
Revealing Intra- and Intermolecular Interactions Determining Physico-Chemical Features of Selected Quinolone Carboxylic Acid Derivatives


Abstract
:
1. Introduction
2. Computational Methodology
2.1. Density Functional Theory (DFT) Procedure for Monomers and Dimers
2.2. Symmetry-Adapted Perturbation Theory (SAPT) Procedure for Dimers
2.3. Car–Parrinello Molecular Dynamics (CPMD) in Vacuo and the Crystalline Phase
2.4. Path Integral Molecular Dynamics (PIMD) in Vacuo and the Crystalline Phase
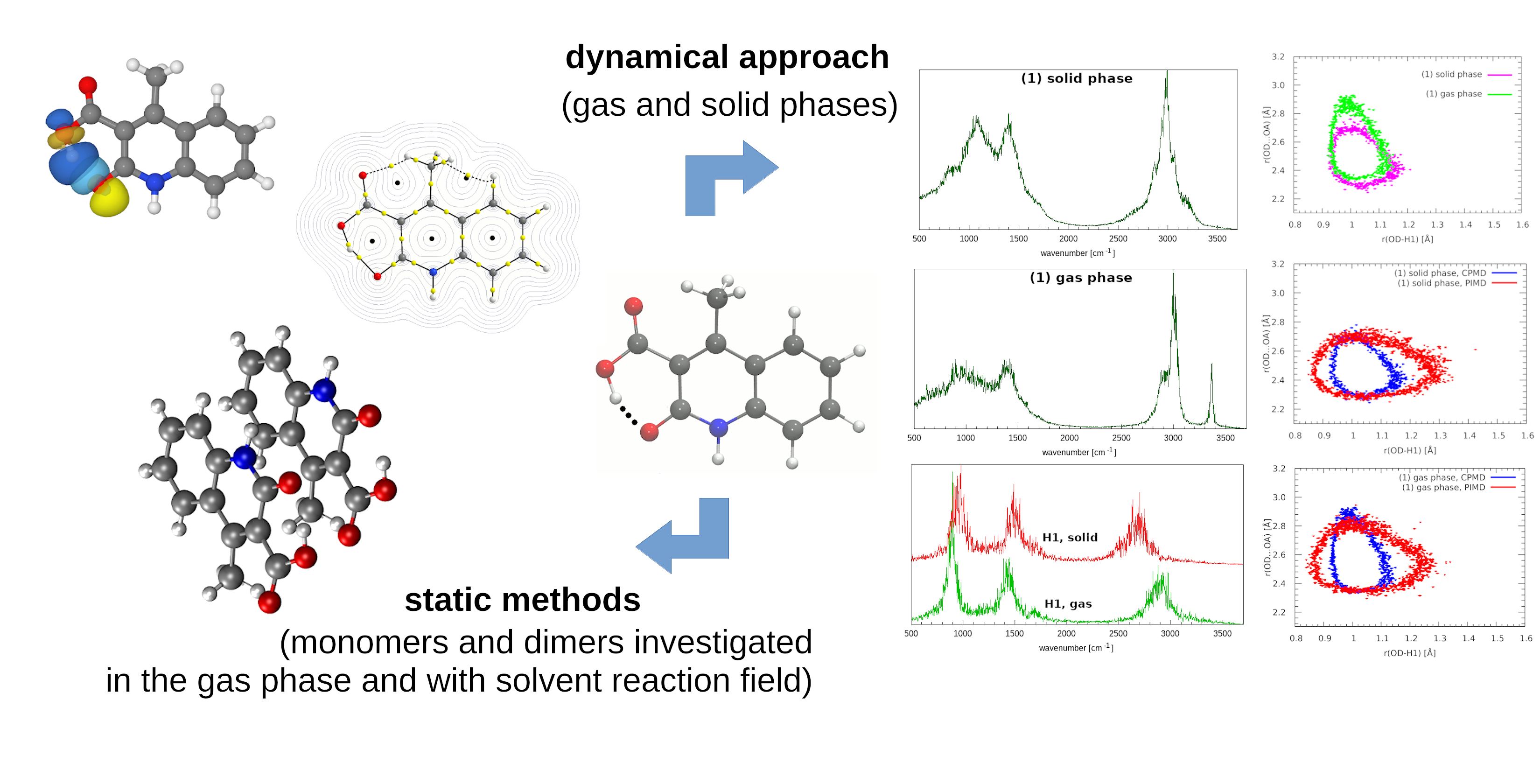
3. Results and Discussion
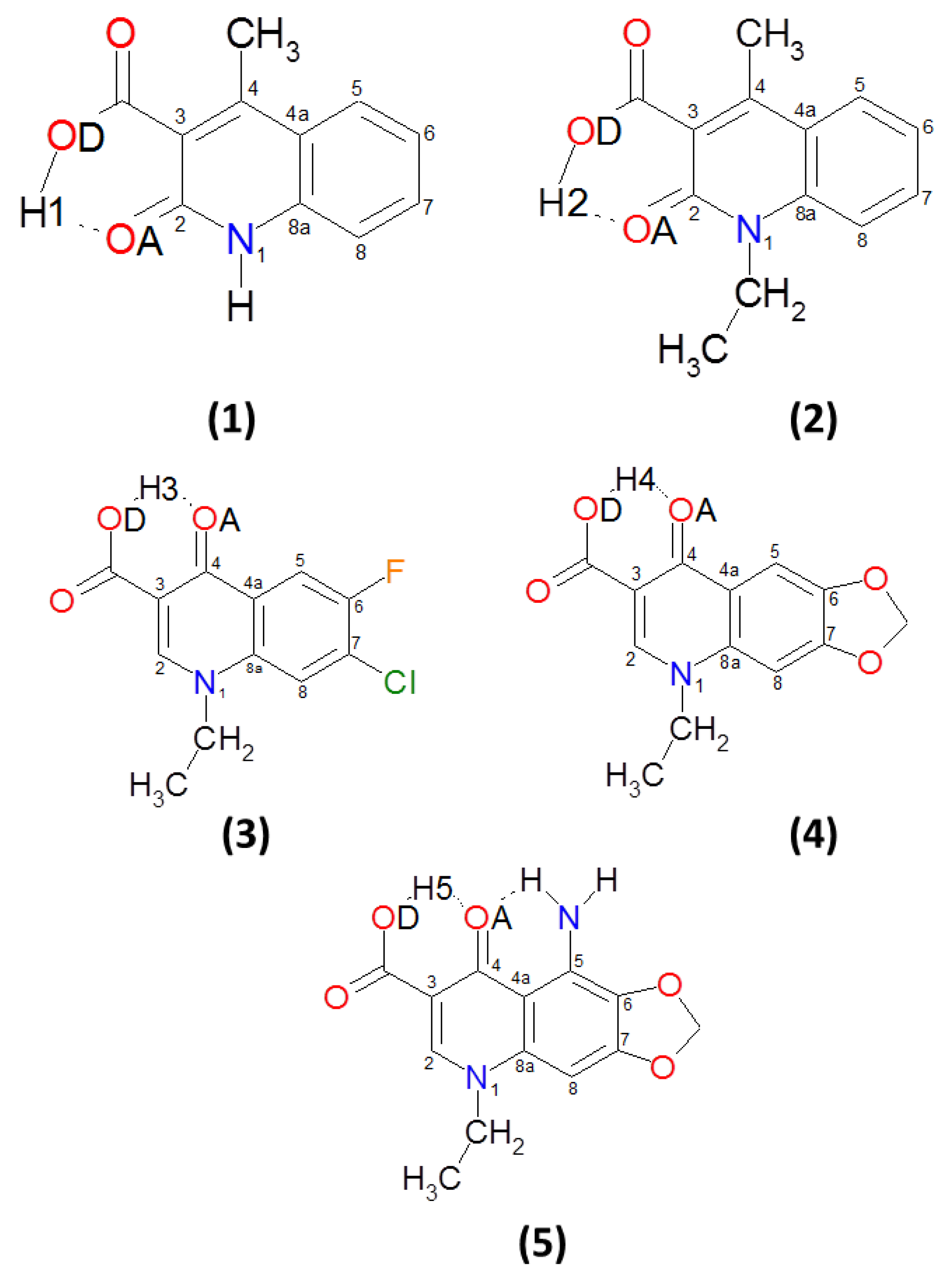
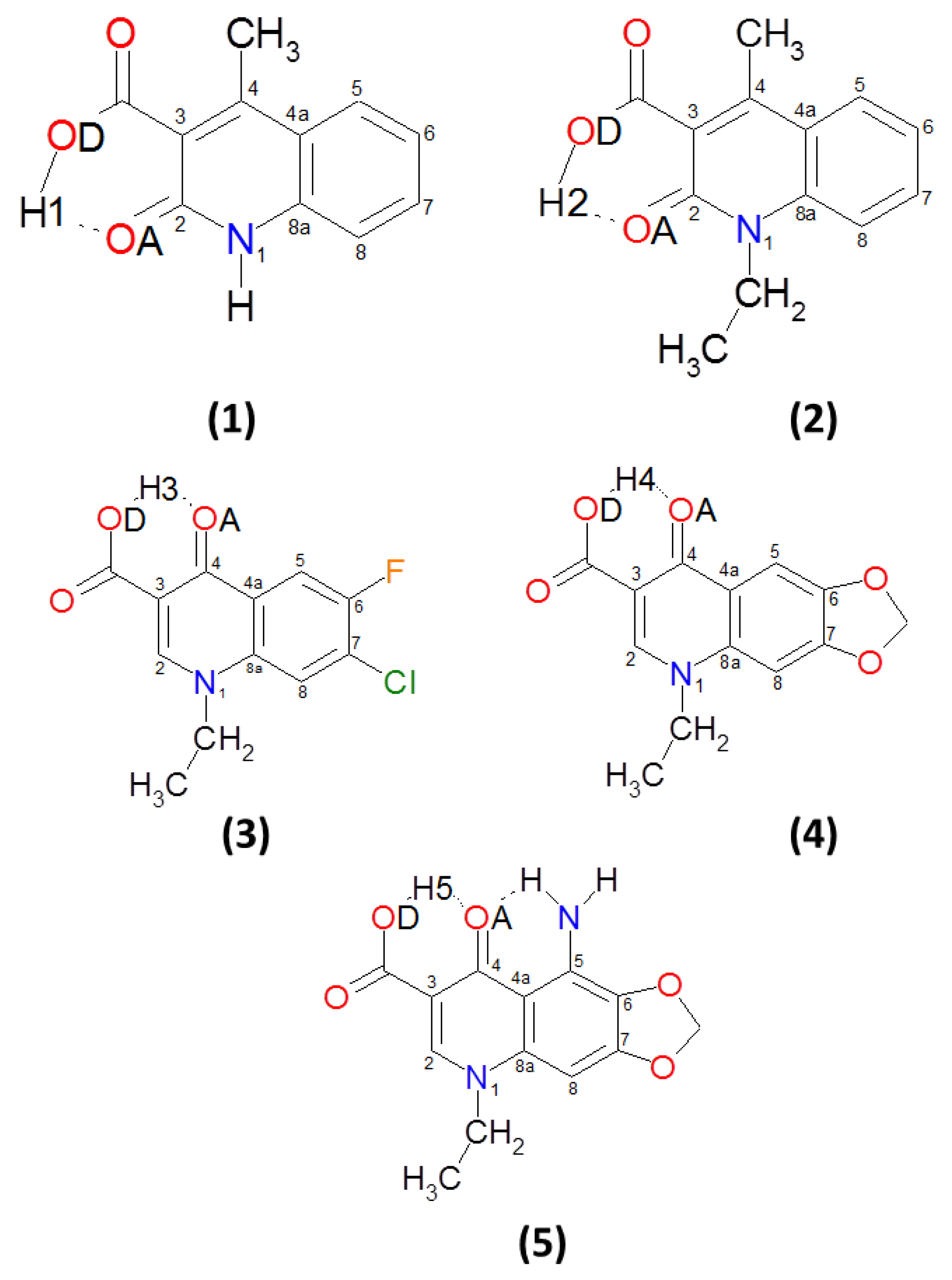
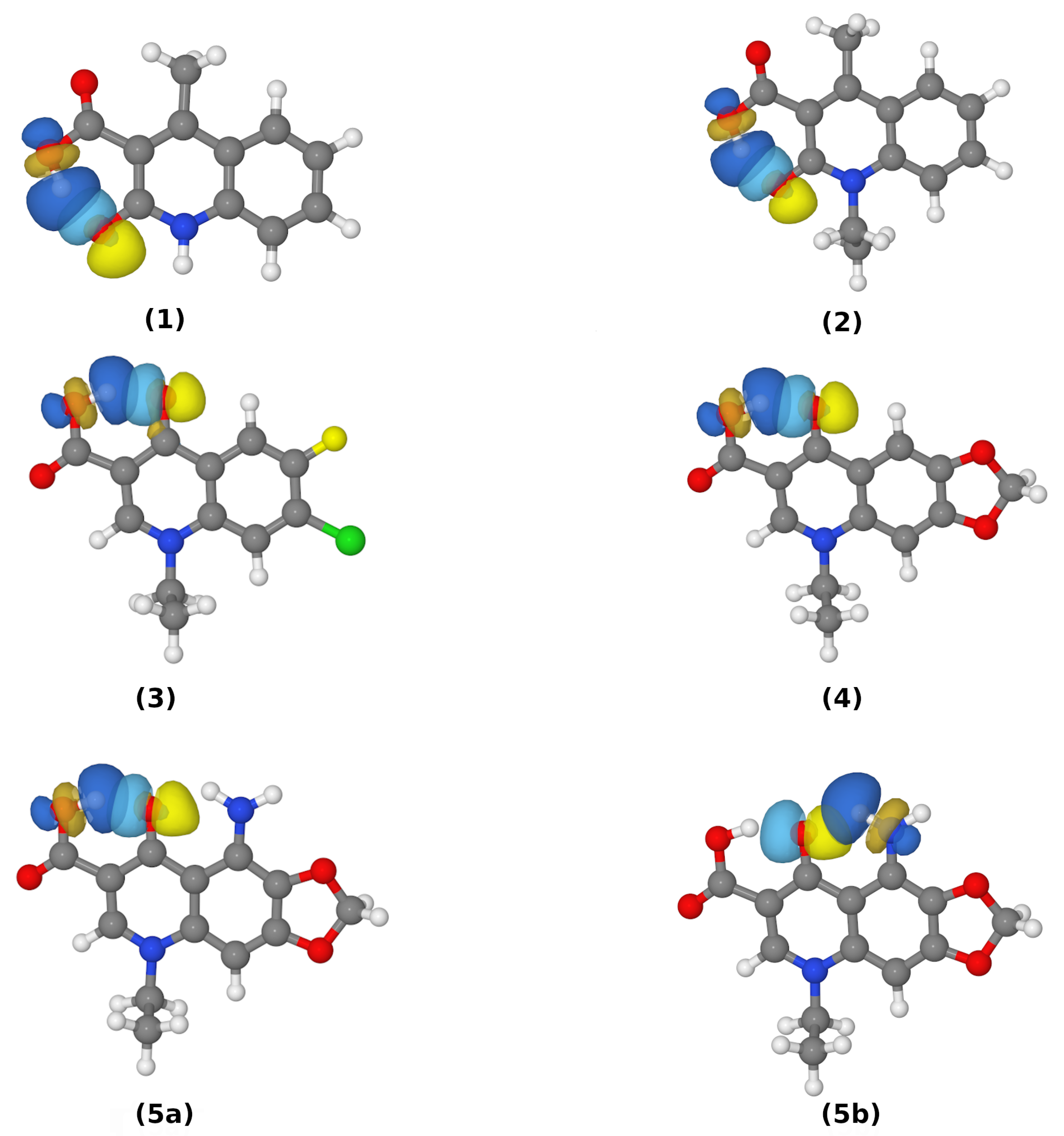
3.1. Structural, Energetic and Electronic Aspects of Intramolecular Hydrogen Bonds in the Investigated Quinolone Carboxylic Acid Derivatives
3.2. Intramolecular Hydrogen Bonds—Energy Estimation
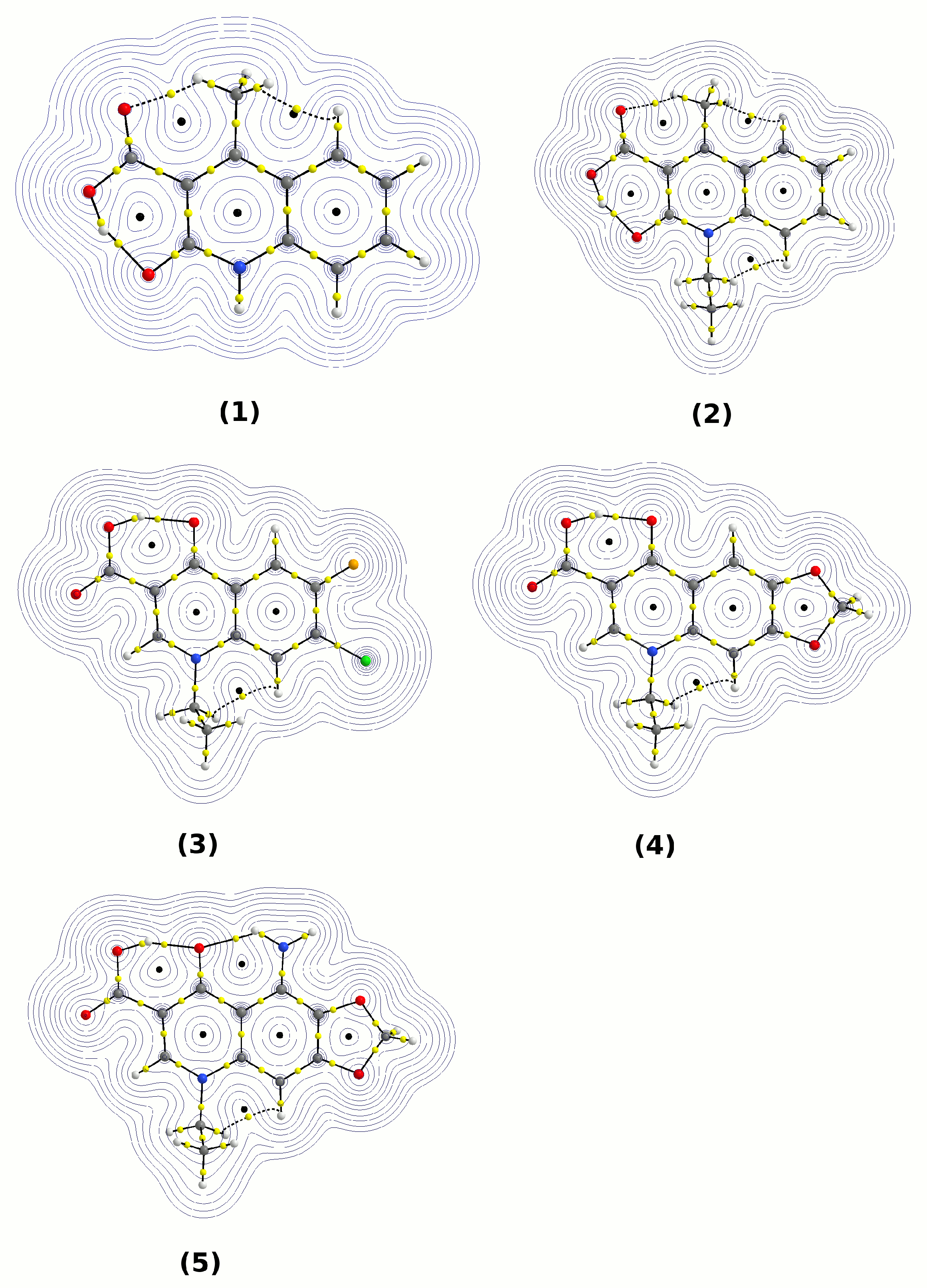
3.3. Atoms in Molecules (AIM) Analysis of the Electronic Structure
3.4. Natural Population Analysis (NPA)
3.5. Natural Bond Orbitals and Wiberg Bond Indices (WBI) Analysis of Hydrogen Bonds of Monomers in the Gas Phase and in the PCM
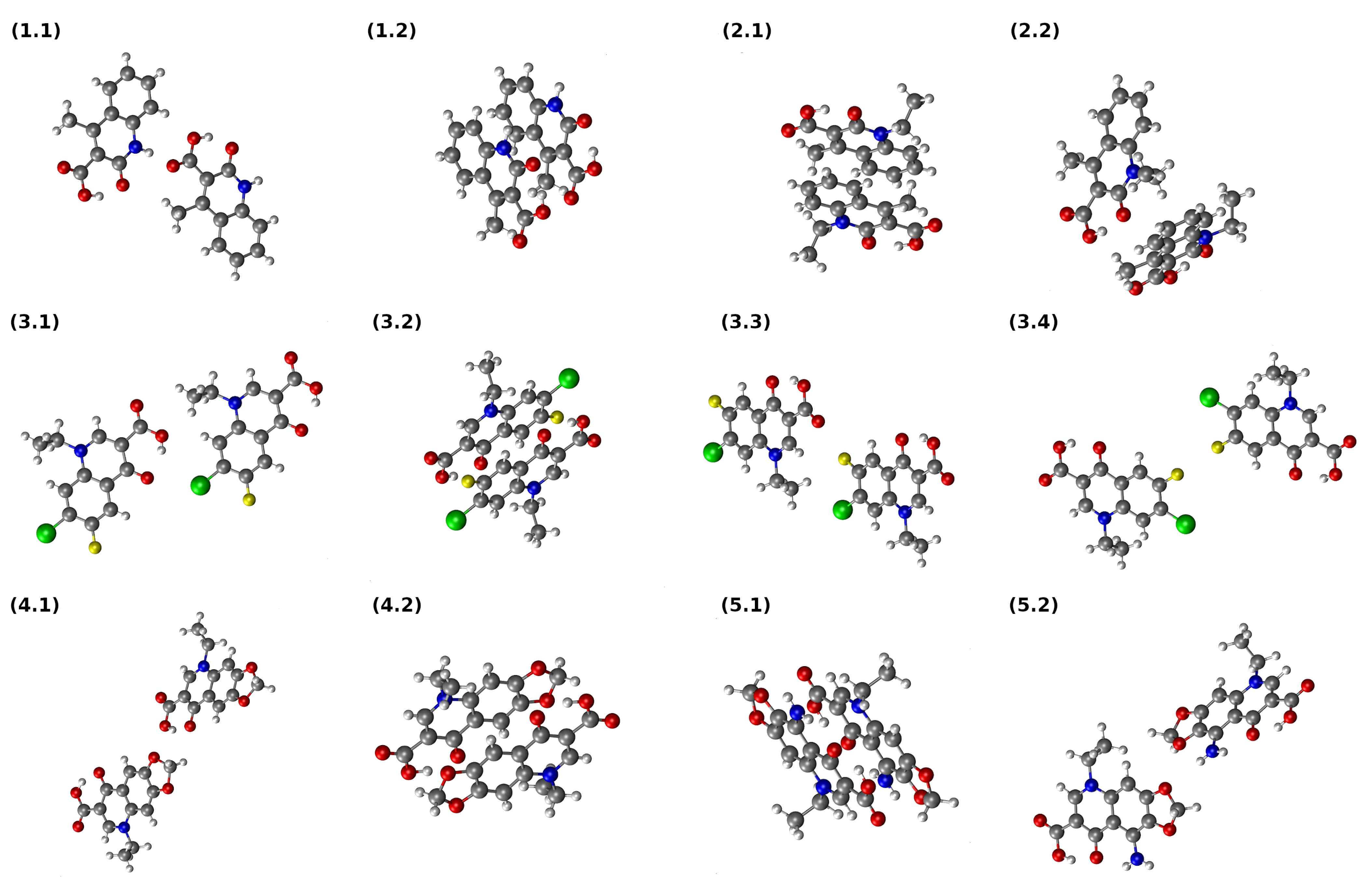
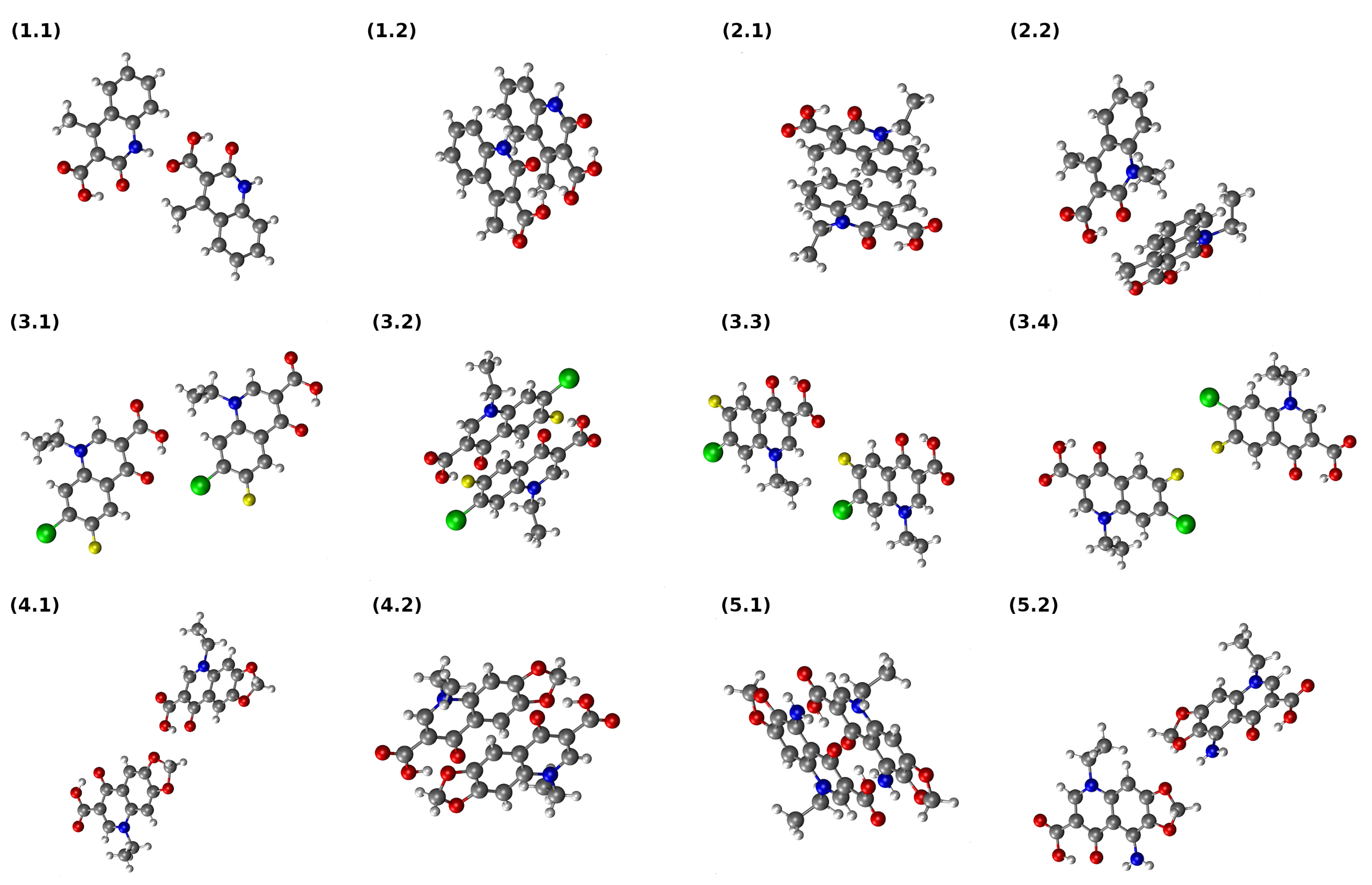
3.6. Interaction Energy in Dimers of Quinolone Acid Derivatives Based on Symmetry-Adapted Perturbation Theory (SAPT) and Atoms in Molecules (AIM) Approaches
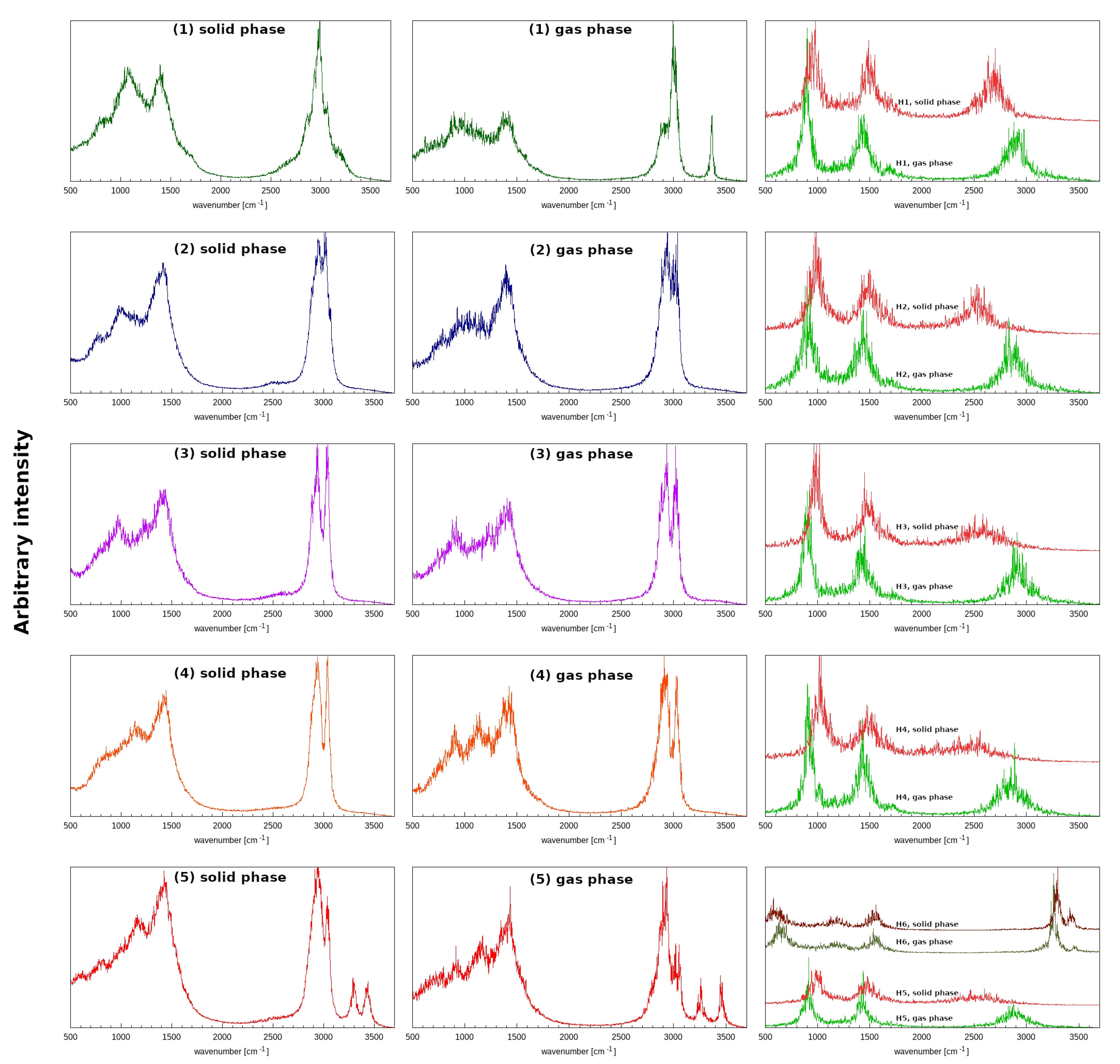
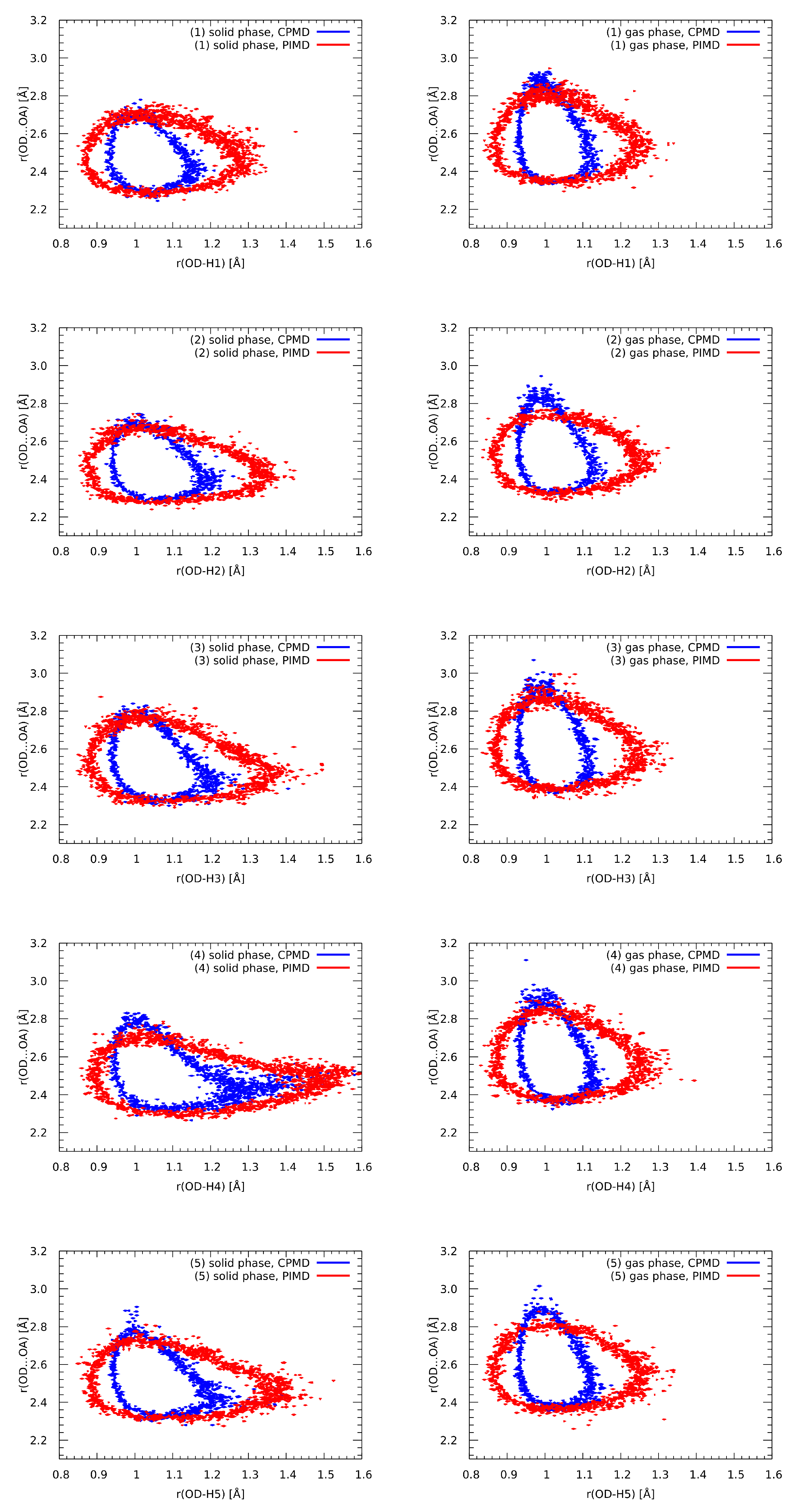
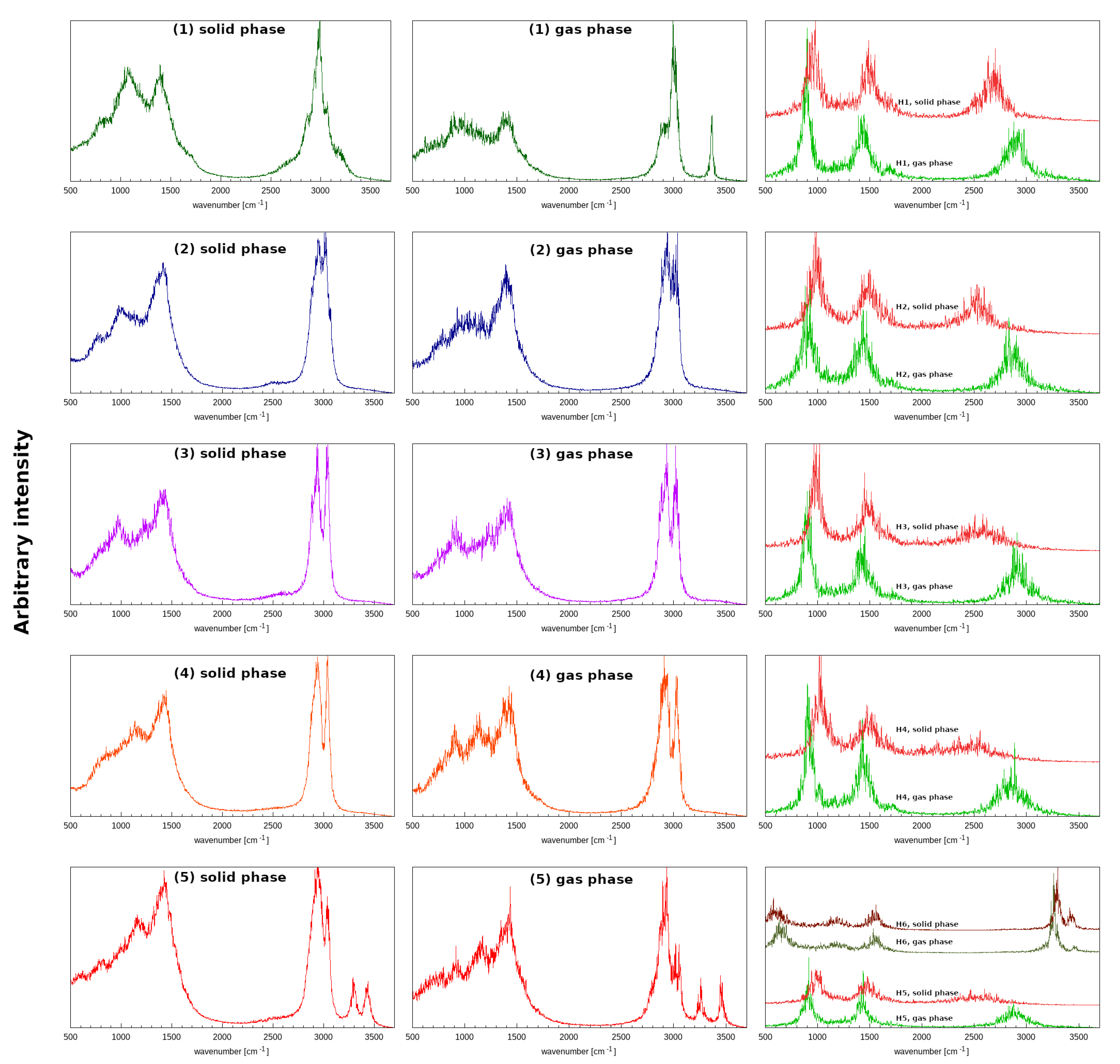
3.7. Geometric and Spectroscopic Characteristics of the Intramolecular Hydrogen Bonds Based on Car–Parrinello Molecular Dynamics (CPMD)
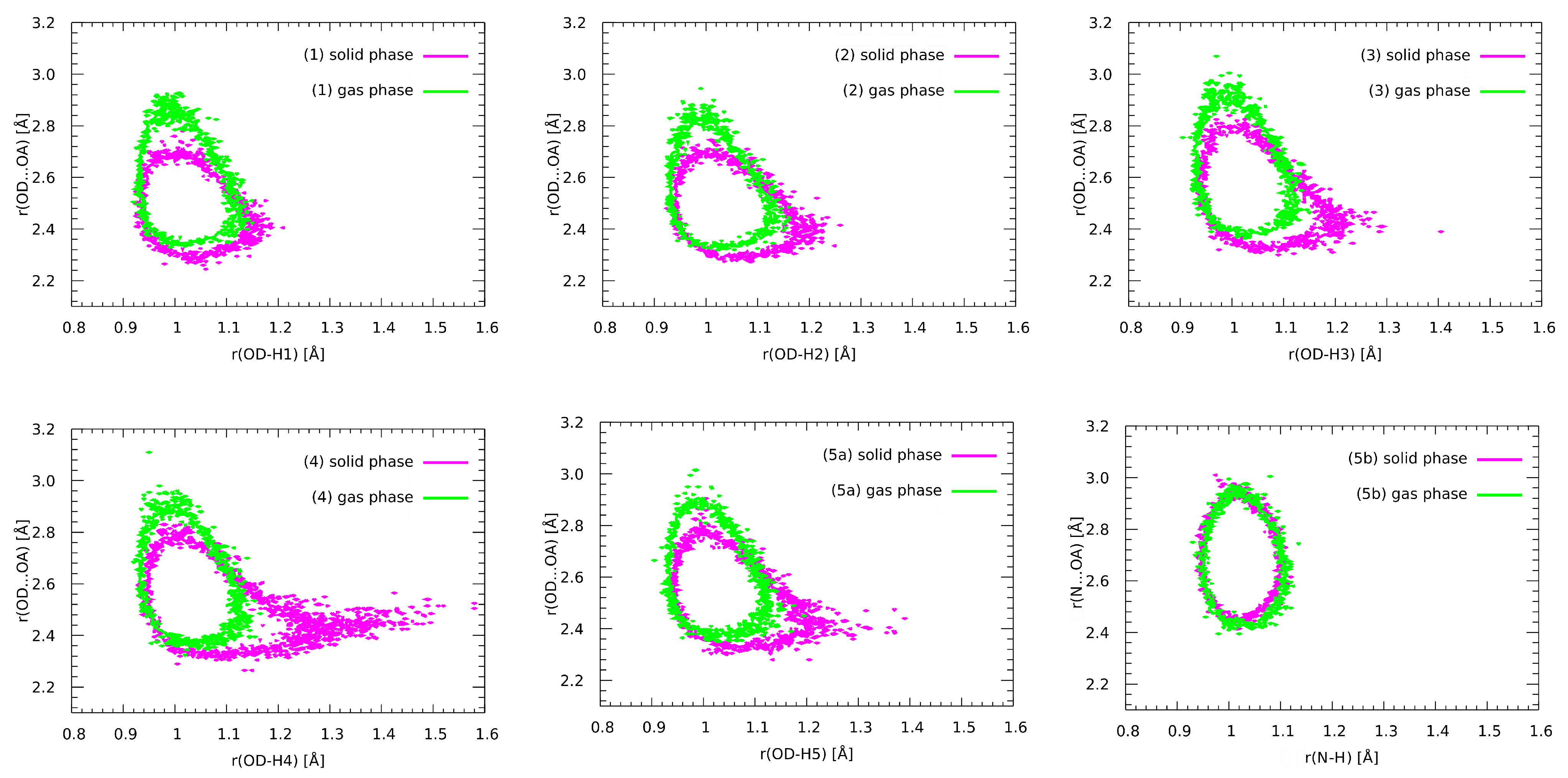
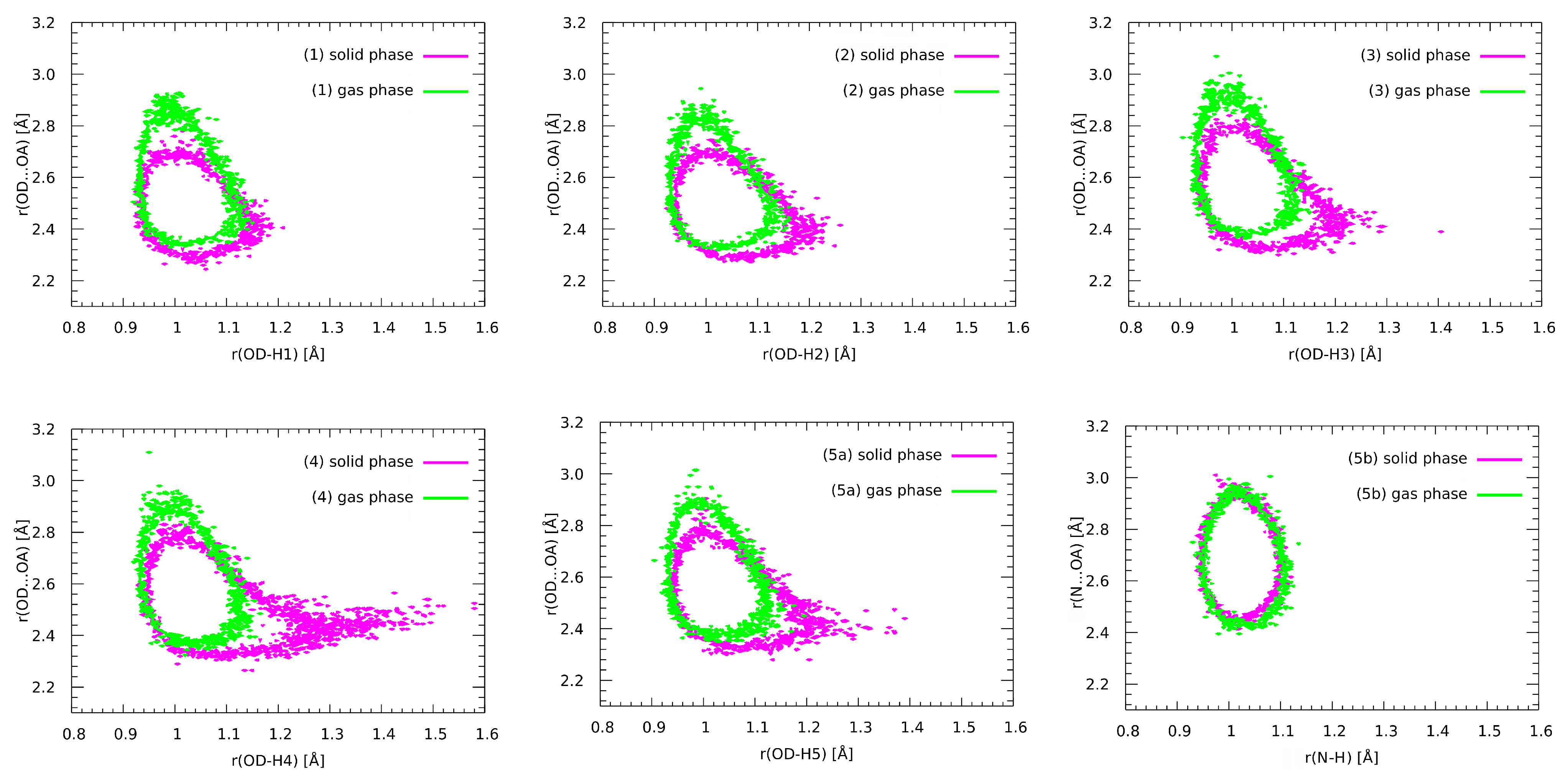
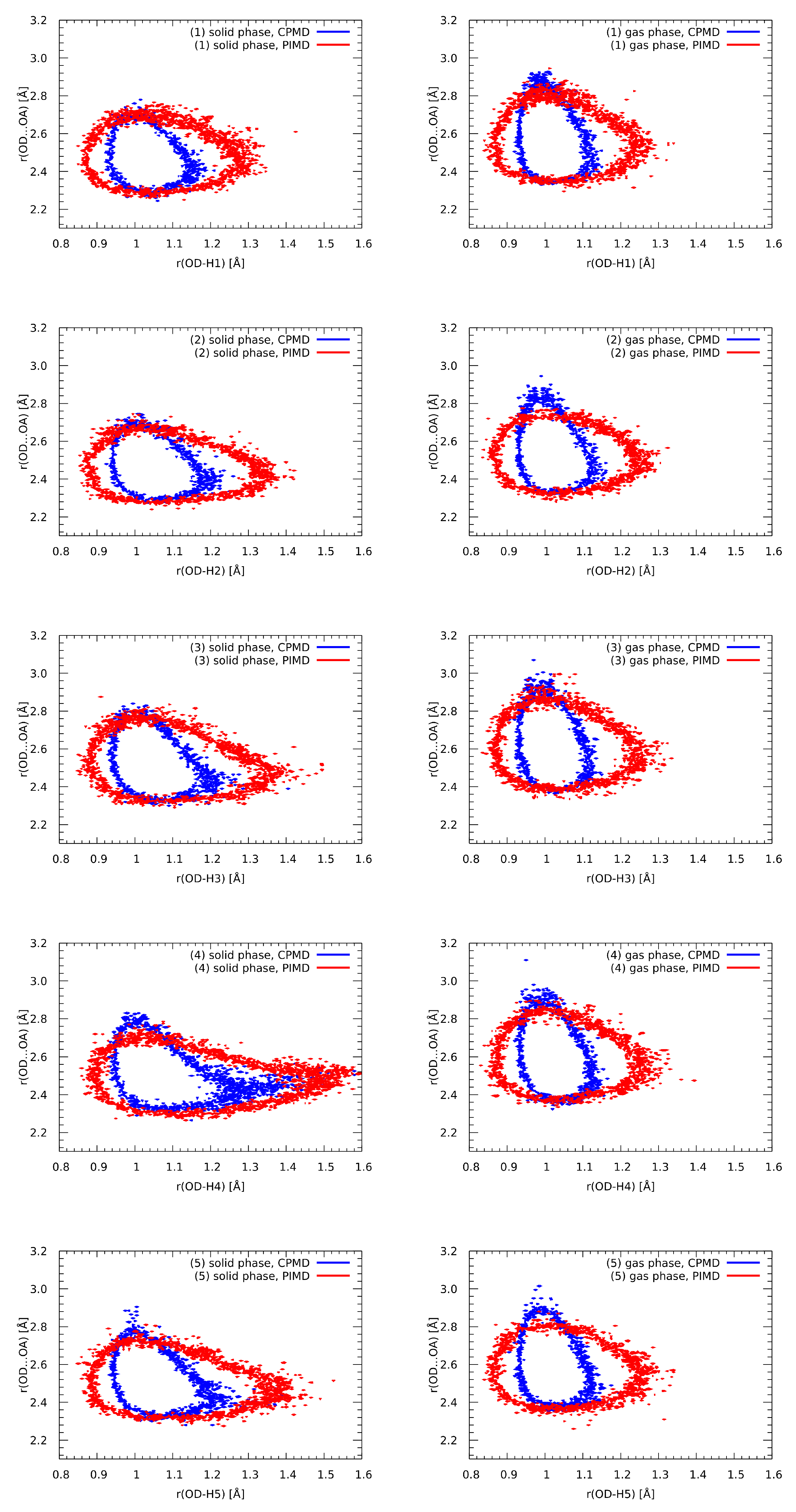
3.8. Nuclear Quantum Effects Inclusion—An Application of Path Integral Molecular Dynamics (PIMD)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density functional theory |
| HB | Hydrogen bond |
| PCM | Polarizable continuum model |
| AIM | Atoms in molecules |
| NBO | Natural bond orbital |
| SAPT | Symmetry-adapted perturbation theory |
| CPMD | Car–Parrinello molecular dynamics |
| PIMD | Path integral molecular dynamics |
| NCI | Non-covalent interactions |
| CCDC | Cambridge Crystallographic Data Centre |
| PES | Potential energy surface |
| BCP | Bond critical point |
| RCP | Ring critical point |
| CCP | Cage critical point |
| RI | Resolution of identity |
| JKI | Coulomb and exchange integrals |
| BSSE | Basis set superposition error |
| NPA | Natural population analysis |
| NAO | Natural atomic orbital |
| WBI | Wiberg bond index |
| IR | Infrared spectroscopy |
References
- Mitscher, L.A. Bacterial Topoisomerase Inhibitors: Quinolone and Pyridone Antibacterial Agents. Chem. Rev. 2005, 105, 559–592. [Google Scholar] [CrossRef] [PubMed]
- Dayam, R.; Al-Mawsawi, L.Q.; Zawahir, Z.; Witvrouw, M.; Debyser, Z.; Neamati, N. Quinolone 3-Carboxylic Acid Pharmacophore: Design of Second Generation HIV-1 Integrase Inhibitors. J. Med. Chem. 2008, 51, 1136–1144. [Google Scholar] [CrossRef]
- Horta, P.; Kuş, N.; Henriques, M.S.C.; Paixão, J.A.; Coelho, L.; Nogueira, F.; O’Neill, P.M.; Fausto, R.; Cristiano, M.L.S. Quinolone–Hydroxyquinoline Tautomerism in Quinolone 3-Esters. Preserving the 4-Oxoquinoline Structure To Retain Antimalarial Activity. J. Org. Chem. 2015, 80, 12244–12257. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.D.; Leung, S.C.; Amporndanai, K.; Davies, J.; Priestley, R.S.; Nixon, G.L.; Berry, N.G.; Hasnain, S.S.; Antonyuk, S.; Ward, S.A.; et al. Potent Antimalarial 2-Pyrazolyl Quinolone bc1 (Qi) Inhibitors with Improved Drug-like Properties. ACS Med. Chem. Lett. 2018, 9, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Zhang, Z.; Gao, D.; Chan, S.; Luo, J.; Tu, Z.C.; Zhang, Z.M.; Ding, K.; Ren, X.; Lu, X. Quinolone antibiotic derivatives as new selective Axl kinase inhibitors. Eur. J. Med. Chem. 2019, 166, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Yang, Y.; Huang, Y.; Gan, T.; Wu, Y.; Gao, H.; Li, Q.; Nie, J.; Huang, W.; Wang, Y.; et al. Novel quinolone derivatives targeting human dihydroorotate dehydrogenase suppress Ebola virus infection in vitro. Antiviral Res. 2021, 194, 105161. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.I. Development of the quinolones. J. Antimicrob. Chemother. 2003, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.; Giménez, M.; Alou, L.; Gómez-Lus, M.; Aguilar, L.; Prieto, J. Ex vivo serum activity (killing rates) after gemifloxacin 320 mg versus trovafloxacin 200 mg single doses against ciprofloxacin-susceptible and -resistant Streptococcus pneumoniae. Int. J. Antimicrob. Agents 2002, 20, 144–146. [Google Scholar] [CrossRef]
- Maciuca, A.M.; Munteanu, A.C.; Mihaila, M.; Badea, M.; Olar, R.; Nitulescu, G.M.; Munteanu, C.V.A.; Bostan, M.; Uivarosi, V. Rare-Earth Metal Complexes of the Antibacterial Drug Oxolinic Acid: Synthesis, Characterization, DNA/Protein Binding and Cytotoxicity Studies. Molecules 2020, 25, 5418. [Google Scholar] [CrossRef]
- Ukrainets, I.V.; Bereznyakova, N.L.; Parshikov, V.A.; Kravchenko, V.N. 4-Hydroxy-2-quinolones 140. Synthesis and diuretic activity of arylalkylamides of 4-methyl-2-oxo-1, 2-dihydro-quinoline-3-carboxylic acid. Chem. Heterocycl. Compd. 2008, 44, 64–72. [Google Scholar] [CrossRef]
- Cygler, M.; Huber, C.P. Structure of oxolinic acid, a potent antibacterial agent. 1-Ethyl-1, 4-dihydro-6, 7-methylenedioxy-4-oxo-3-quinolinecarboxylic acid, C13H11NO5. Acta Cryst. C 1985, 41, 1052–1055. [Google Scholar] [CrossRef]
- Czugler, M.; Argay, G.; Frank, J.; Mészáros, Z.; Kutschabsky, L.; Reck, G. 1-Ethyl-1, 4-dihydro-4-oxo-5-amino-6, 7-methylenedioxy-3-quinolinecarboxylic acid (aminooxolinic acid). Acta Cryst. B 1976, 32, 3124–3126. [Google Scholar] [CrossRef]
- Gleckman, R.; Alvarez, S.; Joubert, D.W.; Matthews, S.J. Drug therapy reviews: Oxolinic acid. Am. J. Hosp. Pharm. 1979, 36, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Tuma, J.; Connors, W.H.; Stitelman, D.H.; Richert, C. On the Effect of Covalently Appended Quinolones on Termini of DNA Duplexes. J. Am. Chem. Soc. 2002, 124, 4236–4246. [Google Scholar] [CrossRef] [PubMed]
- Irgi, E.P.; Geromichalos, G.D.; Balala, S.; Kljun, J.; Kalogiannis, S.; Papadopoulos, A.; Turel, I.; Psomas, G. Cobalt(ii) complexes with the quinolone antimicrobial drug oxolinic acid: Structure and biological perspectives. RSC Adv. 2015, 5, 36353–36367. [Google Scholar] [CrossRef]
- Mjos, K.D.; Cawthray, J.F.; Polishchuk, E.; Abrams, M.J.; Orvig, C. Gallium(iii) and iron(iii) complexes of quinolone antimicrobials. Dalton Trans. 2016, 45, 13146–13160. [Google Scholar] [CrossRef]
- Ukrainets, I.V.; Bereznyakova, N.L.; Parshikov, V.A.; Shishkina, S.V. 4-hydroxy-2-quinolones. 114. Synthesis and structure of 6-R-5-hydroxy-2,4-dioxo-2,3,4,6-tetrahydrobenzo-[c][2,7]naphthyridine-1-carbonitriles. Chem. Heterocycl. Compd. 2007, 43, 608–616. [Google Scholar] [CrossRef]
- Song, J.; Xue, F.H.; Lou, K.X. 7-Chloro-1-ethyl-6-fluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic acid. Acta Cryst. E 2004, 60, o824–o825. [Google Scholar] [CrossRef] [Green Version]
- Cleland, W.W.; Kreevoy, M.M. Low-Barrier Hydrogen Bonds and Enzymic Catalysis. Science 1994, 264, 1887–1890. [Google Scholar] [CrossRef]
- Wu, Z.R.; Ebrahimian, S.; Zawrotny, M.E.; Thornburg, L.D.; Perez-Alvarado, G.C.; Brothers, P.; Pollack, R.M.; Summers, M.F. Solution Structure of 3-Oxo-Δ5-Steroid Isomerase. Science 1997, 276, 415–418. [Google Scholar] [CrossRef]
- Remer, L.C.; Jensen, J.H. Toward a General Theory of Hydrogen Bonding: The Short, Strong Hydrogen Bond [HOH···OH]-. J. Phys. Chem. A 2000, 104, 9266–9275. [Google Scholar] [CrossRef]
- Lane, J.R. CCSDTQ Optimized Geometry of Water Dimer. J. Chem. Theory Comput. 2012, 9, 316–323. [Google Scholar] [CrossRef]
- Alcock, N. Secondary Bonding to Nonmetallic Elements. In Advances in Inorganic Chemistry and Radiochemistry; Elsevier: Amsterdam, The Netherlands, 1972; pp. 1–58. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, G.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1991. [Google Scholar]
- Jeffrey, G. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Mautner, M.M.N. The Ionic Hydrogen Bond. Chem. Rev. 2005, 105, 213–284. [Google Scholar] [CrossRef] [Green Version]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 4. Covalent nature of the strong homonuclear hydrogen bond. Study of the O-H–O system by crystal structure correlation methods. J. Am. Chem. Soc. 1994, 116, 909–915. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Pretto, L.; Ferretti, V.; Gilli, G. Covalent versus Electrostatic Nature of the Strong Hydrogen Bond: Discrimination among Single, Double, and Asymmetric Single-Well Hydrogen Bonds by Variable-Temperature X-ray Crystallographic Methods in β-Diketone Enol RAHB Systems. J. Am. Chem. Soc. 2004, 126, 3845–3855. [Google Scholar] [CrossRef]
- Jabłoński, M. A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules 2020, 25, 5512. [Google Scholar] [CrossRef]
- Grabowski, S. An estimation of strength of intramolecular hydrogen bonds—ab initio and AIM studies. J. Mol. Struct. 2001, 562, 137–143. [Google Scholar] [CrossRef]
- Lane, J.R.; Schrøder, S.D.; Saunders, G.C.; Kjaergaard, H.G. Intramolecular Hydrogen Bonding in Substituted Aminoalcohols. J. Phys. Chem. A 2016, 120, 6371–6378. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals; an Introduction to Modern Structural Chemistry, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, P.J. The halogen bond: Nature and applications. Phys. Sci. Rev. 2017, 2. [Google Scholar] [CrossRef]
- Riley, K.E.; Tran, K.A. Strength, character, and directionality of halogen bonds involving cationic halogen bond donors. Faraday Discuss. 2017, 203, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Engelage, E.; Reinhard, D.; Huber, S.M. Is There a Single Ideal Parameter for Halogen-Bonding-Based Lewis Acidity? Chem. Eur. J. 2020, 26, 3843–3861. [Google Scholar] [CrossRef]
- Portela, S.; Fernández, I. Nature of the Hydrogen Bond Enhanced Halogen Bond. Molecules 2021, 26, 1885. [Google Scholar] [CrossRef]
- Dang, Q.M.; Simpson, J.H.; Parish, C.A.; Leopold, M.C. Evaluating Halogen-Bond Strength as a Function of Molecular Structure Using Nuclear Magnetic Resonance Spectroscopy and Computational Analysis. J. Phys. Chem. A 2021, 125, 9377–9393. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quant. Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Bader, R. Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Natural Bond Orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, D.; Parrinello, M. The Effect of Quantum and Thermal Fluctuations on the Structure of the Floppy Molecule C2H3+. Science 1996, 271, 179–181. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Marx, D.; Klein, M.L.; Parrinello, M. Efficient and general algorithms for path integral Car–Parrinello molecular dynamics. J. Chem. Phys. 1996, 104, 5579–5588. [Google Scholar] [CrossRef]
- CCDC Structural Database. 2021. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 7 May 2021).
- Mercury—Crystal Structure Visualisation. Available online: http://www.ccdc.cam.ac.uk/Solutions/CSDSystem/Pages/Mercury.aspx (accessed on 7 October 2021).
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Keith, T.A. AIMAll (Version 19.10.12), TK Gristmill Software; AIMAll: Overland Park, KS, USA, 2019. [Google Scholar]
- Wiberg, K. Application of the Pople-Santry-Segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1. Available online: https://www.scienceopen.com/document?vid=6652d352-0292-499f-88d6-2221dae56281 (accessed on 7 October 2021).
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D, Version 14.6.4. 2016. Available online: https://sourceforge.net/p/jmol/ (accessed on 24 February 2022).
- Patek, M. Jmol NBO Visualization Helper, Version 2.1. 2015. Available online: https://www.marcelpatek.com/blgdownload/JmolNboVHelper2.1.zip (accessed on 24 February 2022).
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef]
- Schlegel, H.B. Estimating the hessian for gradient-type geometry optimizations. Theor. Chem. Acc. 1984, 66, 333–340. [Google Scholar] [CrossRef]
- Hockney, R.W. Potential Calculation and Some Applications. Methods Comput. Phys. 1970, 9, 135–211. [Google Scholar]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C.; Bersch, C.; Sebald, D.; Campbell, J.; Cunningham, R.; Denholm, D.; Elber, G.; Fearick, R.; Grammes, C.; et al. Gnuplot 5.2.8: An Interactive Plotting Program. 2019. Available online: http://www.gnuplot.info (accessed on 21 February 2022).
- CPMD Version 4.3-4610, Copyright IBM Corp. (1990–2004) Copyright MPI für Festkoerperforschung Stuttgart (1997–2001). Available online: http://www.cpmd.org/ (accessed on 21 February 2022).
- Backus, J.W.; Stern, H.; Ziller, I.; Hughes, R.A.; Nutt, R.; Beeber, R.J.; Best, S.; Goldberg, R.; Haibt, L.M.; Herrick, H.L.; et al. The FORTRAN automatic coding system. In IRE-AIEE-ACM ’57 (Western): Papers Presented at the 26–28 February 1957, Western Joint Computer Conference: Techniques for Reliability; ACM Press: New York, NY, USA, 1957. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Głowiak, T. Proton transfer equilibrium in the intramolecular hydrogen bridge in sterically hindered Schiff bases. J. Mol. Struct. 2002, 615, 97–108. [Google Scholar] [CrossRef]
- Ukrainets, I.V.; Sidorenko, L.V.; Gorokhova, O.V.; Shishkina, S.V. 4-hydroxy-2-quinolones. 96. Synthesis and properties of 4-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid. Chem. Heterocycl. Compd. 2006, 42, 776–781. [Google Scholar] [CrossRef]
- Ukrainets, I.V.; Gorokhova, O.V.; Sidorenko, L.V.; Bereznyakova, N.L. 4-Hydroxy-2-quinolones. 111. Simple synthesis of 1-substituted 4-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acids. Chem. Heterocycl. Compd. 2007, 43, 58–62. [Google Scholar] [CrossRef]
- Dixit, S.K.; Yadav, N.; Kumar, S.; Good, L.; Awasthi, S.K. Synthesis and antibacterial activity of novel fluoroquinolone analogs. Med. Chem. Res. 2014, 23, 5237–5249. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Sokalski, W.A.; Dyguda, E.; Leszczyński, J. Quantitative Classification of Covalent and Noncovalent H-Bonds. J. Phys. Chem. B 2006, 110, 6444–6446. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Martin, F.; Zipse, H. Charge distribution in the water molecule—A comparison of methods. J. Comput. Chem. 2004, 26, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q. Dinitroamino benzene derivatives: A class new potential high energy density compounds. J. Mol. Model. 2013, 19, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Mondal, P.K.; Yadav, H.R.; Choudhury, A.R.; Chopra, D. Quantitative characterization of new supramolecular synthons involving fluorine atoms in the crystal structures of di- and tetrafluorinated benzamides. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Al-Mutairi, A.A.; Katari, B.K.P.; Narasimhan, Y.; Blacque, O.; Al-Wahaibi, L.H.; Al-Alshaikh, M.A.; El-Emam, A.A.; Percino, M.J.; Thamotharan, S. Interplay of weak intermolecular interactions in two Schiff’s bases with organic fluorine derived from 5-nitrothiophene-2-carboxaldehyde: Crystal structures, DFT calculation and in vitro evaluation of bioactivities. J. Mol. Struct. 2020, 1221, 128883. [Google Scholar] [CrossRef]
- El-Emam, A.A.; Kumar, E.S.; Janani, K.; Al-Wahaibi, L.H.; Blacque, O.; El-Awady, M.I.; Al-Shaalan, N.H.; Percino, M.J.; Thamotharan, S. Quantitative assessment of the nature of noncovalent interactions in N-substituted-5-(adamantan-1-yl)-1,3,4-thiadiazole-2-amines: Insights from crystallographic and QTAIM analysis. RSC Adv. 2020, 10, 9840–9853. [Google Scholar] [CrossRef] [Green Version]
- Tariq, S.; Khalid, M.; Raza, A.R.; Rubab, S.L.; de Alcântara Morais, S.F.; Khan, M.U.; Tahir, M.N.; Braga, A.A.C. Experimental and computational investigations of new indole derivatives: A combined spectroscopic, SC-XRD, DFT/TD-DFT and QTAIM analysis. J. Mol. Struct. 2020, 1207, 127803. [Google Scholar] [CrossRef]
- Khalid, M.; Ali, A.; Asim, S.; Tahir, M.N.; Khan, M.U.; Vieira, L.C.C.; de la Torre, A.F.; Usman, M. Persistent prevalence of supramolecular architectures of novel ultrasonically synthesized hydrazones due to hydrogen bonding [X–H⋯O; X=N]: Experimental and density functional theory analyses. J. Phys. Chem. Solids 2021, 148, 109679. [Google Scholar] [CrossRef]
- Neugebauer, U.; Szeghalmi, A.; Schmitt, M.; Kiefer, W.; Popp, J.; Holzgrabe, U. Vibrational spectroscopic characterization of fluoroquinolones. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005, 61, 1505–1517. [Google Scholar] [CrossRef]
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR Investigations of Strong Intramolecular Hydrogen Bonds. Molecules 2017, 22, 552. [Google Scholar] [CrossRef] [Green Version]
- Tuckerman, M.E.; Marx, D. Heavy-Atom Skeleton Quantization and Proton Tunneling in “Intermediate-Barrier” Hydrogen Bonds. Phys. Rev. Lett. 2001, 86, 4946–4949. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | CCDC Code | Unit Cell Data |
|---|---|---|
| 1 | PONNAI [10] | orthorhombic, a = 12.233 Å, b = 18.849 Å, c = 3.816 Å, Z = 4 |
| 2 | MIRJIH [17] | monoclinic, a = 9.086 Å, b = 14.27 Å, c = 9.164 Å, = 112.44, Z = 4 |
| 3 | EVINIG [18] | triclinic, a = 7.145 Å, b = 8.915 Å, c = 9.375 Å, |
| = 71.896, = 80.025, = 85.109, Z = 2 | ||
| 4 | DAHWEO [11] | monoclinic, a = 7.182 Å, b = 10.575 Å, c = 14.758 Å, = 94.26, Z = 4 |
| 5 | AMOXAC [12] | monoclinic, a = 7.485 Å, b = 10.449 Å, c = 15.087 Å, = 99.69, Z = 4 |
| System | H-Bond Energy | |
|---|---|---|
| Gas Phase | PCM | |
| H1•••OA | −6.738 | −6.792 |
| H2•••OA | −6.548 | −6.623 |
| H3•••OA | −9.300 | −9.749 |
| H4•••OA | −10.410 | −10.994 |
| H5•••OA | −9.880 | −10.839 |
| System | Atom | Closed | Open | ||
|---|---|---|---|---|---|
| Gas Phase | PCM | Gas Phase | PCM | ||
| (1) | OD | −0.634 | −0.663 | −0.630 | −0.638 |
| H1 | 0.482 | 0.480 | 0.487 | 0.503 | |
| OA | −0.616 | −0.650 | −0.570 | −0.645 | |
| (2) | OD | −0.636 | −0.664 | −0.634 | −0.641 |
| H2 | 0.483 | 0.480 | 0.486 | 0.502 | |
| OA | −0.629 | −0.652 | −0.583 | −0.646 | |
| (3) | OD | −0.636 | −0.666 | −0.617 | −0.637 |
| H3 | 0.486 | 0.483 | 0.483 | 0.497 | |
| OA | −0.598 | −0.637 | −0.532 | −0.610 | |
| (4) | OD | −0.640 | −0.672 | −0.620 | −0.640 |
| H4 | 0.485 | 0.479 | 0.480 | 0.495 | |
| OA | −0.613 | −0.658 | −0.548 | −0.630 | |
| (5) | OD | −0.640 | −0.672 | −0.664 | −0.640 |
| H5 | 0.486 | 0.482 | 0.514 | 0.494 | |
| OA | −0.664 | −0.685 | −0.611 | −0.651 | |
| System | LP1(O) (O–H) | LP2(O) (O–H) | LP1(O) (O–H) | LP2(O) (O–H) |
|---|---|---|---|---|
| Gas Phase | PCM | |||
| (1) | 5.13 | 31.16 | 6.06 | 41.55 |
| (2) | 5.68 | 31.82 | 6.73 | 43.81 |
| (3) | 3.64 | 24.79 | 4.62 | 35.25 |
| (4) | 3.91 | 27.80 | 5.02 | 41.77 |
| (5a) | 5.12 | 25.67 | 5.93 | 38.70 |
| System | LP1(O) (N–H) | LP2(O) (N–H) | LP1(O) (N–H) | LP2(O) (N–H) |
| Gas Phase | PCM | |||
| (5b) | 4.88 | 4.58 | 4.40 | 3.05 |
| Dimer | Electrostatics | Exchange | Induction | Dispersion | SAPT0 | SAPT2 |
|---|---|---|---|---|---|---|
| X-ray | ||||||
| 1.1 | −13.210 | 13.839 | −5.403 | −5.319 | −13.214 | −10.093 |
| 1.2 | −2.750 | 14.970 | −2.247 | −21.499 | −10.480 | −11.527 |
| 2.1 | −8.162 | 15.608 | −2.094 | −24.792 | −20.580 | −19.441 |
| 2.2 | 0.271 | 5.170 | −1.133 | −9.472 | −5.184 | −5.165 |
| 3.1 | −6.450 | 5.900 | −1.326 | −5.147 | −8.112 | −7.023 |
| 3.2 | −10.802 | 20.367 | −3.908 | −28.922 | −23.880 | −23.265 |
| 3.3 | −1.243 | 3.386 | −1.018 | −4.433 | −4.048 | −3.308 |
| 3.4 | −0.356 | 0.964 | −0.0973 | −1.218 | −0.654 | −0.707 |
| 4.1 | −3.852 | 5.105 | −1.029 | −4.276 | −4.782 | −4.052 |
| 4.2 | −10.309 | 14.439 | −2.250 | −24.401 | −23.429 | −22.522 |
| 5.1 | −13.617 | 19.235 | −3.448 | −28.603 | −27.730 | −26.432 |
| 5.2 | −1.084 | 4.779 | −0.843 | −4.510 | −1.976 | −1.657 |
| Optimized | ||||||
| 1.1 | −14.922 | 15.117 | −6.299 | −6.816 | −17.099 | −12.919 |
| 1.2 | −6.548 | 14.830 | −2.251 | −20.270 | −15.391 | −14.239 |
| 2.1 | −13.272 | 22.225 | −4.115 | −28.418 | −26.131 | −23.580 |
| 2.2 | −12.823 | 23.481 | −4.167 | −28.621 | −25.345 | −22.129 |
| 3.1 | −17.348 | 21.589 | −4.866 | −27.716 | −30.881 | −28.340 |
| 3.2 | −12.679 | 22.836 | −5.124 | −31.014 | −26.965 | −25.981 |
| 3.3 | −12.996 | 20.380 | −4.500 | −26.143 | −24.953 | −23.260 |
| 3.4 | −10.035 | 20.814 | −4.169 | −27.937 | −22.061 | −21.327 |
| 4.1 | −12.958 | 21.469 | −3.736 | −29.782 | −26.509 | −25.008 |
| 4.2 | −21.296 | 29.549 | −6.947 | −31.403 | −32.960 | −30.096 |
| 5.1 | −24.132 | 32.511 | −7.157 | −36.786 | −38.720 | −35.564 |
| 5.2 | −22.811 | 31.007 | −6.949 | −33.766 | −35.697 | −32.518 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojtkowiak, K.; Jezierska, A.; Panek, J.J. Revealing Intra- and Intermolecular Interactions Determining Physico-Chemical Features of Selected Quinolone Carboxylic Acid Derivatives. Molecules 2022, 27, 2299. https://doi.org/10.3390/molecules27072299
Wojtkowiak K, Jezierska A, Panek JJ. Revealing Intra- and Intermolecular Interactions Determining Physico-Chemical Features of Selected Quinolone Carboxylic Acid Derivatives. Molecules. 2022; 27(7):2299. https://doi.org/10.3390/molecules27072299
Chicago/Turabian StyleWojtkowiak, Kamil, Aneta Jezierska, and Jarosław J. Panek. 2022. "Revealing Intra- and Intermolecular Interactions Determining Physico-Chemical Features of Selected Quinolone Carboxylic Acid Derivatives" Molecules 27, no. 7: 2299. https://doi.org/10.3390/molecules27072299
APA StyleWojtkowiak, K., Jezierska, A., & Panek, J. J. (2022). Revealing Intra- and Intermolecular Interactions Determining Physico-Chemical Features of Selected Quinolone Carboxylic Acid Derivatives. Molecules, 27(7), 2299. https://doi.org/10.3390/molecules27072299