel tritiated agonist selective for the human adenosine A3 receptor. Eur. J. Pharmacol. 2007, 556, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Falsini, M.; Ceni, C.; Catarzi, D.; Varano, F.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Marti Navia, A.; Volpini, R.; Colotta, V. New 8-amino-1,2,4-triazolo[4,3-a]pyrazin-3-one derivatives. Evaluation of different moieties on the 6-aryl ring to obtain potent and selective human A2A adenosine receptor antagonists. Bioorg. Med. Chem. Lett. 2020, 30, 127126. [Google Scholar] [CrossRef] [PubMed]
- Buccioni, M.; Marucci, G.; Dal Ben, D.; Giacobbe, D.; Lambertucci, C.; Soverchia, L.; Thomas, A.; Volpini, R.; Cristalli, G. Innovative functional cAMP assay for studying G protein-coupled receptors: Application to the pharmacological characterization of GPR17. Purinergic Signal. 2011, 7, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, K.N.; Kachler, S.; Lambertucci, C.; Vittori, S.; Volpini, R.; Cristalli, G. 9-Ethyladenine derivatives as adenosine receptor antagonists: 2- and 8-substitution results in distinct selectivities. Naunyn Schmiedebergs Arch. Pharm. 2003, 367, 629–634. [Google Scholar] [CrossRef]
- He, J.; Zhu, G.; Wang, G.; Zhang, F. Oxidative Stress and Neuroinflammation Potentiate Each Other to Promote Progression of Dopamine Neurodegeneration. Oxid. Med. Cell. Longev. 2020, 2020, 6137521. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Wu, Y.; Zhang, S.; Ju, Q.; Yang, Y.; Jin, Y.; Shi, H.; Sun, C. CD44 deficiency represses neuroinflammation and rescues dopaminergic neurons in a mouse model of Parkinson’s disease. Pharmacol. Res. 2022, 177, 106133. [Google Scholar] [CrossRef]
- Badanjak, K.; Fixemer, S.; Smajic, S.; Skupin, A.; Grunewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef]
- Tansey, M.G.; McCoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Gyoneva, S.; Shapiro, L.; Lazo, C.; Garnier-Amblard, E.; Smith, Y.; Miller, G.W.; Traynelis, S.F. Adenosine A2A receptor antagonism reverses inflammation-induced impairment of microglial process extension in a model of Parkinson’s disease. Neurobiol. Dis. 2014, 67, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehle, M.S.; West, A.B. M1 and M2 immune activation in Parkinson’s Disease: Foe and ally? Neuroscience 2015, 302, 59–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.M.; Jiang, J.; Wilson, B.; Zhang, W.; Hong, J.S.; Liu, B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. J. Neurochem. 2002, 81, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinson’s Dis. 2017, 3, 30. [Google Scholar] [CrossRef]
- Ding, R.R.; Chen, W.; Guo, C.Y.; Liao, W.T.; Yang, X.; Liao, F.E.; Lin, J.M.; Mei, H.F.; Zeng, Y. Dangguishaoyao-San attenuates LPS-induced neuroinflammation via the TLRs/NF-kappaB signaling pathway. Biomed. Pharmacother. 2018, 105, 187–194. [Google Scholar] [CrossRef]
- Lee, D.G.; Nam, B.R.; Huh, J.W.; Lee, D.S. Isoliquiritigenin Reduces LPS-Induced Inflammation by Preventing Mitochondrial Fission in BV-2 Microglial Cells. Inflammation 2021, 44, 714–724. [Google Scholar] [CrossRef]
- Angeloni, S.; Freschi, M.; Marrazzo, P.; Hrelia, S.; Beghelli, D.; Juan-Garcia, A.; Juan, C.; Caprioli, G.; Sagratini, G.; Angeloni, C. Antioxidant and Anti-Inflammatory Profiles of Spent Coffee Ground Extracts for the Treatment of Neurodegeneration. Oxid. Med. Cell. Longev. 2021, 2021, 6620913. [Google Scholar] [CrossRef]
- Henn, A.; Lund, S.; Hedtjarn, M.; Schrattenholz, A.; Porzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 2009, 26, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Oishi, S.; Takano, R.; Tamura, S.; Tani, S.; Iwaizumi, M.; Hamaya, Y.; Takagaki, K.; Nagata, T.; Seto, S.; Horii, T.; et al. M2 polarization of murine peritoneal macrophages induces regulatory cytokine production and suppresses T-cell proliferation. Immunology 2016, 149, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.K.Y.; Segala, E.; Robertson, N.; Deflorian, F.; Dore, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 2017, 25, 1275–1285.e4. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE 2020.09). Chemical Computing Group, Inc.: Montreal, QC, Canada 1255 University St.: Suite 1600, H3B 3X3. Available online: https://www.chemcomp.com/Products.htm (accessed on 7 January 2022).
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Marucci, G.; Santinelli, C.; Spinaci, A.; Thomas, A.; Volpini, R. Simulation and Comparative Analysis of Different Binding Modes of Non-nucleoside Agonists at the A2A Adenosine Receptor. Mol. Inform. 2016, 35, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Marucci, G.; Thomas, A.; Volpini, R.; Cristalli, G. Molecular modeling study on potent and selective adenosine A3 receptor agonists. Bioorg. Med. Chem. 2010, 18, 7923–7930. [Google Scholar] [CrossRef]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Thomas, A.; Volpini, R. Simulation and comparative analysis of binding modes of nucleoside and non-nucleoside agonists at the A2B adenosine receptor. In Silico Pharmacol. 2013, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Cpd | R1 | R2 | A1AR 1 | A2AAR 2 | A2BAR 3 | A3AR 4 |
| ZM241385 (4) | pHO-Ph(CH2)2NH | 2-Furyl | 774 [38] | 1.6 | 75 | 743 [38] |
| ANR 94 (5) | H | OCH2CH3 | 2400 | 46 | >30,000 | 21,000 [39] |
| 7 | pCH3O-Ph(CH2)2NH | H | 5840 (±558) | 1460 (±255) | >30,000 | 23,600 (±3500) |
| 8 | pCH3O-Ph(CH2)2NH | Br | 545 (±114) | 34 (±7.6) | 4260 (±1795) | 4030 (±825) |
| 9 | pHO-Ph(CH2)2NH | Br | 411 (±104) | 44 (±7.03) | 393 (±133) | 3810 (±205) |
| 10 | pCH3O-Ph(CH2)2NH | OH | 1390 (±280) | >30,000 | >30,000 | 777 (±578) |
| 11 | pHO-Ph(CH2)2NH | OH | 7054 (±419) | >30,000 | >30,000 | >30,000 |
| 12 | pCH3O-Ph(CH2)2NH | 2-Furyl | 2.6 (±0.34) | 1.0 (±0.06) | >30,000 | 16 (±5.02) |
| 13 | pHO-Ph(CH2)2NH | 2-Furyl | 51 (±11) | 1.8 (±0.01) | 1434 (±728) | 12 (±2.1) |
| 17 | SCH3 | SO2CH3 | 5720 (±318) | 3238 (±743) | n.d. 5 | 14,710 (±1600) |
| 18 | SCH3 | OCH2CH3 | 4568 (±953) | 583 (±88) | n.d. 5 | 9716 (±775) |
| 19 | OCH2CH3 | OCH2CH3 | 6360 (±1298) | 1061 (±217) | n.d. 5 | 4786 (±929) |
| 20 | H | OCH2CH3 | 9769 (±185) | 2029 (±477) | >30,000 | 1019 (±172) |
| 21 | pHO-Ph(CH2)2NH | OCH2CH3 | 5507 (±364) | 178 (±49) | >30,000 | 6130 (±190) |
| Gene | 5′-Forward-3′ | 5′-Reverse-3′ |
|---|---|---|
| IL-1β | GTTCCCATTAGACAACTGCACTACAG | GTCGTTGCTTGGTTCTCCTTGTA |
| IL-6 | TCCTTCAGAGAGATACAGAAAC | TTCTGTGACTCCAGCTTATC |
| COX-2 | TGGGGTGATGAGCAACTATT | AAGGAGCTCTGGGTCAAACT |
| iNOS | CCTCCTCCACCCTACCAAGT | CACCCAAAGTGCTTCAGTCA |
| NLRP3 | GATGCTGGAATTAGACAACTG | GTACATTTCACCCAACTGTAG |
| IL-10 | CAGGACTTTAAGGGTTACTTG | ATTTTCACAGGGGAGAAATC |
| GAPDH | ACCACAGTCCATGCCATCAC | TCCACCACCCTGTTGCTGTA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinaci, A.; Lambertucci, C.; Buccioni, M.; Dal Ben, D.; Graiff, C.; Barbalace, M.C.; Hrelia, S.; Angeloni, C.; Tayebati, S.K.; Ubaldi, M.; et al. A2A Adenosine Receptor Antagonists: Are Triazolotriazine and Purine Scaffolds Interchangeable? Molecules 2022, 27, 2386. https://doi.org/10.3390/molecules27082386
Spinaci A, Lambertucci C, Buccioni M, Dal Ben D, Graiff C, Barbalace MC, Hrelia S, Angeloni C, Tayebati SK, Ubaldi M, et al. A2A Adenosine Receptor Antagonists: Are Triazolotriazine and Purine Scaffolds Interchangeable? Molecules. 2022; 27(8):2386. https://doi.org/10.3390/molecules27082386
Chicago/Turabian StyleSpinaci, Andrea, Catia Lambertucci, Michela Buccioni, Diego Dal Ben, Claudia Graiff, Maria Cristina Barbalace, Silvana Hrelia, Cristina Angeloni, Seyed Khosrow Tayebati, Massimo Ubaldi, and et al. 2022. "A2A Adenosine Receptor Antagonists: Are Triazolotriazine and Purine Scaffolds Interchangeable?" Molecules 27, no. 8: 2386. https://doi.org/10.3390/molecules27082386
APA StyleSpinaci, A., Lambertucci, C., Buccioni, M., Dal Ben, D., Graiff, C., Barbalace, M. C., Hrelia, S., Angeloni, C., Tayebati, S. K., Ubaldi, M., Masi, A., Klotz, K.-N., Volpini, R., & Marucci, G. (2022). A2A Adenosine Receptor Antagonists: Are Triazolotriazine and Purine Scaffolds Interchangeable? Molecules, 27(8), 2386. https://doi.org/10.3390/molecules27082386