
Quinazoline Based HDAC Dual Inhibitors as Potential Anti-Cancer Agents


Abstract
:1. Introduction
2. History and Significance of Quinazolines
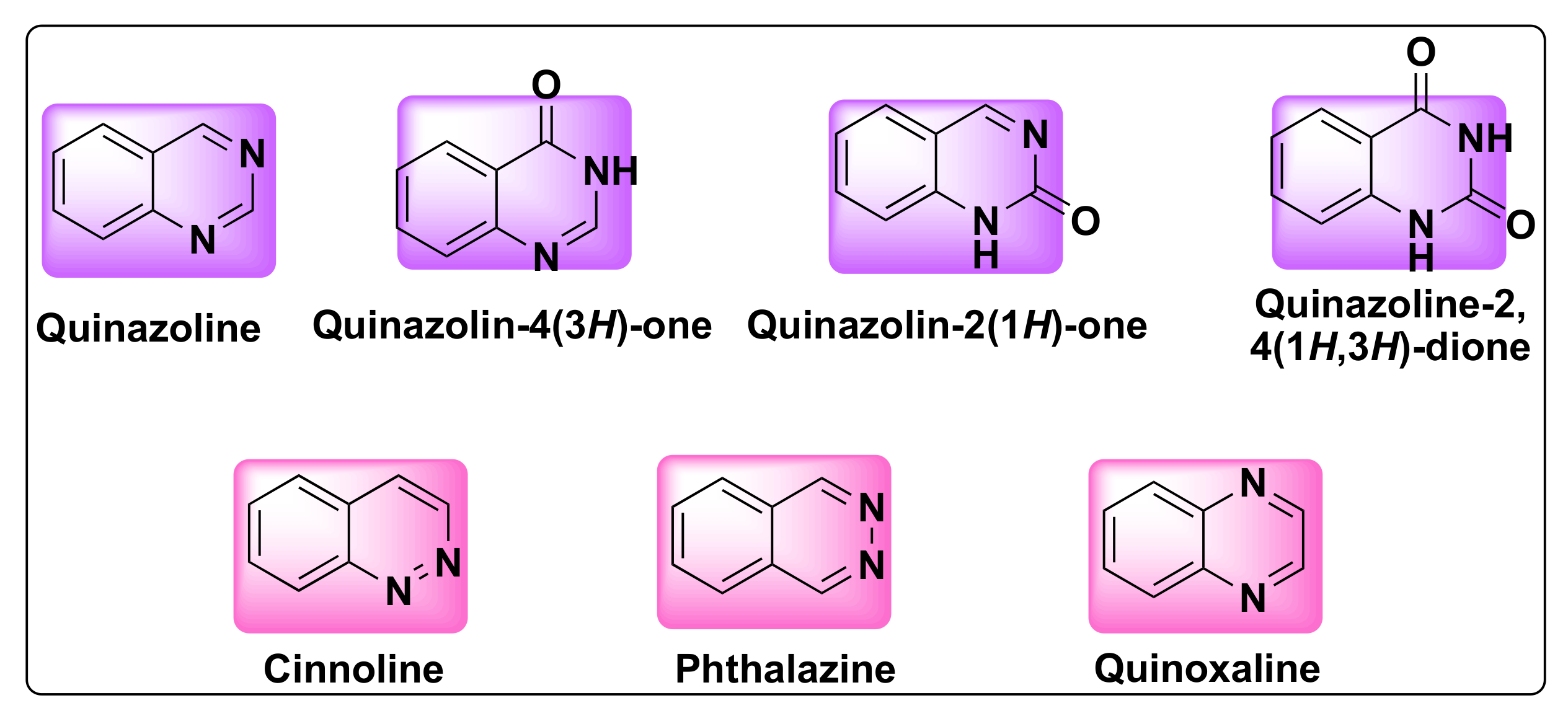
3. Structure, Isomerism, and Synthesis of Quinazolines
4. Quinazolines as Anti-Cancer Agents
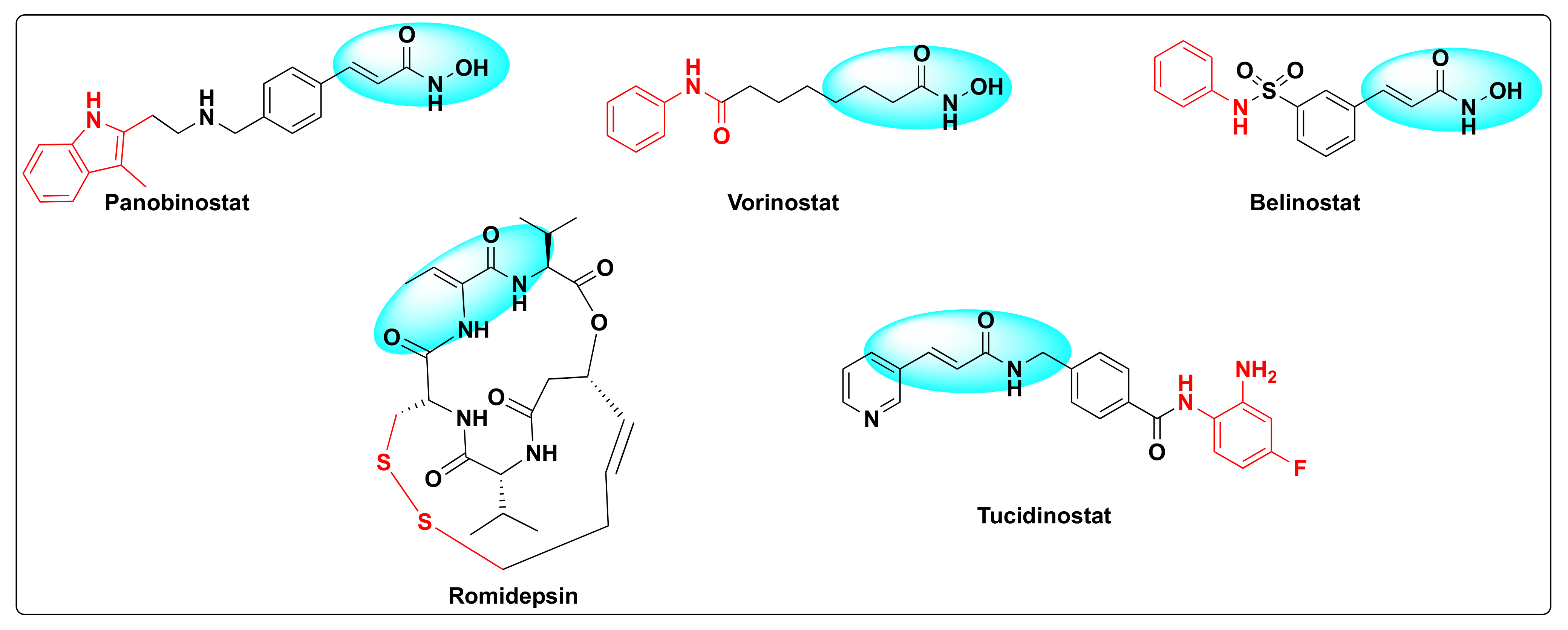
5. Histone Deacetylase Inhibition as Anti-Cancer Agents
6. Multi-Targeted Therapy and Multi-Drug Resistance
7. Synergistic Effect of the Dual Inhibition/Hybrid Approach
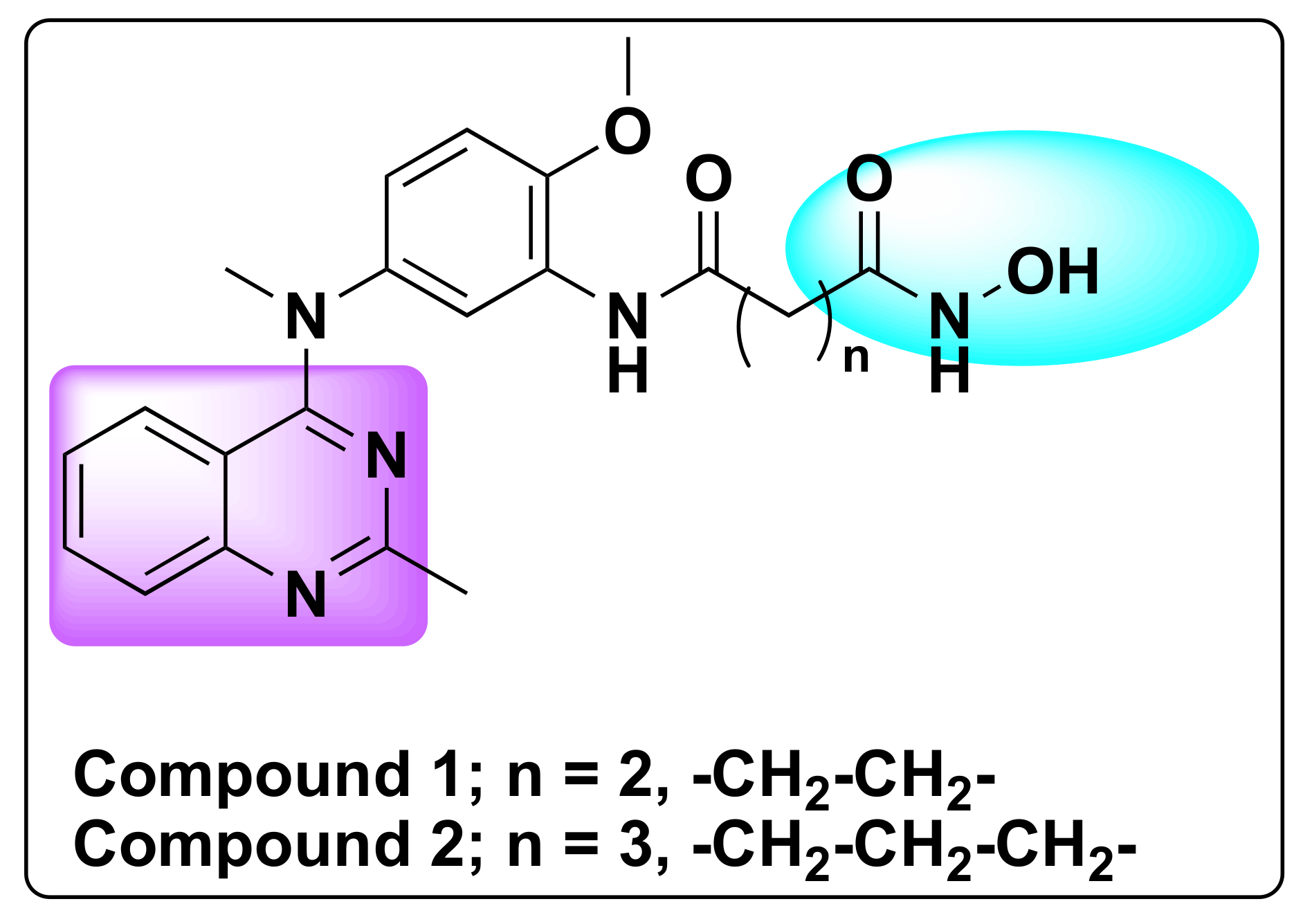
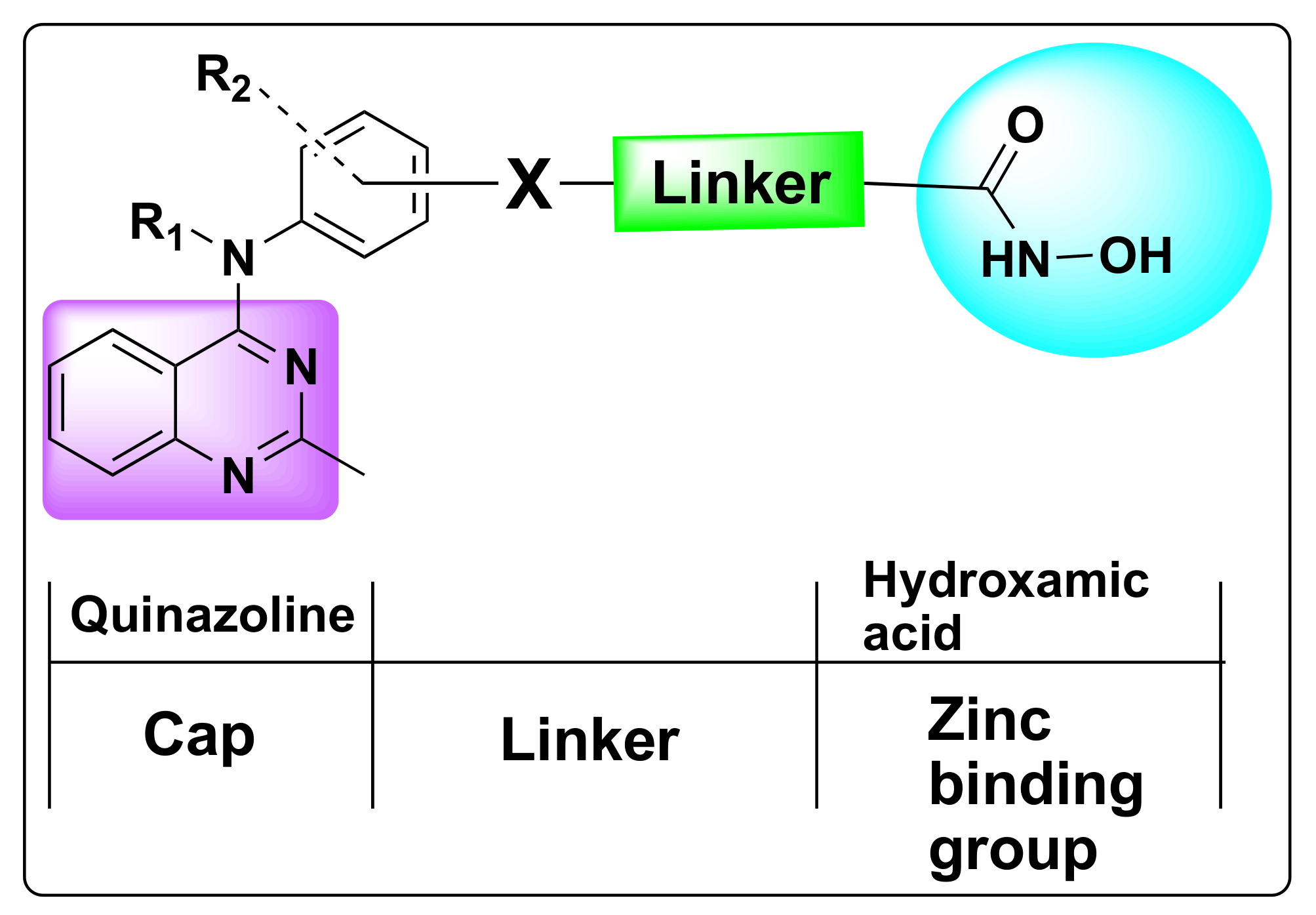
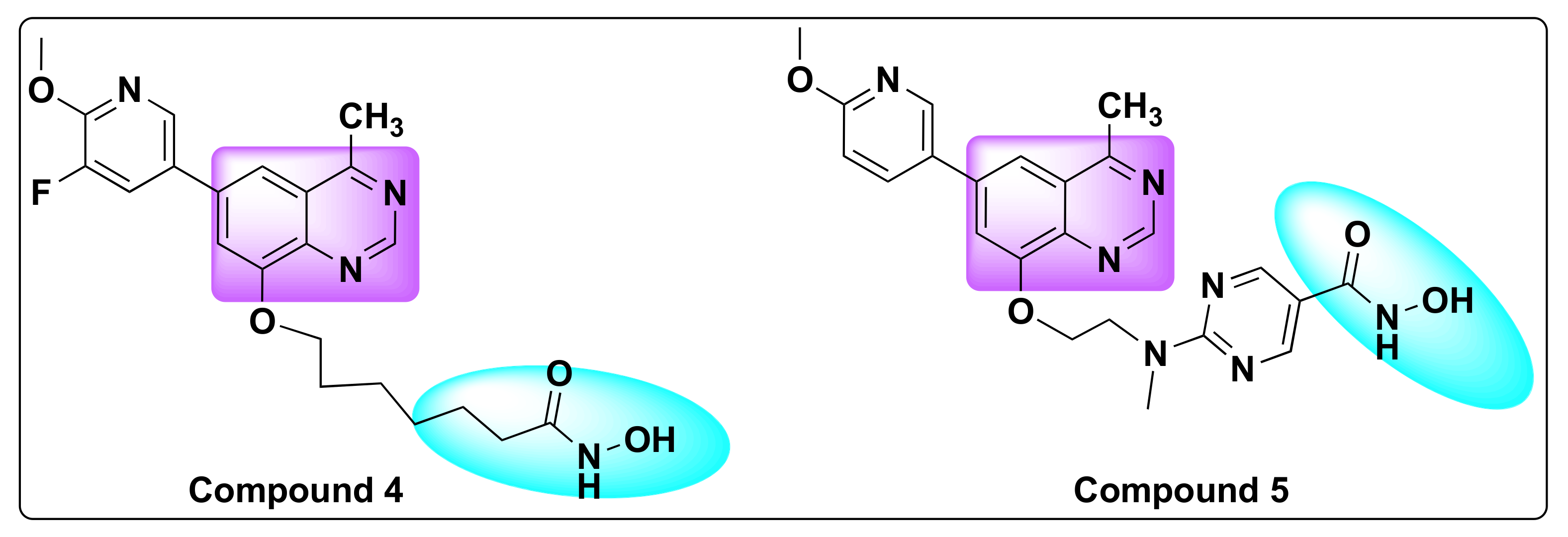
7.1. Quinazoline Based Dual Inhibition of HDAC1 and HDAC6 Enzymes
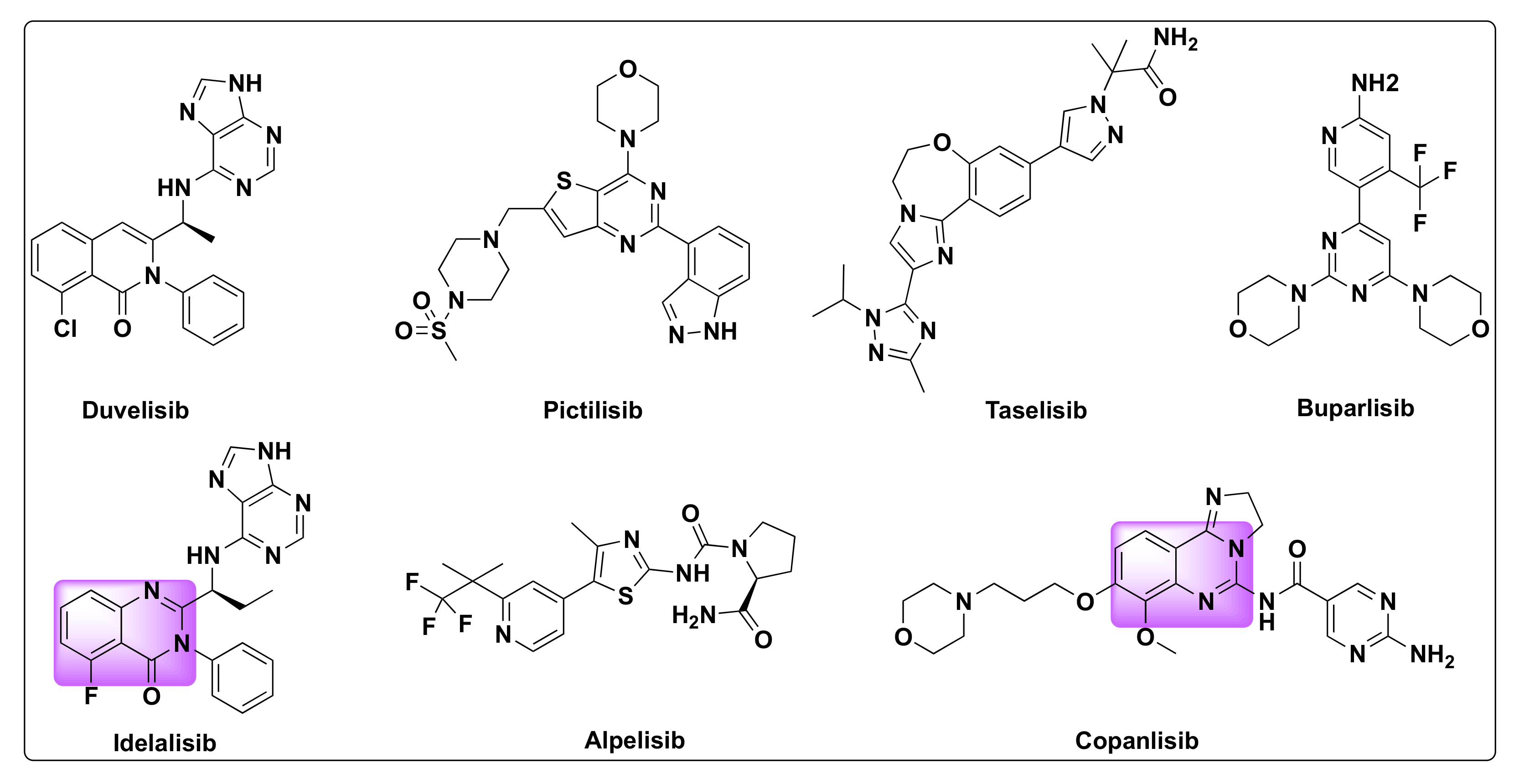
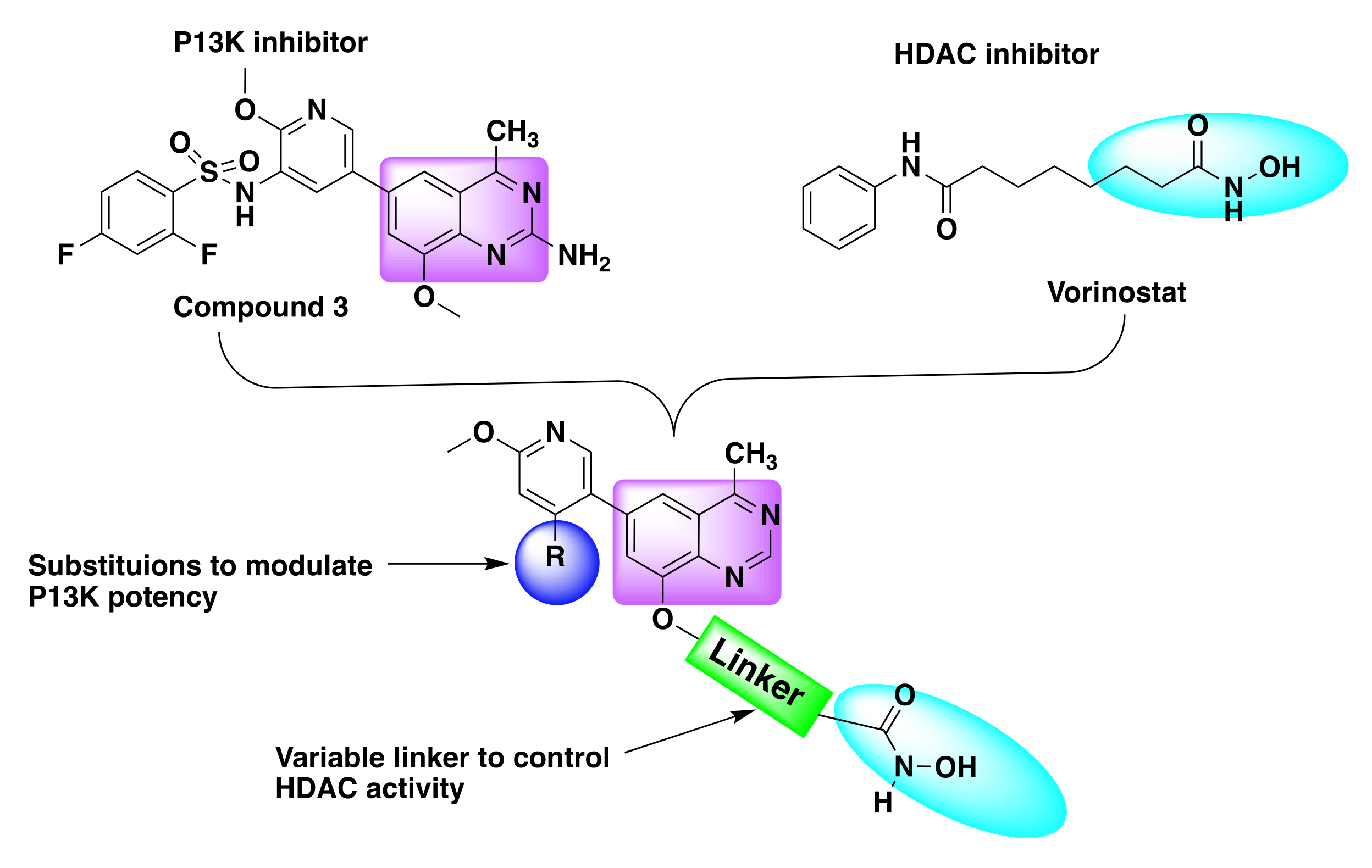
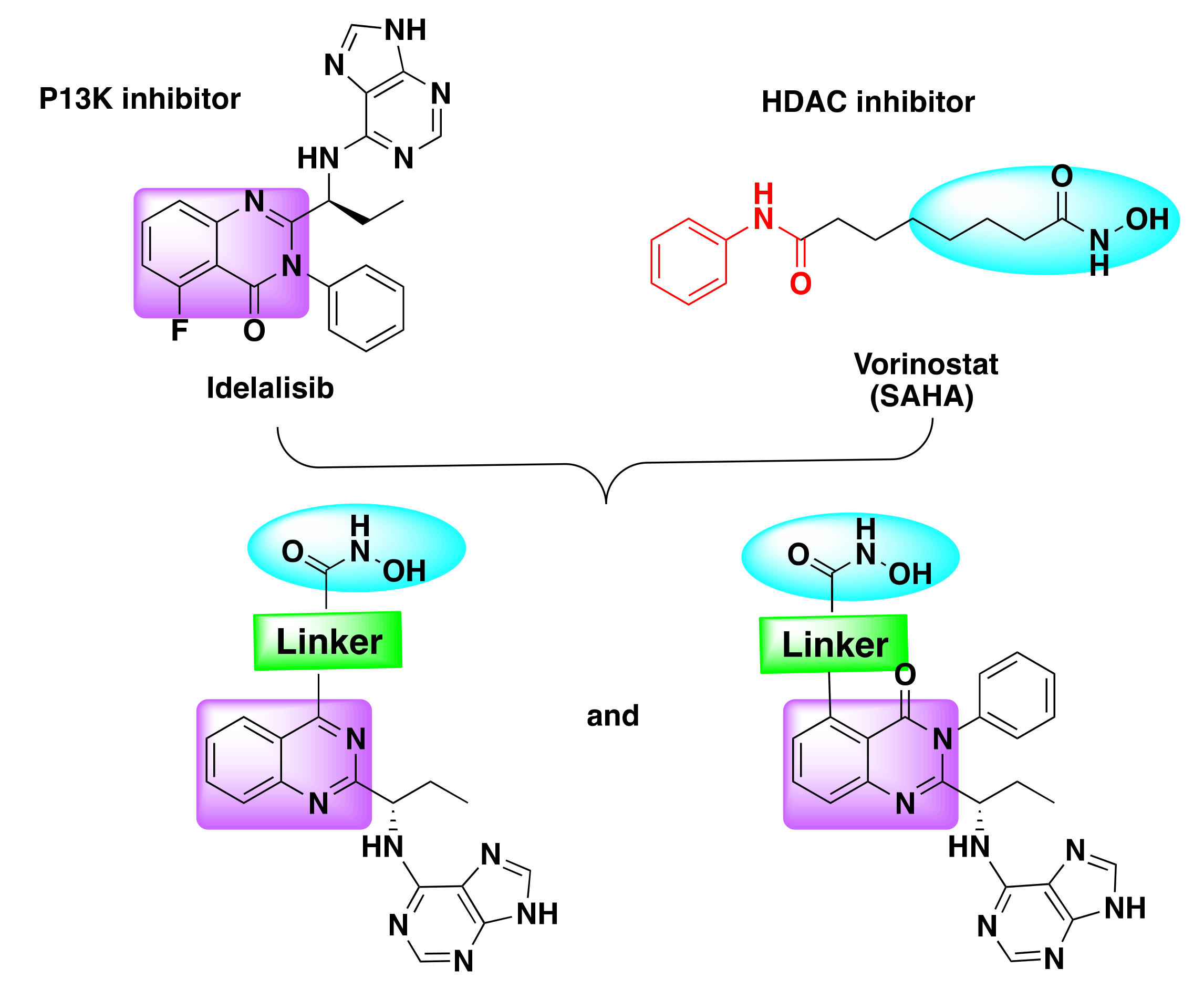
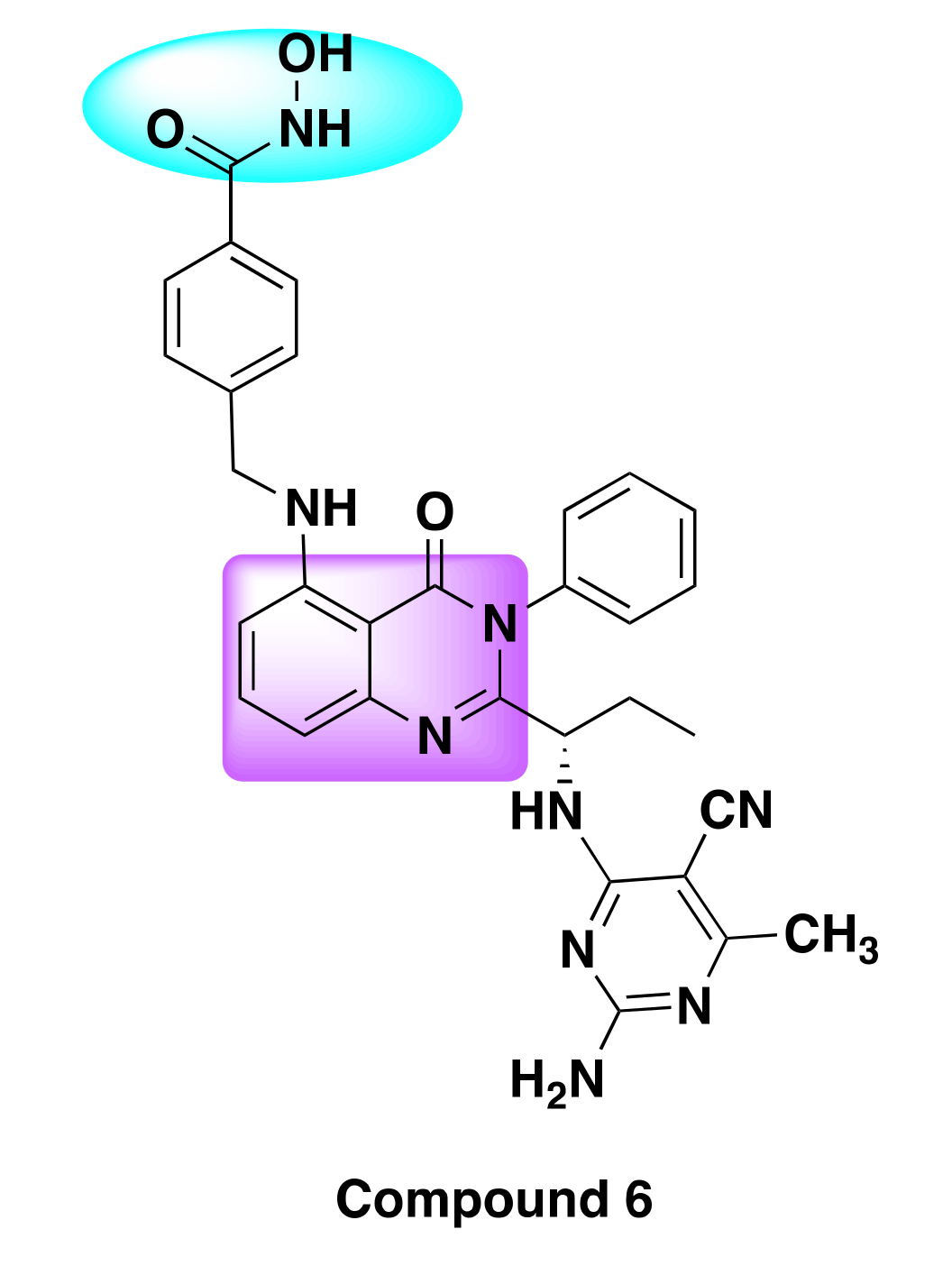
7.2. Quinazoline Based Dual Inhibition of HDAC and PI3K
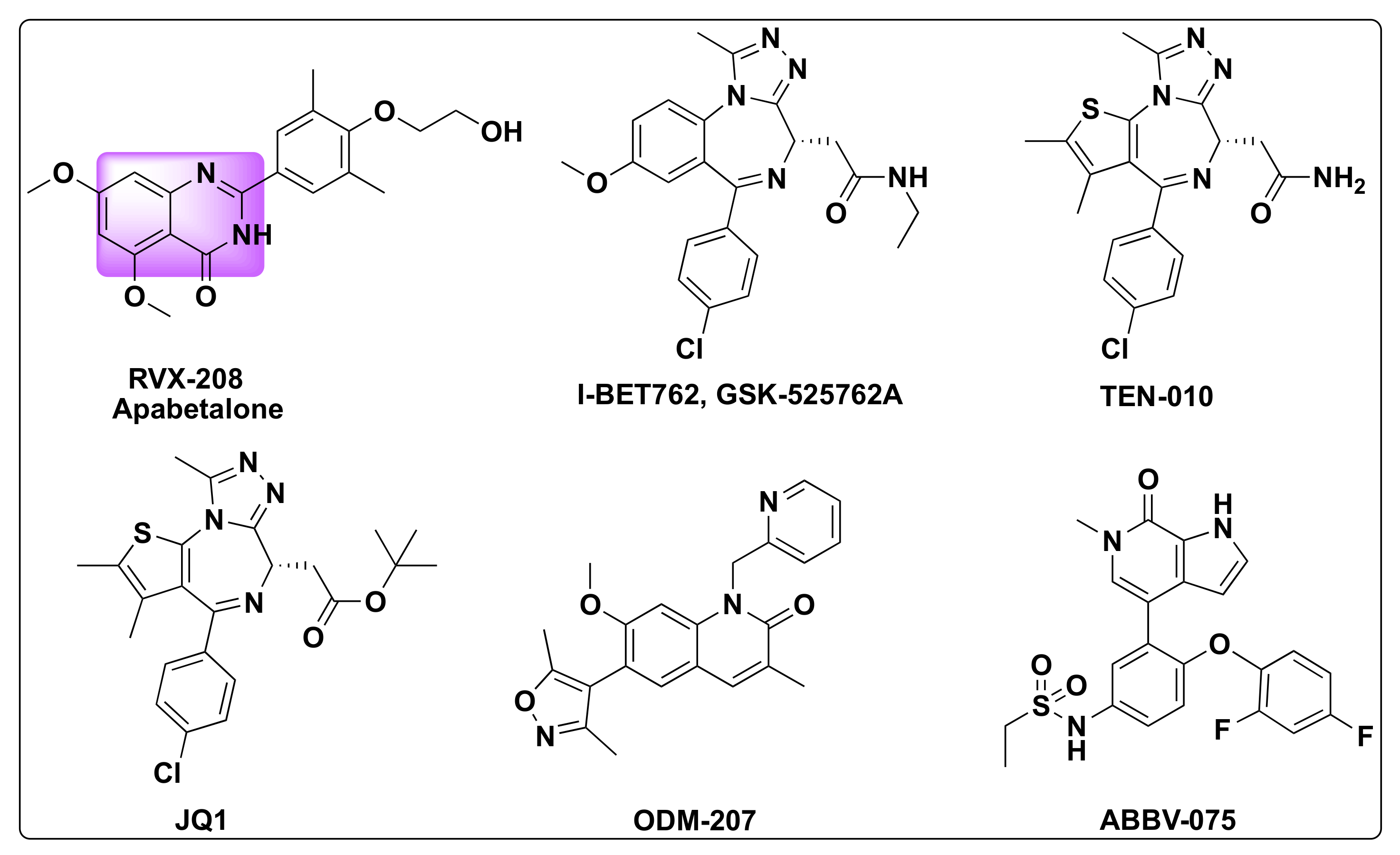
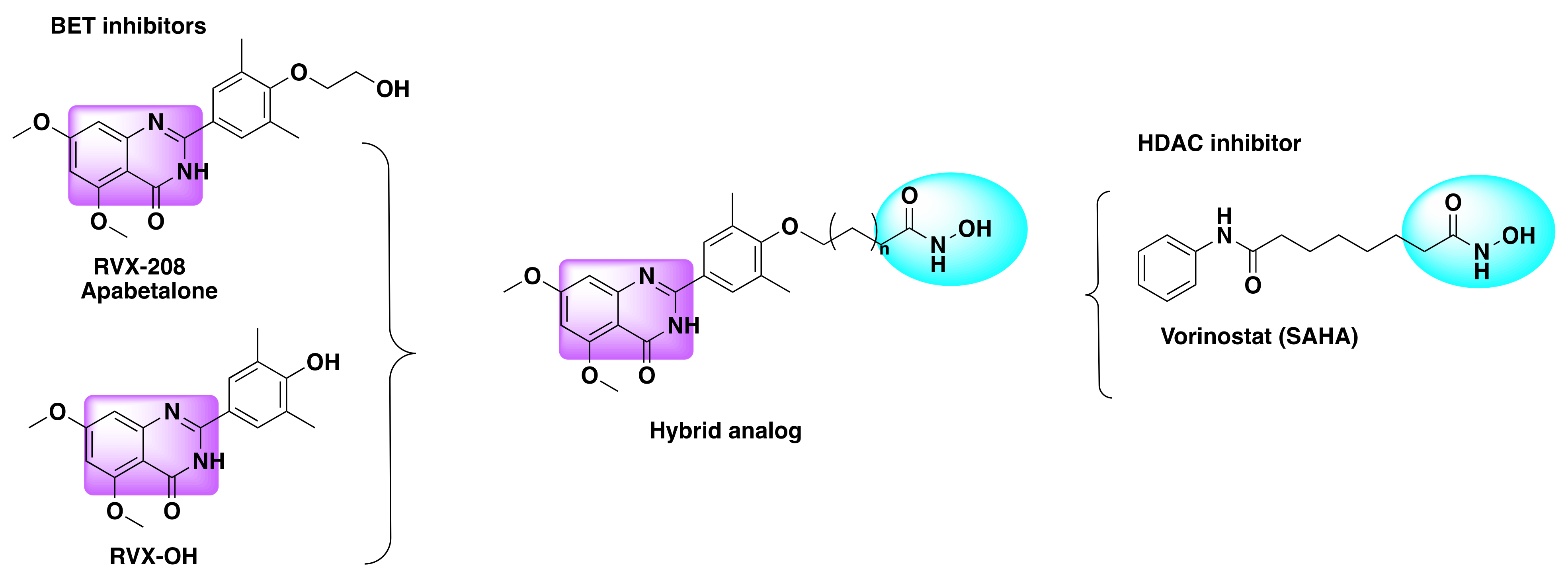
7.3. Quinazoline Based Dual Inhibition of HDAC and BET
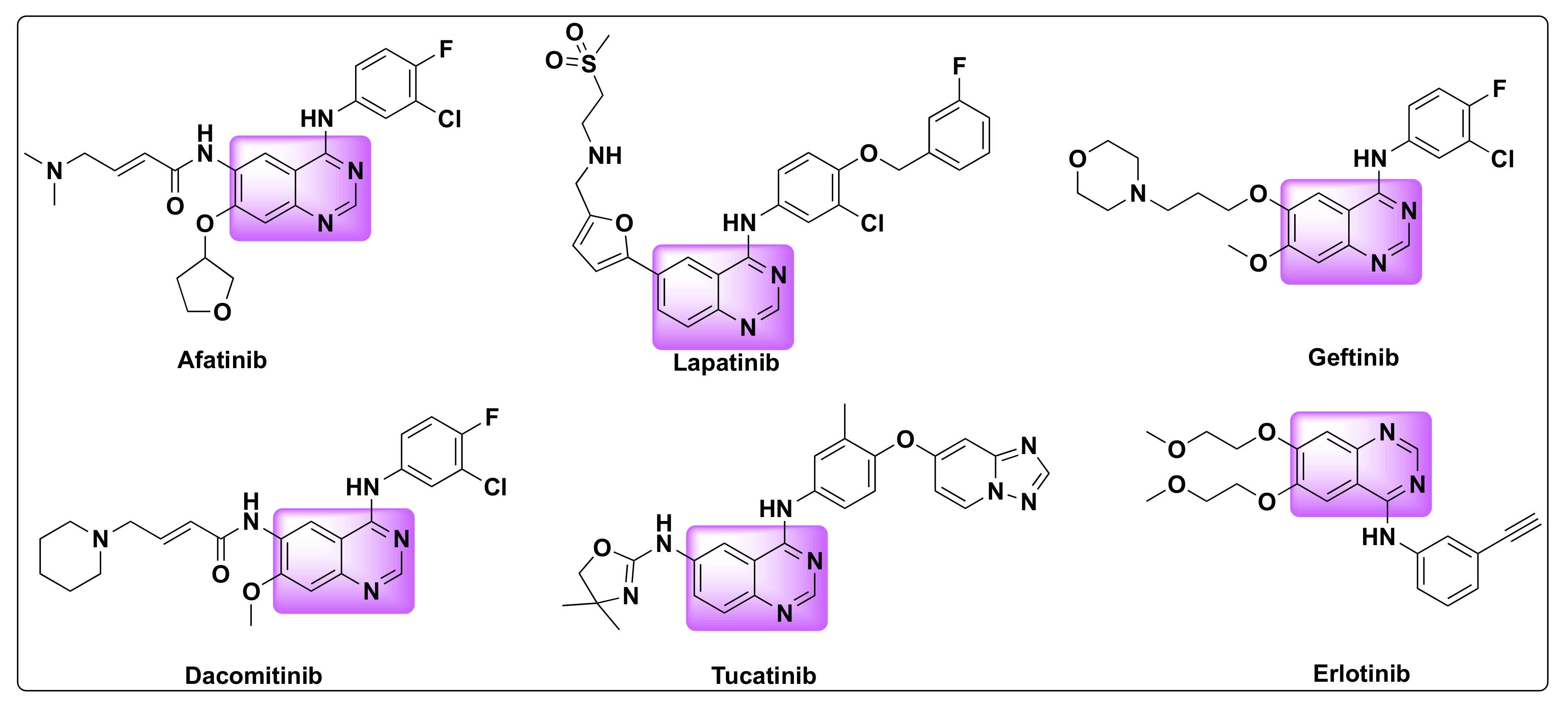
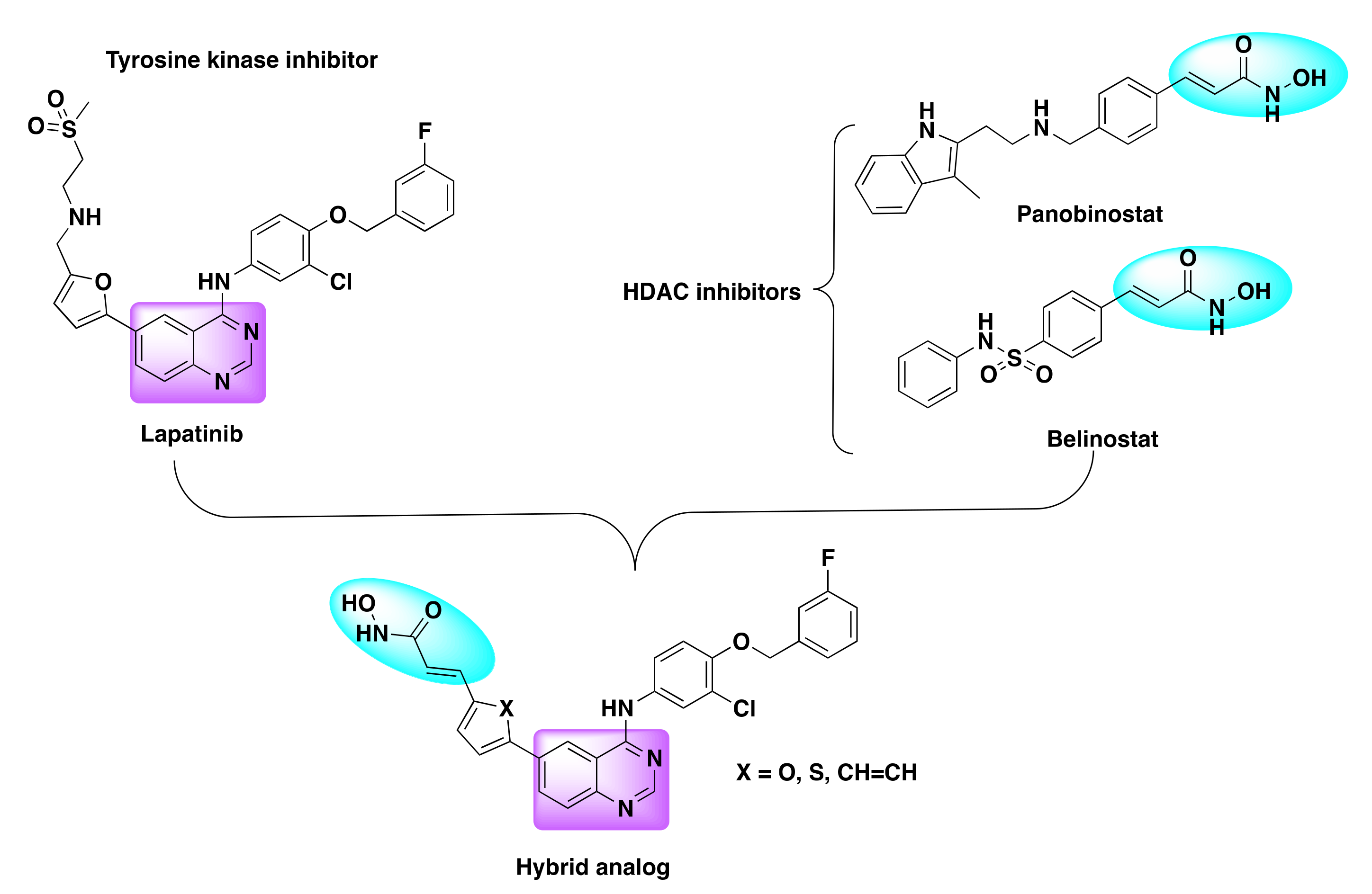
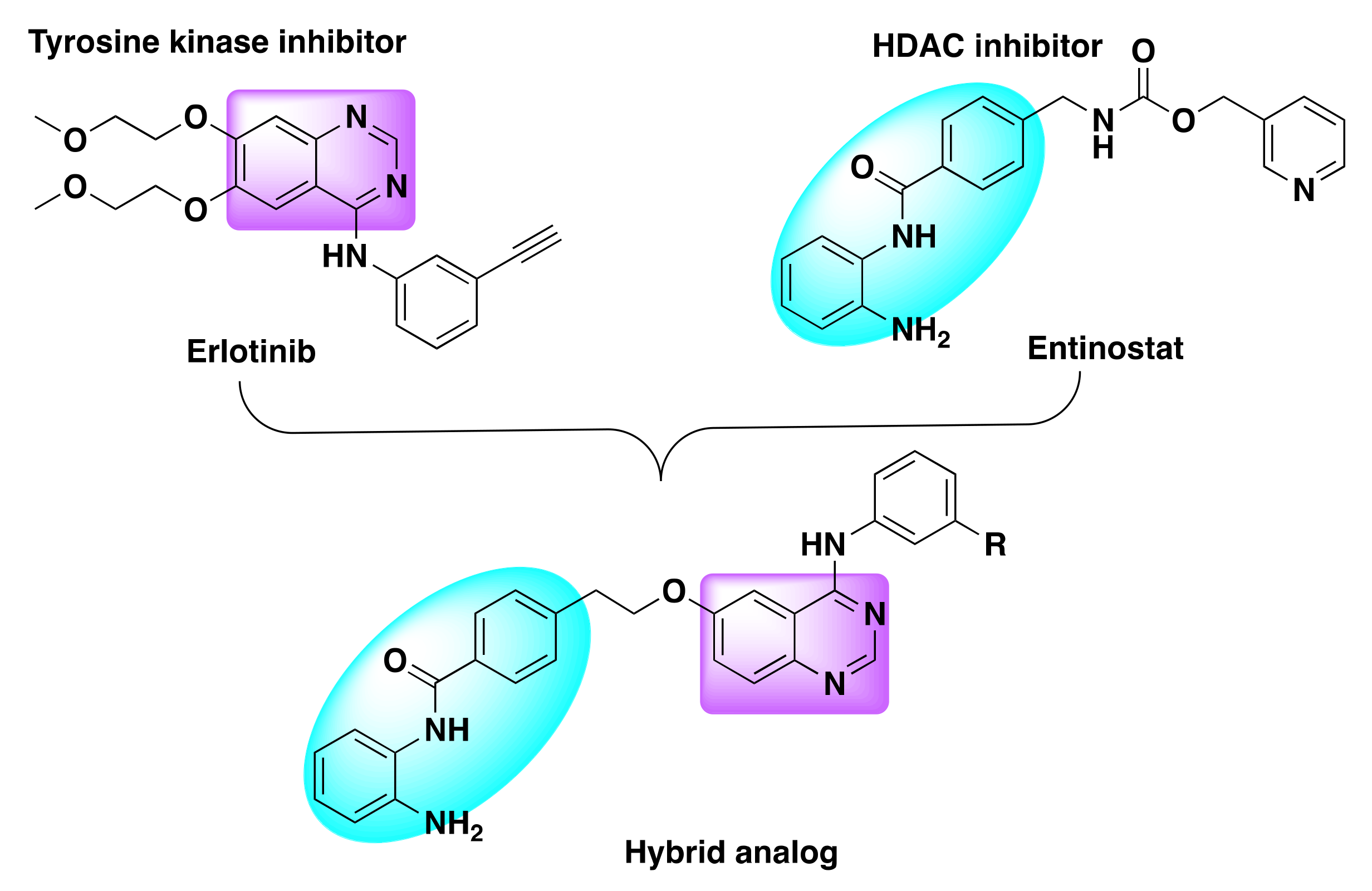
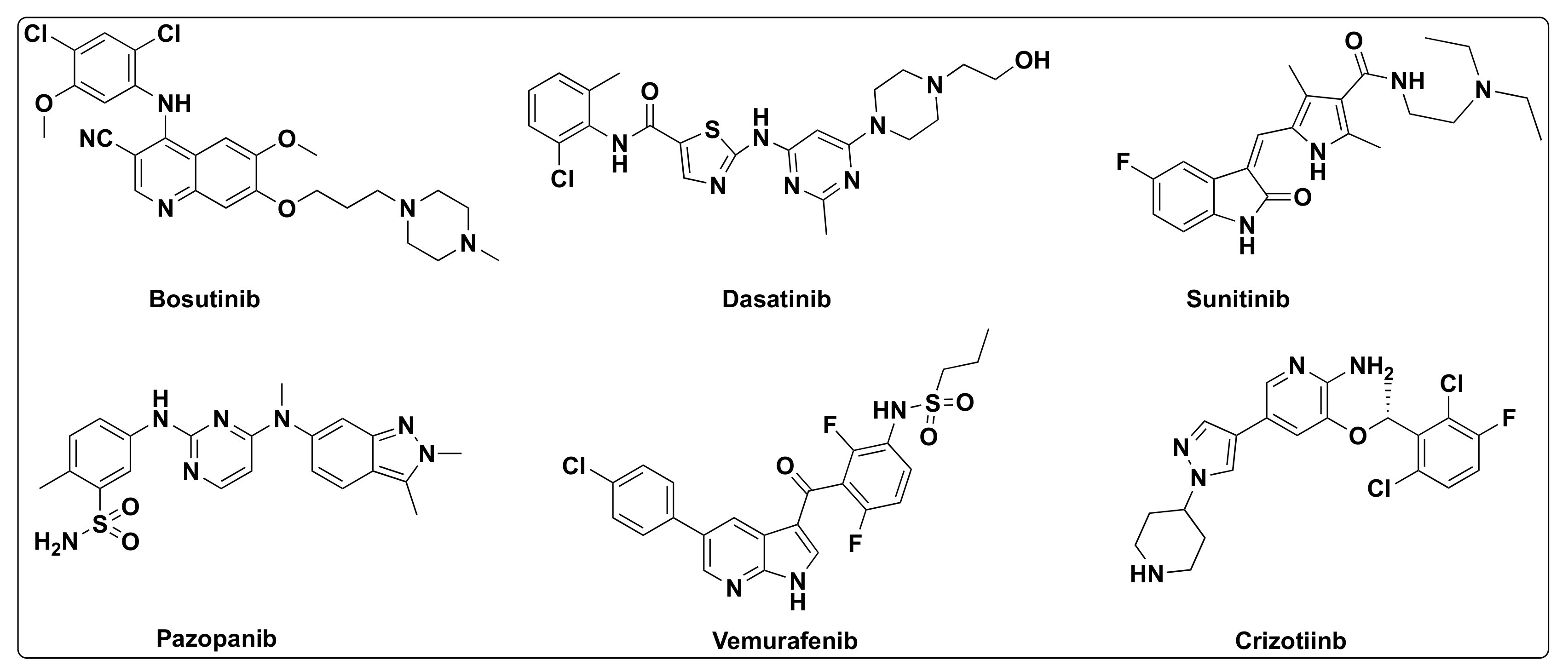
7.4. Quinazoline Based Dual Inhibition of HDAC and EGFR
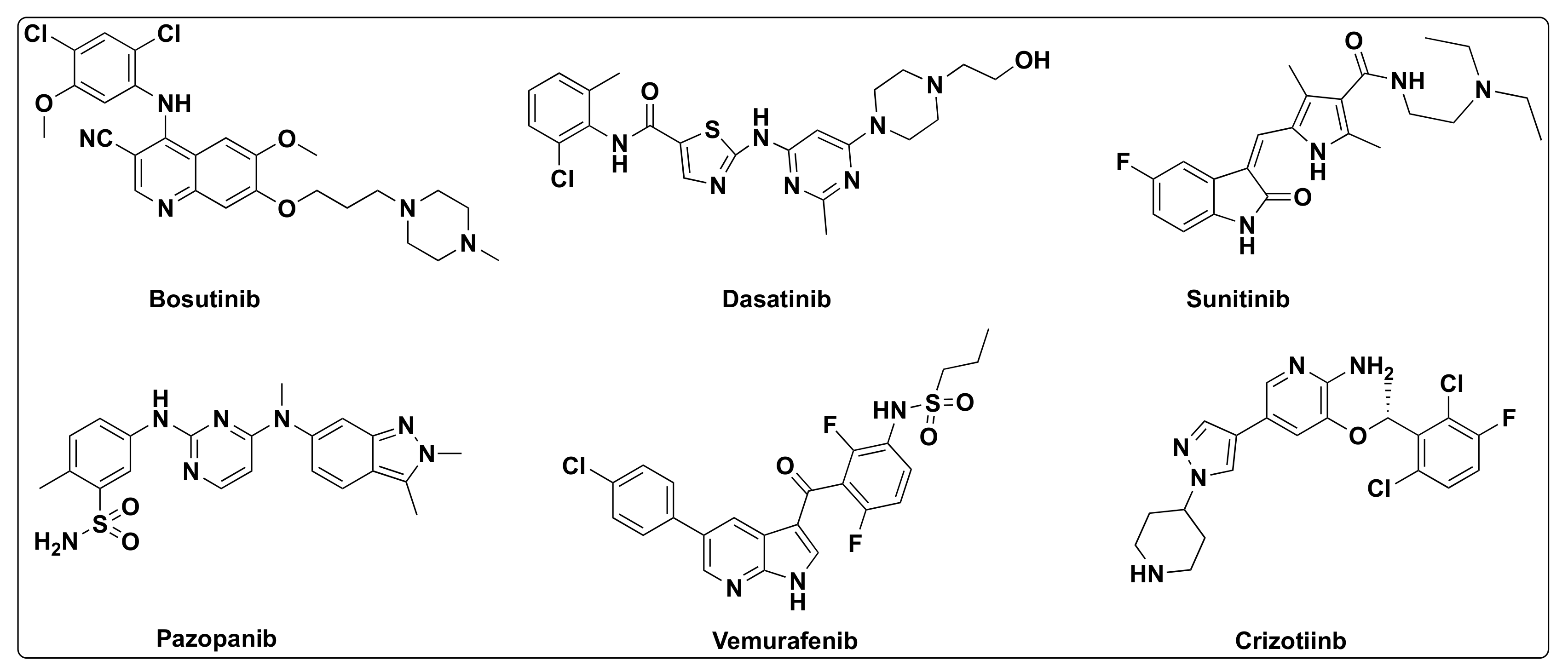
7.5. Quinazoline Based Dual Inhibition of HDAC and VEGFR
7.6. Quinazoline Based Dual Inhibitors of HDACs and Histone Methyltransferases (G9a)
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/20-Breast-fact-sheet.pdf (accessed on 9 March 2022).
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, S.A.; Arshia; Ishtiaq, M.; Khan, K.M. Quinazoline and Quinazolinone as Important Medicinal Scaffolds: A Comparative Patent Review (2011–2016). Expert Opin. Ther. Pat. 2018, 28, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Zviagin, E.; Skouta, R. FDA-Approved Oximes and Their Significance in Medicinal Chemistry. Pharmaceuticals 2022, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, O.M.; Bill, A.; Dhuguru, J.; Szollosi, D.E.; Edafiogho, I.O. Design, Synthesis and Biological Evaluation of Piperazino-Enaminones as Novel Suppressants of pro-Inflammatory Cytokines. Bioorgan. Med. Chem. 2018, 26, 3890–3898. [Google Scholar] [CrossRef]
- Szollosi, D.E.; Ghoneim, O.A.M.; Manzoor, M.K.; Dhuguru, J.; Edafiogho, I.O. Novel Piperazino-Enaminones Suppress Pro-Inflammatory Cytokines and Inhibit Chemokine Receptor CCR2. Inflammation 2016, 39, 2053–2061. [Google Scholar] [CrossRef]
- Khan, I.; Zaib, S.; Batool, S.; Abbas, N.; Ashraf, Z.; Iqbal, J.; Saeed, A. Quinazolines and Quinazolinones as Ubiquitous Structural Fragments in Medicinal Chemistry: An Update on the Development of Synthetic Methods and Pharmacological Diversification. Bioorgan. Med. Chem. 2016, 24, 2361–2381. [Google Scholar] [CrossRef]
- Karan, R.; Agarwal, P.; Sinha, M.; Mahato, N. Recent Advances on Quinazoline Derivatives: A Potential Bioactive Scaffold in Medicinal Chemistry. ChemEngineering 2021, 5, 73. [Google Scholar] [CrossRef]
- El-Azab, A.S.; ElTahir, K.E.H. Design and Synthesis of Novel 7-Aminoquinazoline Derivatives: Antitumor and Anticonvulsant Activities. Bioorgan. Med. Chem. Lett. 2012, 22, 1879–1885. [Google Scholar] [CrossRef]
- Liu, T.; Peng, F.; Cao, X.; Liu, F.; Wang, Q.; Liu, L.; Xue, W. Design, Synthesis, Antibacterial Activity, Antiviral Activity, and Mechanism of Myricetin Derivatives Containing a Quinazolinone Moiety. ACS Omega 2021, 6, 30826–30833. [Google Scholar] [CrossRef]
- Wang, H.-X.; Liu, H.-Y.; Li, W.; Zhang, S.; Wu, Z.; Li, X.; Li, C.-W.; Liu, Y.-M.; Chen, B.-Q. Design, Synthesis, Antiproliferative and Antibacterial Evaluation of Quinazolinone Derivatives. Med. Chem. Res. 2019, 28, 203–214. [Google Scholar] [CrossRef]
- Shagufta, S.; Ahmad, I. An Insight into the Therapeutic Potential of Quinazoline Derivatives as Anticancer Agents. Med. Chem. Commun. 2017, 8, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Sicard, R.; Dhuguru, J.; Liu, W.; Patel, N.; Landgraf, R.; Wilson, J.N. A Fluorescent Reporter of ATP Binding-Competent Receptor Kinases. Bioorgan. Med. Chem. Lett. 2012, 22, 5532–5535. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Liu, W.; Gonzalez, W.G.; Babinchak, W.M.; Miksovska, J.; Landgraf, R.; Wilson, J.N. Emission Tuning of Fluorescent Kinase Inhibitors: Conjugation Length and Substituent Effects. J. Org. Chem. 2014, 79, 4940–4947. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, P.; Matysiak, J.; Kuszta, P.; Czarnek, K.; Niezabitowska, E.; Baj, T. Quinazoline Derivatives as Potential Therapeutic Agents in Urinary Bladder Cancer Therapy. Front. Chem. 2021, 9, 765552. [Google Scholar] [CrossRef]
- Asif, M. Chemical Characteristics, Synthetic Methods, and Biological Potential of Quinazoline and Quinazolinone Derivatives. Int. J. Med. Chem. 2014, 2014, 395637. [Google Scholar] [CrossRef]
- Fishman, M.; Cruickshank, P.A. Febrifugine Antimalarial Agents. I. Pyridine Analogs of Febrifugine. J. Med. Chem. 1970, 13, 155–156. [Google Scholar] [CrossRef]
- Sharma, S.; Kumar, M.; Kumar, V.; Kumar, N. Metal-Free Transfer Hydrogenation of Nitroarenes in Water with Vasicine: Revelation of Organocatalytic Facet of an Abundant Alkaloid. J. Org. Chem. 2014, 79, 9433–9439. [Google Scholar] [CrossRef]
- Faisal, M.; Saeed, A. Chemical Insights into the Synthetic Chemistry of Quinazolines: Recent Advances. Front. Chem. 2021, 8, 594717. [Google Scholar] [CrossRef]
- Jafari, E.; Khajouei, M.R.; Hassanzadeh, F.; Hakimelahi, G.H.; Khodarahmi, G.A. Quinazolinone and Quinazoline Derivatives: Recent Structures with Potent Antimicrobial and Cytotoxic Activities. Res. Pharm. Sci. 2016, 11, 1–14. [Google Scholar]
- Mirgany, T.O.; Abdalla, A.N.; Arifuzzaman, M.; Motiur Rahman, A.F.M.; Al-Salem, H.S. Quinazolin-4(3H)-One Based Potential Multiple Tyrosine Kinase Inhibitors with Excellent Cytotoxicity. J. Enzym. Inhib. Med. Chem. 2021, 36, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R. Vincent Allfrey’s Work on Histone Acetylation. J. Biol. Chem. 2012, 287, 2270–2271. [Google Scholar] [CrossRef] [Green Version]
- Hesham, H.M.; Lasheen, D.S.; Abouzid, K.A.M. Chimeric HDAC Inhibitors: Comprehensive Review on the HDAC-Based Strategies Developed to Combat Cancer. Med. Res. Rev. 2018, 38, 2058–2109. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 1004. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.-S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone Deacetylase 3 (HDAC3) and Other Class I HDACs Regulate Colon Cell Maturation and P21 Expression and Are Deregulated in Human Colon Cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Al-Janadi, A.; Chandana, S.R.; Conley, B.A. Histone Deacetylation: An Attractive Target for Cancer Therapy? Drugs R D 2008, 9, 369–383. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. IJMS 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Dhuguru, J.; Skouta, R. Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef] [Green Version]
- Smalley, J.P.; Cowley, S.M.; Hodgkinson, J.T. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules 2020, 25, 4394. [Google Scholar] [CrossRef]
- Persidis, A. Cancer Multidrug Resistance. Nat. Biotechnol. 2000, 18, IT18–IT20. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of Conventional Anti-Cancer Drugs as New Tools against Multidrug Resistant Tumors. Drug Resist. Updates 2020, 50, 100682. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.A.; Fawell, S.; Floc’h, N.; Flemington, V.; McKerrecher, D.; Smith, P.D. Challenges and Opportunities in Cancer Drug Resistance. Chem. Rev. 2021, 121, 3297–3351. [Google Scholar] [CrossRef]
- Szumilak, M.; Wiktorowska-Owczarek, A.; Stanczak, A. Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance? Molecules 2021, 26, 2601. [Google Scholar] [CrossRef]
- Teiten, M.-H.; Dicato, M.; Diederich, M. Hybrid Curcumin Compounds: A New Strategy for Cancer Treatment. Molecules 2014, 19, 20839–20863. [Google Scholar] [CrossRef]
- Cheng, J.; Qin, J.; Guo, S.; Qiu, H.; Zhong, Y. Design, Synthesis and Evaluation of Novel HDAC Inhibitors as Potential Antitumor Agents. Bioorgan. Med. Chem. Lett. 2014, 24, 4768–4772. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, J.; Jia, Y.; Wang, X.; Zhang, L.; Liu, C.; Fang, H.; Xu, W. Development of Tetrahydroisoquinoline-Based Hydroxamic Acid Derivatives: Potent Histone Deacetylase Inhibitors with Marked in Vitro and in Vivo Antitumor Activities. J. Med. Chem. 2011, 54, 2823–2838. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Chao, M.-W.; Tu, H.-J.; Chen, L.-C.; Hsu, K.-C.; Liou, J.-P.; Yang, C.-R.; Yen, S.-C.; HuangFu, W.-C.; Pan, S.-L. A Novel Dual HDAC and HSP90 Inhibitor, MPT0G449, Downregulates Oncogenic Pathways in Human Acute Leukemia in Vitro and in Vivo. Oncogenesis 2021, 10, 39. [Google Scholar] [CrossRef]
- Chen, J.; Sang, Z.; Jiang, Y.; Yang, C.; He, L. Design, Synthesis, and Biological Evaluation of Quinazoline Derivatives as Dual HDAC1 and HDAC6 Inhibitors for the Treatment of Cancer. Chem. Biol Drug Des. 2019, 93, 232–241. [Google Scholar] [CrossRef]
- Akinleye, A.; Avvaru, P.; Furqan, M.; Song, Y.; Liu, D. Phosphatidylinositol 3-Kinase (PI3K) Inhibitors as Cancer Therapeutics. J. Hematol. Oncol. 2013, 6, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Wang, C.; Ji, M.; Wu, D.; Lv, Y.; Zhang, K.; Dong, Y.; Jin, J.; Chen, J.; Zhang, J.; et al. Discovery and Optimization of 2-Amino-4-Methylquinazoline Derivatives as Highly Potent Phosphatidylinositol 3-Kinase Inhibitors for Cancer Treatment. J. Med. Chem. 2018, 61, 6087–6109. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The Phosphatidylinositol 3-Kinase–AKT Pathway in Human Cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K Signalling in Cancer: Opportunities, Challenges and Limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K Inhibitors Are Finally Coming of Age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Copanlisib: First Global Approval. Drugs 2017, 77, 2057–2062. [Google Scholar] [CrossRef]
- Zhang, K.; Lai, F.; Lin, S.; Ji, M.; Zhang, J.; Zhang, Y.; Jin, J.; Fu, R.; Wu, D.; Tian, H.; et al. Design, Synthesis, and Biological Evaluation of 4-Methyl Quinazoline Derivatives as Anticancer Agents Simultaneously Targeting Phosphoinositide 3-Kinases and Histone Deacetylases. J. Med. Chem. 2019, 62, 6992–7014. [Google Scholar] [CrossRef]
- Yan, Z.; Zhang, K.; Ji, M.; Xu, H.; Chen, X. A Dual PI3K/HDAC Inhibitor Downregulates Oncogenic Pathways in Hematologic Tumors In Vitro and In Vivo. Front. Pharmacol. 2021, 12, 741697. [Google Scholar] [CrossRef]
- Thakur, A.; Tawa, G.J.; Henderson, M.J.; Danchik, C.; Liu, S.; Shah, P.; Wang, A.Q.; Dunn, G.; Kabir, M.; Padilha, E.C.; et al. Design, Synthesis, and Biological Evaluation of Quinazolin-4-One-Based Hydroxamic Acids as Dual PI3K/HDAC Inhibitors. J. Med. Chem. 2020, 63, 4256–4292. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET Family in Immunity and Disease. Signal Transduct. Target. Ther. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Sanchez, R.; Zhou, M.-M. Scaling the Druggability Landscape of Human Bromodomains, a New Class of Drug Targets. J. Med. Chem. 2012, 55, 7342–7345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadury, J.; Nilsson, L.M.; Veppil Muralidharan, S.; Green, L.C.; Li, Z.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. BET and HDAC Inhibitors Induce Similar Genes and Biological Effects and Synergize to Kill in Myc-Induced Murine Lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and Extra-Terminal Motif Inhibitors: A Review of Preclinical and Clinical Advances in Cancer Therapy. Future Sci. 2019, 5, FSO372. [Google Scholar] [CrossRef] [Green Version]
- Mita, M.M.; Mita, A.C. Bromodomain Inhibitors a Decade Later: A Promise Unfulfilled? Br. J. Cancer 2020, 123, 1713–1714. [Google Scholar] [CrossRef]
- Qi, J.; Shi, Y. Selective Targeting of Different Bromodomains by Small Molecules. Cancer Cell 2020, 37, 764–766. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving Clinical Success with BET Inhibitors as Anti-Cancer Agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.; Bamborough, P.; Petretich, M.; et al. Selective Targeting of BD1 and BD2 of the BET Proteins in Cancer and Immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective Inhibition of the BD2 Bromodomain of BET Proteins in Prostate Cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) Reduces Vascular Inflammation in Vitro and in CVD Patients by a BET-Dependent Epigenetic Mechanism. Clin. Epigenetics 2019, 11, 102. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, P.; Chen, H.; Wold, E.A.; Tian, B.; Brasier, A.R.; Zhou, J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem. 2017, 60, 4533–4558. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Gao, W. Current Status in the Discovery of Dual BET/HDAC Inhibitors. Bioorgan. Med. Chem. Lett. 2021, 31, 127671. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Sharma, S.; Qi, J.; Valenta, J.A.; Schaub, L.J.; Shah, B.; Peth, K.; Portier, B.P.; Rodriguez, M.; Devaraj, S.G.T.; et al. Highly Active Combination of BRD4 Antagonist and Histone Deacetylase Inhibitor against Human Acute Myelogenous Leukemia Cells. Mol. Cancer Ther. 2014, 13, 1142–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, M.; He, L.; Zheng, L.; Huang, L.; Zhou, Y.; Wang, T.; Chen, Y.; Shen, M.; Wang, F.; Yang, Z.; et al. Structure-Based Design, Synthesis and in Vitro Antiproliferative Effects Studies of Novel Dual BRD4/HDAC Inhibitors. Bioorgan. Med. Chem. Lett. 2017, 27, 4051–4055. [Google Scholar] [CrossRef]
- Ismail, R.S.M.; Ismail, N.S.M.; Abuserii, S.; Abou El Ella, D.A. Recent Advances in 4-Aminoquinazoline Based Scaffold Derivatives Targeting EGFR Kinases as Anticancer Agents. Future J. Pharm. Sci. 2016, 2, 9–19. [Google Scholar] [CrossRef]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in Solid Tumours: Diagnostic Strategies, Targeted Therapy, and Acquired Resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic Kinase Signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Eroglu, Z.; Tagawa, T.; Somlo, G. Human Epidermal Growth Factor Receptor Family-Targeted Therapies in the Treatment of HER2-Overexpressing Breast Cancer. Oncologist 2014, 19, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Opdam, F.L.; Guchelaar, H.; Beijnen, J.H.; Schellens, J.H.M. Lapatinib for Advanced or Metastatic Breast Cancer. Oncologist 2012, 17, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Novel Chimeric Histone Deacetylase Inhibitors: A Series of Lapatinib Hybrides as Potent Inhibitors of Epidermal Growth Factor Receptor (EGFR), Human Epidermal Growth Factor Receptor 2 (HER2), and Histone Deacetylase Activity. J. Med. Chem. 2010, 53, 8546–8555. [Google Scholar] [CrossRef] [PubMed]
- Voigtlaender, M.; Schneider-Merck, T.; Trepel, M. Lapatinib. In Small Molecules in Oncology; Martens, U.M., Ed.; Recent Results in Cancer Research; Springer International Publishing: Cham, Switzerland, 2018; Volume 211, pp. 19–44. [Google Scholar] [CrossRef]
- Beckers, T.; Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Maier, T.; Ciossek, T.; Baer, T.; Kelter, G.; et al. Chimerically Designed HDAC- and Tyrosine Kinase Inhibitors. A Series of Erlotinib Hybrids as Dual-Selective Inhibitors of EGFR, HER2 and Histone Deacetylases. Med. Chem. Commun. 2012, 3, 829. [Google Scholar] [CrossRef]
- Mahboobi, S.; Dove, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Design of Chimeric Histone Deacetylase- and Tyrosine Kinase-Inhibitors: A Series of Imatinib Hybrides as Potent Inhibitors of Wild-Type and Mutant BCR-ABL, PDGF-Rβ, and Histone Deacetylases. J. Med. Chem. 2009, 52, 2265–2279. [Google Scholar] [CrossRef]
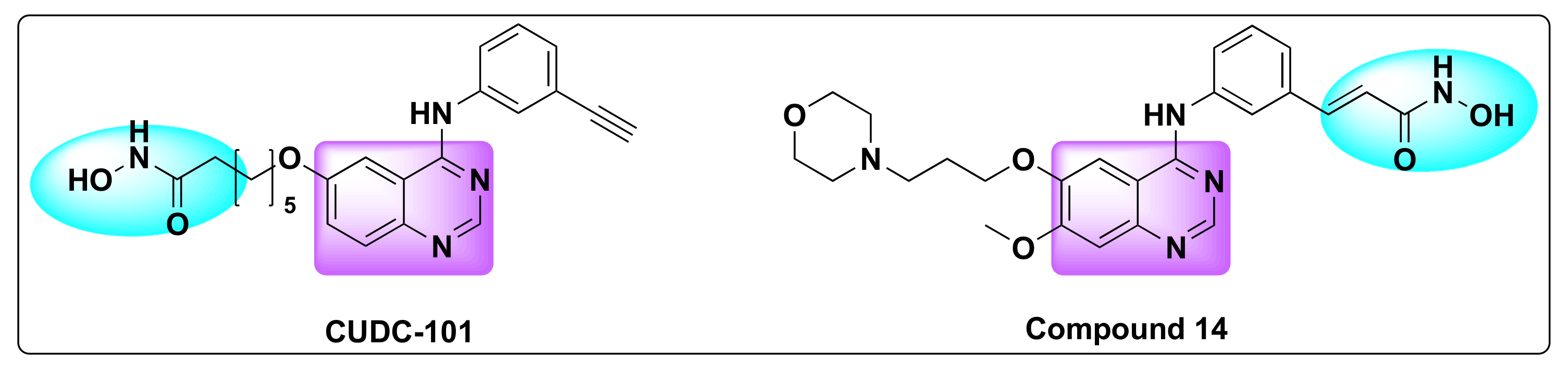
- Cai, X.; Zhai, H.-X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.-J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-Methoxyquinazolin-6-Yloxy)-N-Hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Wang, J.; Pursell, N.W.; Samson, M.E.S.; Atoyan, R.; Ma, A.W.; Selmi, A.; Xu, W.; Cai, X.; Voi, M.; Savagner, P.; et al. Potential Advantages of CUDC-101, a Multitargeted HDAC, EGFR, and HER2 Inhibitor, in Treating Drug Resistance and Preventing Cancer Cell Migration and Invasion. Mol. Cancer Ther. 2013, 12, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Ma, D.; Wei, D.; Lu, T.; Yu, K.; Zhang, Z.; Wang, W.; Fang, Q.; Wang, J. CUDC-101 Overcomes Arsenic Trioxide Resistance via Caspase-Dependent Promyelocytic Leukemia-Retinoic Acid Receptor Alpha Degradation in Acute Promyelocytic Leukemia. Anti-Cancer Drugs 2020, 31, 158–168. [Google Scholar] [CrossRef]
- Galloway, T.J.; Wirth, L.J.; Colevas, A.D.; Gilbert, J.; Bauman, J.E.; Saba, N.F.; Raben, D.; Mehra, R.; Ma, A.W.; Atoyan, R.; et al. A Phase I Study of CUDC-101, a Multitarget Inhibitor of HDACs, EGFR, and HER2, in Combination with Chemoradiation in Patients with Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1566–1573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Su, M.; Chen, Y.; Li, J.; Lu, W. The Design and Synthesis of a New Class of RTK/HDAC Dual-Targeted Inhibitors. Molecules 2013, 18, 6491–6503. [Google Scholar] [CrossRef]
- Ding, C.; Li, D.; Wang, Y.-W.; Han, S.-S.; Gao, C.-M.; Tan, C.-Y.; Jiang, Y.-Y. Discovery of ErbB/HDAC Inhibitors by Combining the Core Pharmacophores of HDAC Inhibitor Vorinostat and Kinase Inhibitors Vandetanib, BMS-690514, Neratinib, and TAK-285. Chin. Chem. Lett. 2017, 28, 1220–1227. [Google Scholar] [CrossRef]
- Goehringer, N.; Biersack, B.; Peng, Y.; Schobert, R.; Herling, M.; Ma, A.; Nitzsche, B.; Höpfner, M. Anticancer Activity and Mechanisms of Action of New Chimeric EGFR/HDAC-Inhibitors. IJMS 2021, 22, 8432. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Zhang, D.; Wang, L.; Lu, T.; Jiao, Y. Discovery of Novel Potent VEGFR-2 Inhibitors Exerting Significant Antiproliferative Activity against Cancer Cell Lines. J. Med. Chem. 2018, 61, 140–157. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.R.; Baker, D.; James, N.H.; Ratcliffe, K.; Jenkins, M.; Ashton, S.E.; Sproat, G.; Swann, R.; Gray, N.; Ryan, A.; et al. Vascular Endothelial Growth Factor Receptors VEGFR-2 and VEGFR-3 Are Localized Primarily to the Vasculature in Human Primary Solid Cancers. Clin. Cancer Res. 2010, 16, 3548–3561. [Google Scholar] [CrossRef] [Green Version]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomäki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellström, M.; Schomber, T.; Peltonen, R.; et al. Blocking VEGFR-3 Suppresses Angiogenic Sprouting and Vascular Network Formation. Nature 2008, 454, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Neves, K.B.; Rios, F.J.; van der Mey, L.; Alves-Lopes, R.; Cameron, A.C.; Volpe, M.; Montezano, A.C.; Savoia, C.; Touyz, R.M. VEGFR (Vascular Endothelial Growth Factor Receptor) Inhibition Induces Cardiovascular Damage via Redox-Sensitive Processes. Hypertension 2018, 71, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C. A Deeper Understanding of VEGFR Inhibitors. Nat. Rev. Drug Discov. 2012, 11, 831. [Google Scholar] [CrossRef]
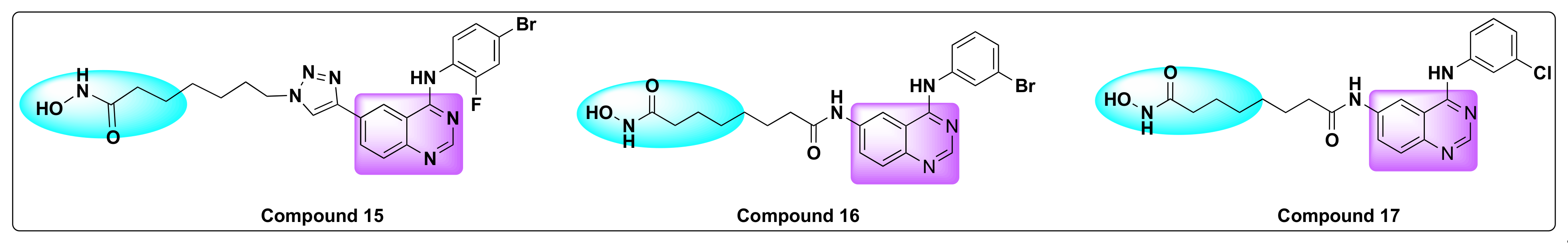
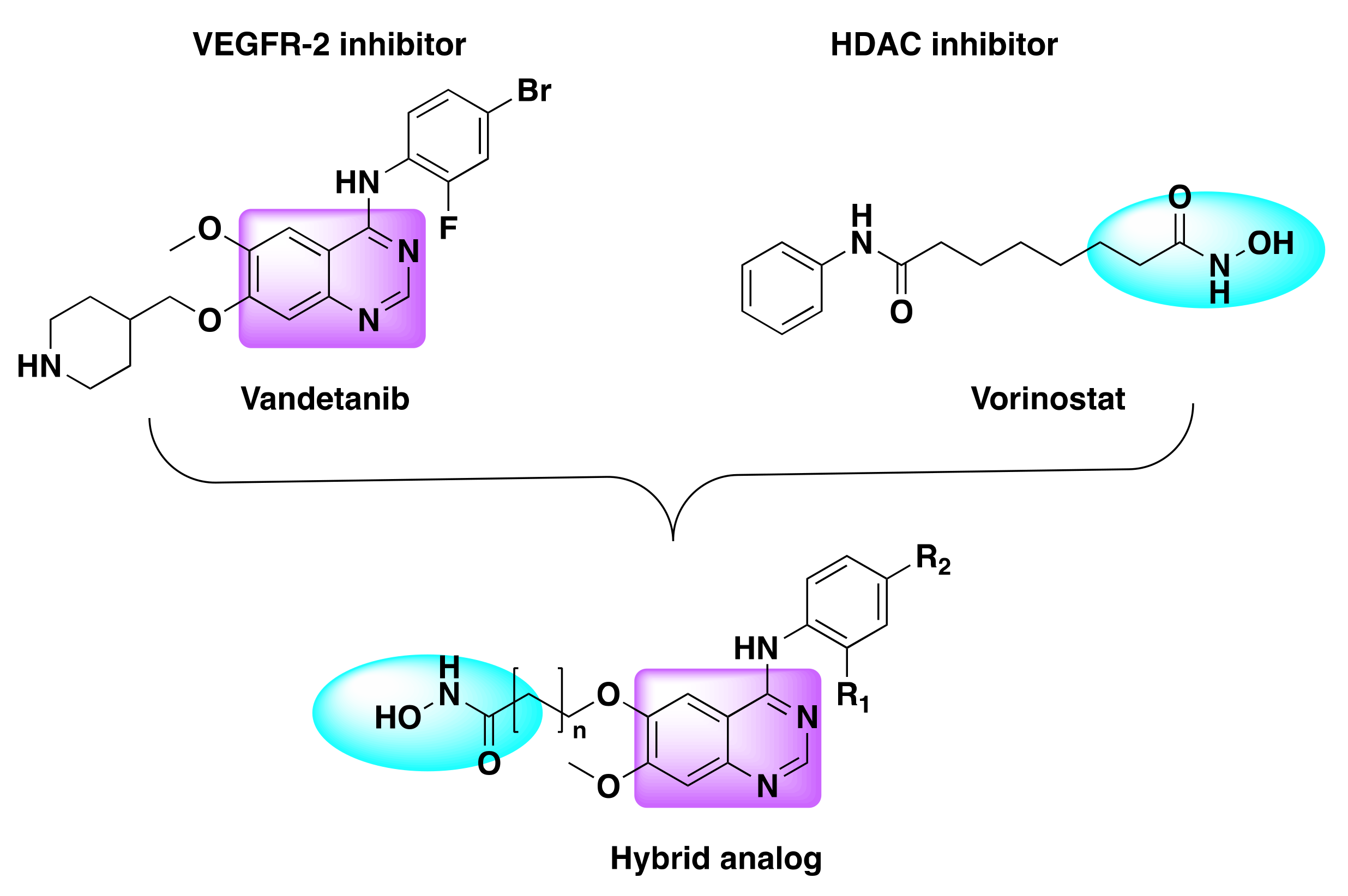
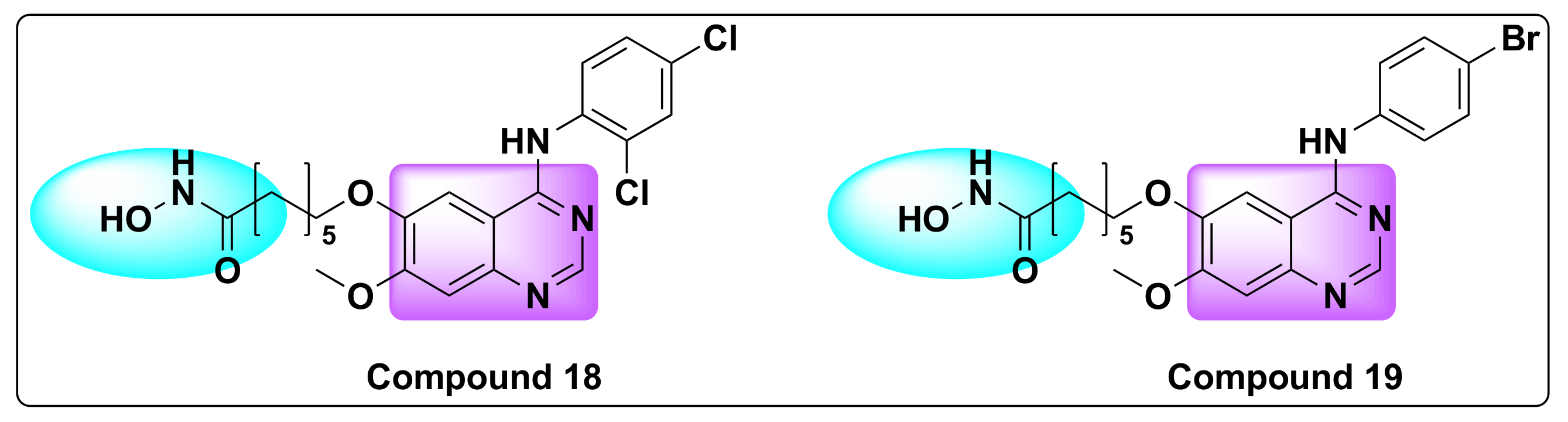
- Peng, F.-W.; Wu, T.-T.; Ren, Z.-W.; Xue, J.-Y.; Shi, L. Hybrids from 4-Anilinoquinazoline and Hydroxamic Acid as Dual Inhibitors of Vascular Endothelial Growth Factor Receptor-2 and Histone Deacetylase. Bioorgan. Med. Chem. Lett. 2015, 25, 5137–5141. [Google Scholar] [CrossRef]
- Peng, F.-W.; Xuan, J.; Wu, T.-T.; Xue, J.-Y.; Ren, Z.-W.; Liu, D.-K.; Wang, X.-Q.; Chen, X.-H.; Zhang, J.-W.; Xu, Y.-G.; et al. Design, Synthesis and Biological Evaluation of N-Phenylquinazolin-4-Amine Hybrids as Dual Inhibitors of VEGFR-2 and HDAC. Eur. J. Med. Chem. 2016, 109, 1–12. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Huang, M.; Wang, T.; Li, Z.; Chen, Y.; Liu, C.; Lei, Z.; Chu, X. Lysine Methylation of Transcription Factors in Cancer. Cell Death Dis. 2019, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Riahi, H.; Brekelmans, C.; Foriel, S.; Merkling, S.H.; Lyons, T.A.; Itskov, P.M.; Kleefstra, T.; Ribeiro, C.; van Rij, R.P.; Kramer, J.M.; et al. The Histone Methyltransferase G9a Regulates Tolerance to Oxidative Stress–Induced Energy Consumption. PLoS Biol. 2019, 17, e2006146. [Google Scholar] [CrossRef] [PubMed]
- Watson, Z.L.; Yamamoto, T.M.; McMellen, A.; Kim, H.; Hughes, C.J.; Wheeler, L.J.; Post, M.D.; Behbakht, K.; Bitler, B.G. Histone Methyltransferases EHMT1 and EHMT2 (GLP/G9A) Maintain PARP Inhibitor Resistance in High-Grade Serous Ovarian Carcinoma. Clin. Epigenetics 2019, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A.; Przanowski, P.; Jackl, J.; Wojtas, B.; Kaminska, B. BIX01294, an Inhibitor of Histone Methyltransferase, Induces Autophagy-Dependent Differentiation of Glioma Stem-like Cells. Sci. Rep. 2016, 6, 38723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Li, F.; Babault, N.; Dong, A.; Zeng, H.; Wu, H.; Chen, X.; Arrowsmith, C.H.; Brown, P.J.; Liu, J.; et al. Discovery of Potent and Selective Inhibitors for G9a-Like Protein (GLP) Lysine Methyltransferase. J. Med. Chem. 2017, 60, 1876–1891. [Google Scholar] [CrossRef] [Green Version]
- Haebe, J.R.; Bergin, C.J.; Sandouka, T.; Benoit, Y.D. Emerging Role of G9a in Cancer Stemness and Promises as a Therapeutic Target. Oncogenesis 2021, 10, 76. [Google Scholar] [CrossRef]
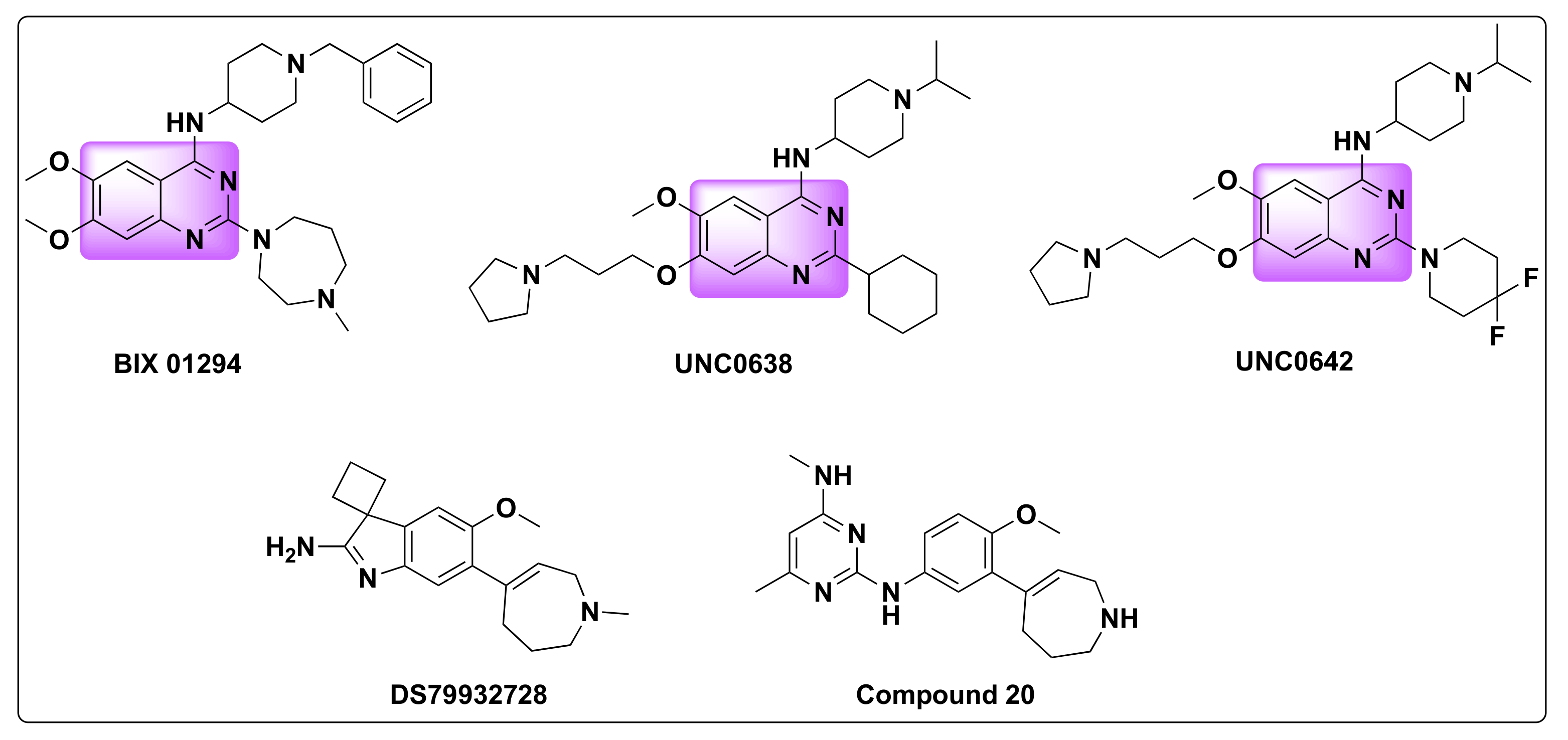
- Katayama, K.; Ishii, K.; Terashima, H.; Tsuda, E.; Suzuki, M.; Yotsumoto, K.; Hiramoto, K.; Yasumatsu, I.; Torihata, M.; Ishiyama, T.; et al. Discovery of DS79932728: A Potent, Orally Available G9a/GLP Inhibitor for Treating β-Thalassemia and Sickle Cell Disease. ACS Med. Chem. Lett. 2021, 12, 121–128. [Google Scholar] [CrossRef]
- Katayama, K.; Ishii, K.; Tsuda, E.; Yotsumoto, K.; Hiramoto, K.; Suzuki, M.; Yasumatsu, I.; Igarashi, W.; Torihata, M.; Ishiyama, T.; et al. Discovery of Novel Histone Lysine Methyltransferase G9a/GLP (EHMT2/1) Inhibitors: Design, Synthesis, and Structure-Activity Relationships of 2,4-Diamino-6-Methylpyrimidines. Bioorgan. Med. Chem. Lett. 2020, 30, 127475. [Google Scholar] [CrossRef]
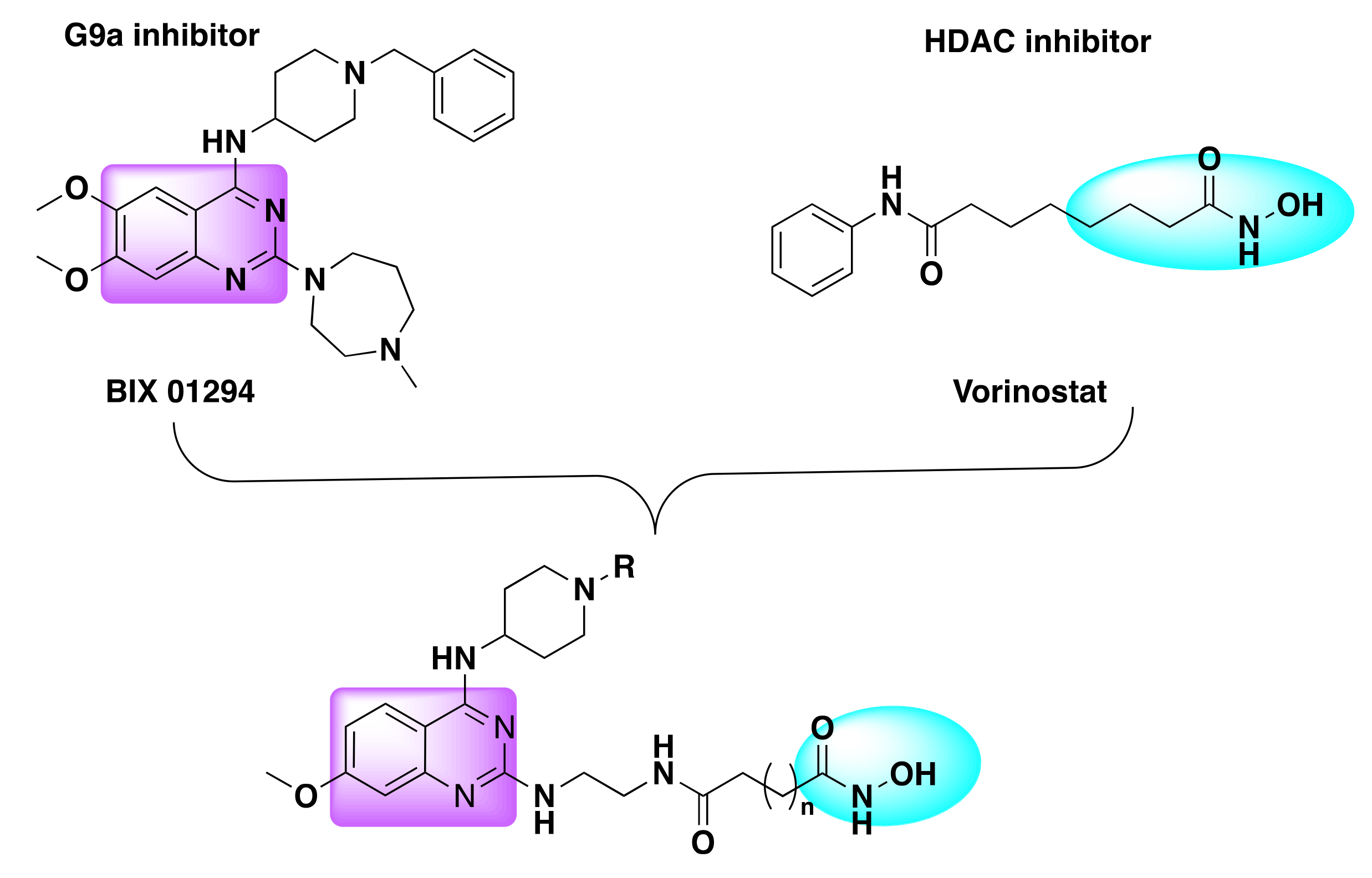
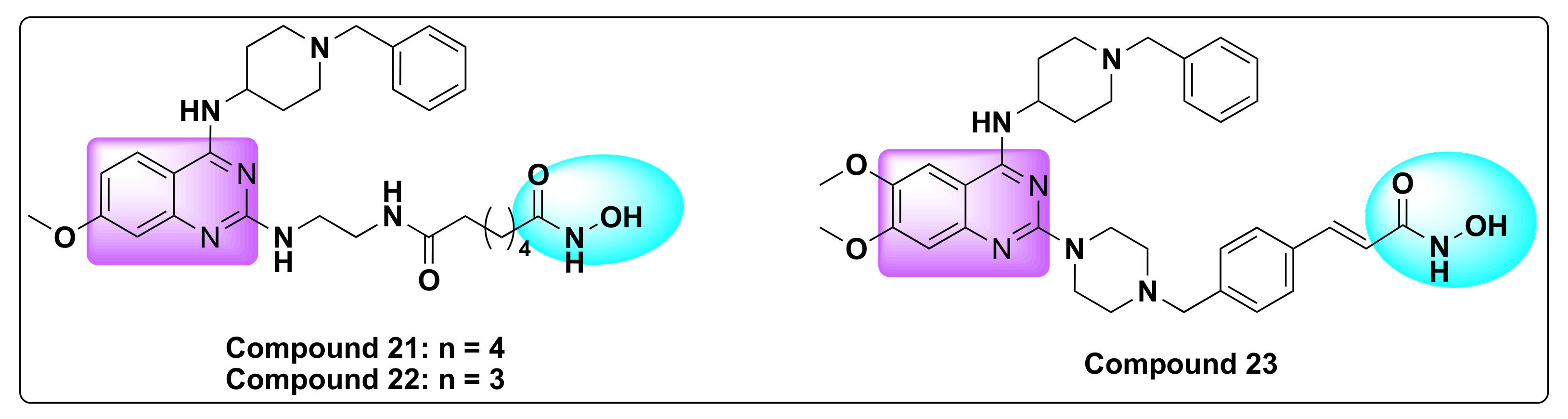
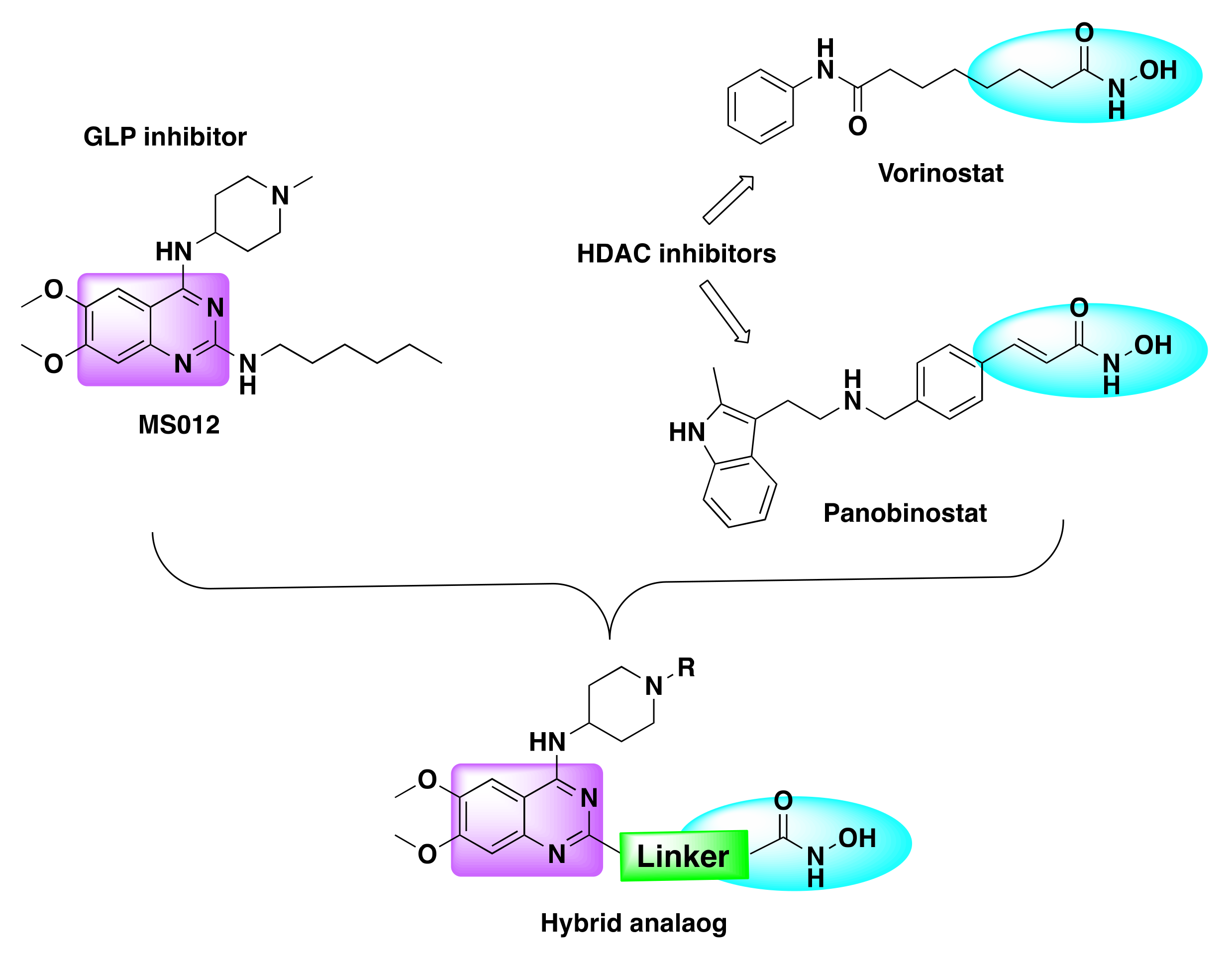
- Zang, L.; Kondengaden, S.M.; Zhang, Q.; Li, X.; Sigalapalli, D.K.; Kondengadan, S.M.; Huang, K.; Li, K.K.; Li, S.; Xiao, Z.; et al. Structure Based Design, Synthesis and Activity Studies of Small Hybrid Molecules as HDAC and G9a Dual Inhibitors. Oncotarget 2017, 8, 63187–63207. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Dai, Q.; Yuan, Z.; Fan, T.; Zhang, C.; Liu, Z.; Chu, B.; Sun, Q.; Chen, Y.; Jiang, Y. Quinazoline-Based Hydroxamic Acid Derivatives as Dual Histone Methylation and Deacetylation Inhibitors for Potential Anticancer Agents. Bioorgan. Med. Chem. 2022, 53, 116524. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | IC50 (nM) | |
|---|---|---|
| HDAC1 | HDAC6 | |
| 1 | 31 ± 0.37 | 16.15 ± 0.62 |
| 2 | 37 ± 0.24 | 35 ± 0.71 |
| SAHA | 11.4 ± 0.39 | 16.01 ± 1.37 |
| Compd | IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| HepG2 | M-M-231 | MCF-7 | H1975 | H460 | Hela | U266 | RPMI8226 | |
| 1 | 3.5 ± 0.48 | 2.65 ± 0.71 | 2.55 ± 0.40 | 2.88 ± 0.69 | 1.05 ± 0.02 | 2.41 ± 0.59 | <0.01 | <0.01 |
| 2 | 4.51 ± 0.33 | 7.41 ± 0.81 | 8.62 ± 1.01 | 39.51 ± 1.00 | 37.59 ± 2.62 | 3.16 ± 0.15 | 0.15 ± 0.01 | 0.37 ± 0.21 |
| SAHA | >1000 | >1000 | >1000 | >1000 | >1000 | 720 ± 13.02 | 569 ± 21.00 | 420 ± 9.37 |
| Compd | IC50 (nM) | IC50 (µM) HCT116 | ||
|---|---|---|---|---|
| PI3Kα | HDAC1 | mTOR | ||
| 4 | 42 | 1.4 | 2861 | 0.15 |
| 5 | 226 | 1.1 | 6262 | 0.007 |
| CUDC-907 | 69 | 0.36 | 431 | 0.005 |
| BKM120 | 20 | — | — | 1.3 |
| SAHA | — | 32 | — | 1.9 |
| Cancer Type | Cell Line | IC50 (µM) | ||||
|---|---|---|---|---|---|---|
| Compd 4 | Compd 5 | SAHA | BKM120 | CUDC-907 | ||
| AML | THP-1 | 5.4 | 0.91 | 9.5 | 12 | — |
| CML | K562 | 6.8 | 1.2 | 10 | 7.9 | 0.28 |
| Breast cancer | MCF-7 | 2.0 | 1.1 | 4.1 | 5.7 | 0.041 |
| MDA-MB-453 | 0.30 | 0.004 | 0.37 | 0.37 | 0.009 | |
| Colon | HCT-8 | 0.32 | 0.059 | 0.35 | 1.7 | 0.005 |
| HCT116 | 0.15 | 0.007 | 1.9 | 1.3 | 0.005 | |
| Brain cancer | U87 | 3.2 | 0.34 | 10 | 4.8 | 0.007 |
| NSCLC | NCI-H460 | 3.0 | 2.7 | >100 | 3.2 | 0.15 |
| Lung cancer | NCI-H1299 | 1.0 | 0.18 | 5.1 | 3.6 | 0.025 |
| Pancreas cancer | Capan2 | 0.43 | 0.27 | 3.0 | 42 | 0.007 |
| SW1990 | 0.9 | 0.23 | 3.8 | 1.3 | 0.18 | |
| Prostrate | DU145 | 0.54 | 0.066 | 1.8 | 45 | 0.56 |
| Stomach | HGC-27 | 0.11 | 0.014 | 0.35 | 0.99 | 0.041 |
| Liver | HepG2 | 1.1 | 0.076 | 2.0 | 3.2 | 0.015 |
| Huh7 | 2.5 | 0.24 | 4.3 | 2.0 | 0.56 | |
| BEL-7402 | 5.2 | 1.6 | 7.1 | 13 | 0.013 | |
| Compd | IC50 (nM) | |||||
|---|---|---|---|---|---|---|
| PI3Kα | PI3Kβ | PI3Kγ | PI3Kδ | HDAC6 | HDAC Isoform Selectivity | |
| 6 | 47 | — | 9 | 7.0 | 12 | >39 |
| PI-103 | 4 | 7 | 55 | 5 | — | — |
| TSA | — | — | — | — | 6 | — |
| Cancer Type | Cell Line | GI50 (µM) | ||
|---|---|---|---|---|
| Compd 6 | Idelalisib | SAHA | ||
| Leukemia | CCRF-CEM | 0.7 | 22.3 | 0.7 |
| SR | 0.7 | 0.4 | ||
| NSCLC | HOP-62 | 2.4 | >100 | 1.6 |
| HOP-92 | 0.9 | 14.1 | 2.9 | |
| Colon cancer | HCT-2998 | 2.1 | 38.5 | 1.9 |
| KM-12 | 1.7 | 1.2 | 0.8 | |
| CNS cancer | SNB-75 | 0.7 | 1.2 | 0.8 |
| U251 | 1.7 | 53.2 | 1.6 | |
| Melanoma | LOX IMV1 | 1.6 | 33.5 | 1.2 |
| M14 | 1.8 | 37.8 | 1.3 | |
| Ovarian cancer | IGROV1 | 1.5 | 4.8 | 1.1 |
| OVCAR3 | 1.2 | 17.7 | 1.4 | |
| Renal cancer | A498 | 1.0 | 1.1 | 1.4 |
| CAKI-1 | 1.3 | 20.5 | 1.2 | |
| RXF 393 | 1.2 | 1.4 | 1.3 | |
| Breast cancer | MDA-MB-232/ATCC | 1.8 | 42.3 | 2.5 |
| HS 578T | 2.0 | 6.0 | 3.6 | |
| T-47D | 1.2 | 5.2 | 0.5 | |
| Compd | IC50 (nM) | ||
|---|---|---|---|
| BRD4/BD1 | BRD4/BD2 | HDAC1 | |
| 7 | >5000 | 225 ± 32 | 32 ± 10 |
| 8 | >5000 | 401 ± 21 | 204 ± 21 |
| RVX-208 | 1985 ± 233 | 67 ± 9 | — |
| SAHA | — | — | 10 ± 3 |
| Compd | IC50 (µM) | ||
|---|---|---|---|
| MV4-11 | OCI-AML2 | OCI-AML3 | |
| 7 | 1.67 ± 0.21 | 1.52 ± 0.21 | 1.09 ± 0.18 |
| 8 | 0.56 ± 0.09 | 0.38 ± 0.08 | 0.43 ± 0.18 |
| RVX-208 | 4.48 ± 0.21 | 8.31 ± 0.32 | 7.17 ± 0.34 |
| SAHA | 0.98 ± 0.27 | 0.78 ± 0.32 | 0.85 ± 0.29 |
| Compd | IC50 (µM) | |||||
|---|---|---|---|---|---|---|
| HDAC | rHDAC1 | rHDAC3 | rHDAC6 | rHDAC8 | H3K (EC50) | |
| 9 | 0.63 | 0.61 | 6.30 | 0.23 | 11 | 3.65 |
| 10 | 0.047 | 0.035 | 0.066 | 0.086 | 0.63 | 4.70 |
| SNDX/MS 275 | 11 | 0.082 | 0.62 | >30 | ≥25 | 2.35 |
| Compd | IC50 (µM) | |||||||
|---|---|---|---|---|---|---|---|---|
| EGFR | HER2 | PDGF-Rβ | InsR | Ab11 | CDK2 | PKA | PIK1 | |
| 9 | 0.025 | 0.0041 | 3.3 | 7.34 | 7.7 | 0.44 | 120 | 12 |
| 10 | 0.018 | 0.011 | 2.5 | 3.6 | 5.3 | 0.92 | >100 | 6.80 |
| Lapatinib | 0.0029 | 0.0045 | 8.5 | 17 | 23 | 11 | >100 | >100 |
| Compd | IC50 (µM) | |||||
|---|---|---|---|---|---|---|
| A549 | HeLa | A431 | Cal27 | SKOV3 | SKBR3 | |
| 9 | 3.9 | 3.2 | 1.9 | 0.82 | 2.1 | 0.51 |
| 10 | 1.2 | 0.97 | 0.72 | 0.59 | 1.0 | 0.35 |
| Lapatinib | 8.5 | 5.9 | 0.97 | 0.007 | 0.003 | 0.002 |
| SAHA | 1.8 | 1.5 | 4.2 | 3.2 | 2.2 | 2.6 |
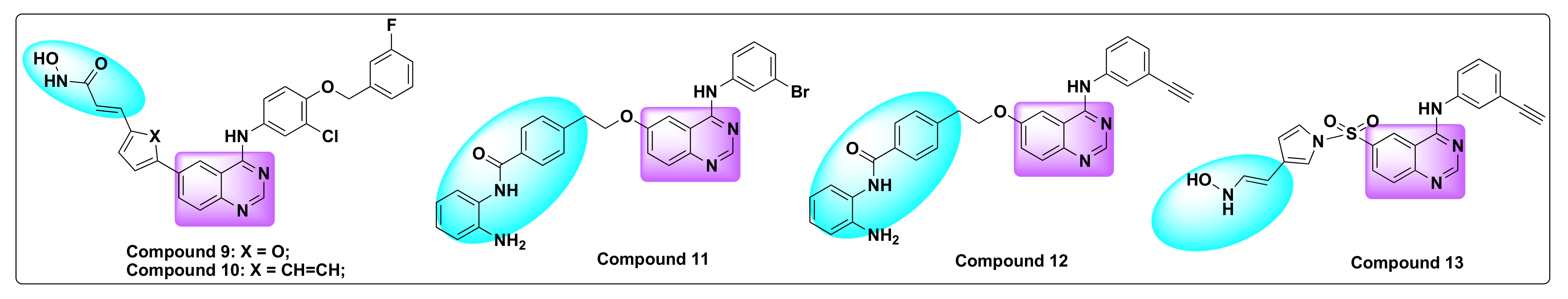
| Compd | IC50 (µM) | |||||
|---|---|---|---|---|---|---|
| nHDAC | cHDAC | rHDAC1 | rHDAC3 | rHDAC6 | rHDAC8 | |
| 11 | 0.25 | 2.46 | 0.074 | 0.51 | >32 | >32 |
| 12 | 0.20 | 1.85 | 0.041 | 0.55 | >32 | >32 |
| 13 | 0.0064 | 0.034 | 0.0065 | 0.015 | 0.0073 | 0.59 |
| SAHA | 0.079 | 0.063 | 0.013 | 0.037 | 0.025 | 1.2 |
| Erlotinib | >32 | >15.8 | >32 | >32 | >32 | >32 |
| Compd | IC50 (mM) | |||||||
|---|---|---|---|---|---|---|---|---|
| EGFR | HER2 | PDGF-Rβ | InsR | Ab11 | CDK2 | PKA | PIK1 | |
| 11 | 0.033 | 0.039 | 28.0 | 150 | 11.0 | 22.0 | >100 | >100 |
| 12 | 0.078 | 0.066 | 7.2 | 36 | 7.0 | 13.0 | >100 | >100 |
| 13 | 1.2 | 3.4 | 70.0 | 65.0 | 27.0 | >100 | >100 | >100 |
| Erlotinib | 0.005 | 0.12 | 2.3 | >100 | 2.1 | >100 | >100 | >100 |
| Compd | IC50 (µM) | |||||
|---|---|---|---|---|---|---|
| A549 | HeLa | A431 | Cal27 | SKOV3 | SKBR3 | |
| 11 | 1.3 | 1.45 | 1.7 | 0.62 | 5.1 | 6.3 |
| 12 | 0.9 | 1.35 | 1.6 | 0.61 | 3.4 | 5.1 |
| 13 | 0.24 | 0.5 | 0.26 | 0.16 | 0.19 | 0.56 |
| SAHA | 1.8 | 1.5 | 4.2 | 3.2 | 2.6 | 2.2 |
| Erlotinib | >50 | 8.3 | 2.5 | 0.074 | 15 | 31 |
| Compd | IC50 (nM) | ||
|---|---|---|---|
| HDAC | EGFR | HER2 | |
| CUDC101 | 4.4 | 2.4 | 15.7 |
| SAHA | 40.0 | — | — |
| Erlotinib | — | 48.0 | 134.8 |
| Lapatinib | — | 11.2 | 10.2 |
| Compd | IC50 (µM) | ||
|---|---|---|---|
| HDAC | HDAC3 | HDAC6 | |
| 14 | 0.16 ± 0.02 | 0.18 ± 0.05 | 0.56 ± 0.06 |
| SAHA | 0.25 ± 0.04 | 0.17 ± 0.02 | 0.23 ± 0.06 |
| Lapatinib | — | — | — |
| Compd | Invitro RTK Inhibition (%) | |
|---|---|---|
| EGFR (%) | HER2 (%) | |
| 14 | 9.7 | 47.2 |
| SAHA | 0 | 0 |
| Lapatinib | 92.7 | 92.0 |
| Compd | VEGFR2 (%) | HER2 (%) | EGFR (%) |
|---|---|---|---|
| 15 | 14.57 ± 26.24 | −76.87 ± 4.49 | 99.26 ± 0.48 |
| Lapatinib | — | 91.38 ± 1.28 | 99.46 ± 1.14 |
| Staurosporine | 97.90 ± 1.78 | — | — |
| Compd | IC50 (nM) | |||
|---|---|---|---|---|
| EGFR | HER2 | HDAC1 | HDAC6 | |
| 15 | 10.3 ± 0.8 | >1000 | 1.1 ± 0.1 | 4.3 ± 0.2 |
| Erlotinib | 13.3 ± 0.9 | — | — | — |
| Lapatinib | — | 23.9 ± 1.4 | — | |
| SAHA | — | — | 12.2 ± 0.6 | 11.4 ± 1.1 |
| Compd | IC50 (µM) | ||||
|---|---|---|---|---|---|
| A549 | BT-474 | A431 | SK-BR-3 | NCI-H1975 | |
| 15 | 0.71 ± 0.04 | 5.13 ± 0.62 | 0.26 ± 0.02 | 0.91 ± 0.05 | 7.85 ± 0.62 |
| Erlotinib | 16.83 ± 1.46 | 2.26 ± 0.17 | 1.85 ± 0.21 | 3.43 ± 0.19 | 23.76 ± 1.58 |
| SAHA | 2.57 ± 0.37 | 2.67 ± 0.38 | 2.29 ± 0.04 | 2.58 ± 0.13 | 1.90 ± 0.09 |
| Compd | IC50 (µM) | |||||
|---|---|---|---|---|---|---|
| DU145 | Hep-G2 | Jurkat | Hut78 | SupT11 | SMZ1 | |
| 16 | 3.53 ± 0.23 | 4.94 ± 0.38 | — | — | — | — |
| 17 | 3.23 ± 0.18 | 3.92 ± 0.25 | 1.40 ± 0.12 | 1.18 ± 0.22 | 6.22 ± 0.25 | 2.24 ± 0.17 |
| SAHA | 0.68 ± 0.04 | 3.22 ± 0.44 | 1.7 ± 0.17 | 5.07 ± 0.42 | 4.67 ± 0.31 | 2.87 ± 0.33 |
| Gefitinib | 11.88 ± 2.13 | 18.53 ± 1.78 | 10.95 ± 0.28 | >20 | >20 | >20 |
| Compd | IC50 | ||
|---|---|---|---|
| VEGFR2 (nM) | HDAC (nM) | MCF-7 (µM) | |
| 18 | 84 | 2.8 | 1.2 |
| Vandetanib | 62 | >10,000 | 18.5 |
| SAHA | >10,000 | 12 | 4.5 |
| Compd | IC50 | ||
|---|---|---|---|
| VEGFR-2 (nM) | HDAC (nM) | MCF-7 (µM) | |
| 19 | 74 | 2.2 | 0.85 |
| Vandetanib | 54 | >10,000 | 18.5 |
| SAHA | >10,000 | 15 | 4.2 |
| Compd | IC50 (nM) | |||
|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC6 | HDAC8 | |
| 19 | 1.8 | 3.3 | 16.4 | 4.6 |
| Compd | IC50 (µM) | |||
|---|---|---|---|---|
| MDA-MB-231 | MCF-7 | A549 | HEK293 | |
| 21 | 89.33 ± 1.23 | 79.43 ± 2.72 | >100 | 56.96 ± 1.12 |
| 22 | 10.02 ± 1.66 | 37.36 ± 2.20 | 36.24 ± 1.76 | 19.95 ± 0.19 |
| BIX-01294 | 2.155 ± 0.88 | 8.103 ± 1.99 | 21.74 ± 2.73 | 2.048 ± 0.98 |
| SAHA | 2.874 ± 0.84 | 8.124 ± 4.98 | 19.31 ± 1.26 | 2.482 ± 1.13 |
| Compd | IC50 (µM) | ||
|---|---|---|---|
| G9a * | Hela ** | K562 ** | |
| 21 | 37.79 ± 2.80 | 15.33 ± 0.79 | 27.75 ± 0.59 |
| 22 | 7.136 ± 1.62 | 13.8 ± 1.22 | 5.735 ± 1.23 |
| BIX-01294 | 4.563 ± 1.2 | — | — |
| SAHA | — | 5.044 ± 0.53 | 2.056 ± 0.59 |
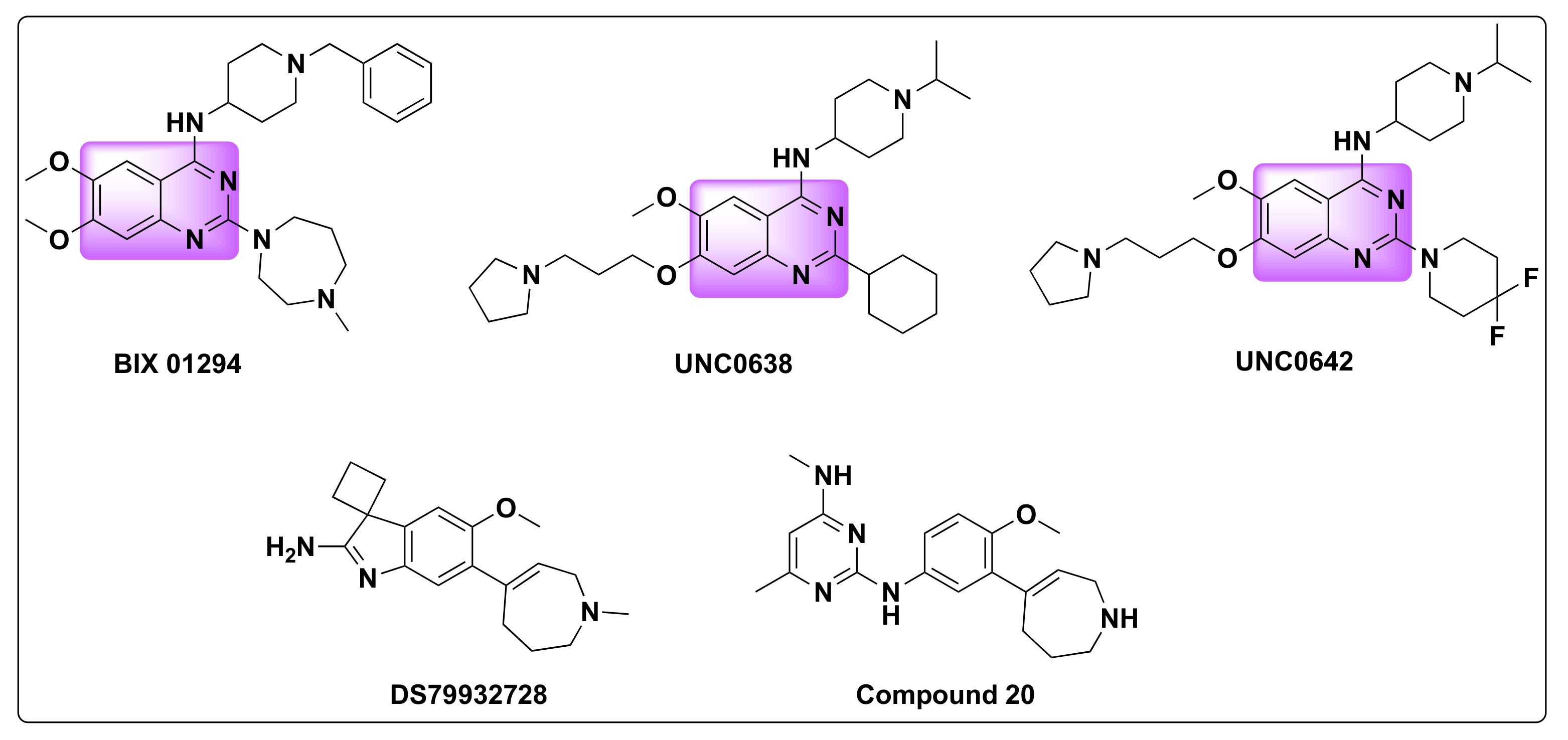
| Compd | IC50 (µM) | |||||||
|---|---|---|---|---|---|---|---|---|
| A431 | HT-29 | HeLa | K562 | MDA-MB-231 | HCT-116 | HEPG2 | HL-7702 | |
| 23 | 2.38 ± 1.10 | 6.99 ± 2.77 | 3.34 ± 0.17 | 2.6 ± 0.95 | 9.3 ± 0.14 | 11.05 ± 0.04 | 2.74 ± 0.05 | 39.81 ± 8.86 |
| SAHA | 0.63 ± 2.03 | 0.56 ± 0.14 | 0.48 ± 0.57 | 0.68 ± 0.7 | 2.36 ± 0.36 | 1.08 ± 0.1 | >50 | 1.35 ± 1.03 |
| UNC0642 | 3.38 ± 1.22 | 1.35 ± 2.4 | 1.74 ± 0.07 | 0.92 ± 0.56 | 3.23 ± 0.28 | 5.01 ± 0.88 | 2.69 ± 0.06 | 3.23 ± 0.19 |
| Compd | IC50 (nM) | ||
|---|---|---|---|
| HDAC1 | HDAC6 | GLP | |
| 23 | 89 | 13 | 1.3 |
| SAHA | 16 | 13 | — |
| UNC0642 | — | — | 2.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhuguru, J.; Ghoneim, O.A. Quinazoline Based HDAC Dual Inhibitors as Potential Anti-Cancer Agents. Molecules 2022, 27, 2294. https://doi.org/10.3390/molecules27072294
Dhuguru J, Ghoneim OA. Quinazoline Based HDAC Dual Inhibitors as Potential Anti-Cancer Agents. Molecules. 2022; 27(7):2294. https://doi.org/10.3390/molecules27072294
Chicago/Turabian StyleDhuguru, Jyothi, and Ola A. Ghoneim. 2022. "Quinazoline Based HDAC Dual Inhibitors as Potential Anti-Cancer Agents" Molecules 27, no. 7: 2294. https://doi.org/10.3390/molecules27072294
APA StyleDhuguru, J., & Ghoneim, O. A. (2022). Quinazoline Based HDAC Dual Inhibitors as Potential Anti-Cancer Agents. Molecules, 27(7), 2294. https://doi.org/10.3390/molecules27072294