
Discovery of Novel Epoxyketone Peptides as Lipase Inhibitors

 , ,
, ,  ,
, 
Abstract
:
1. Introduction
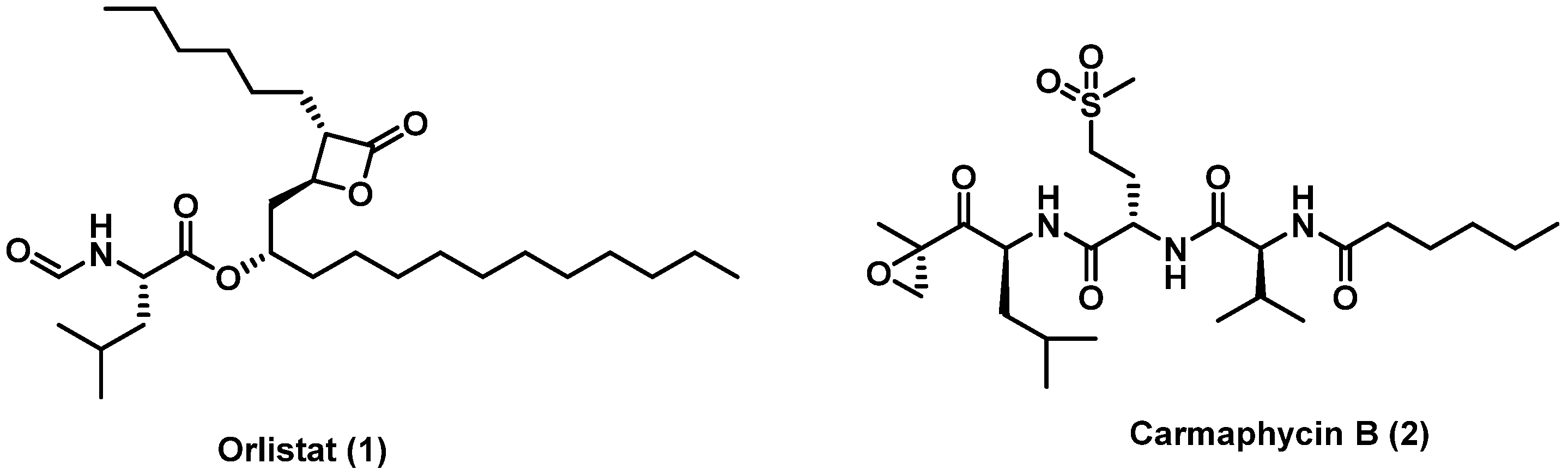
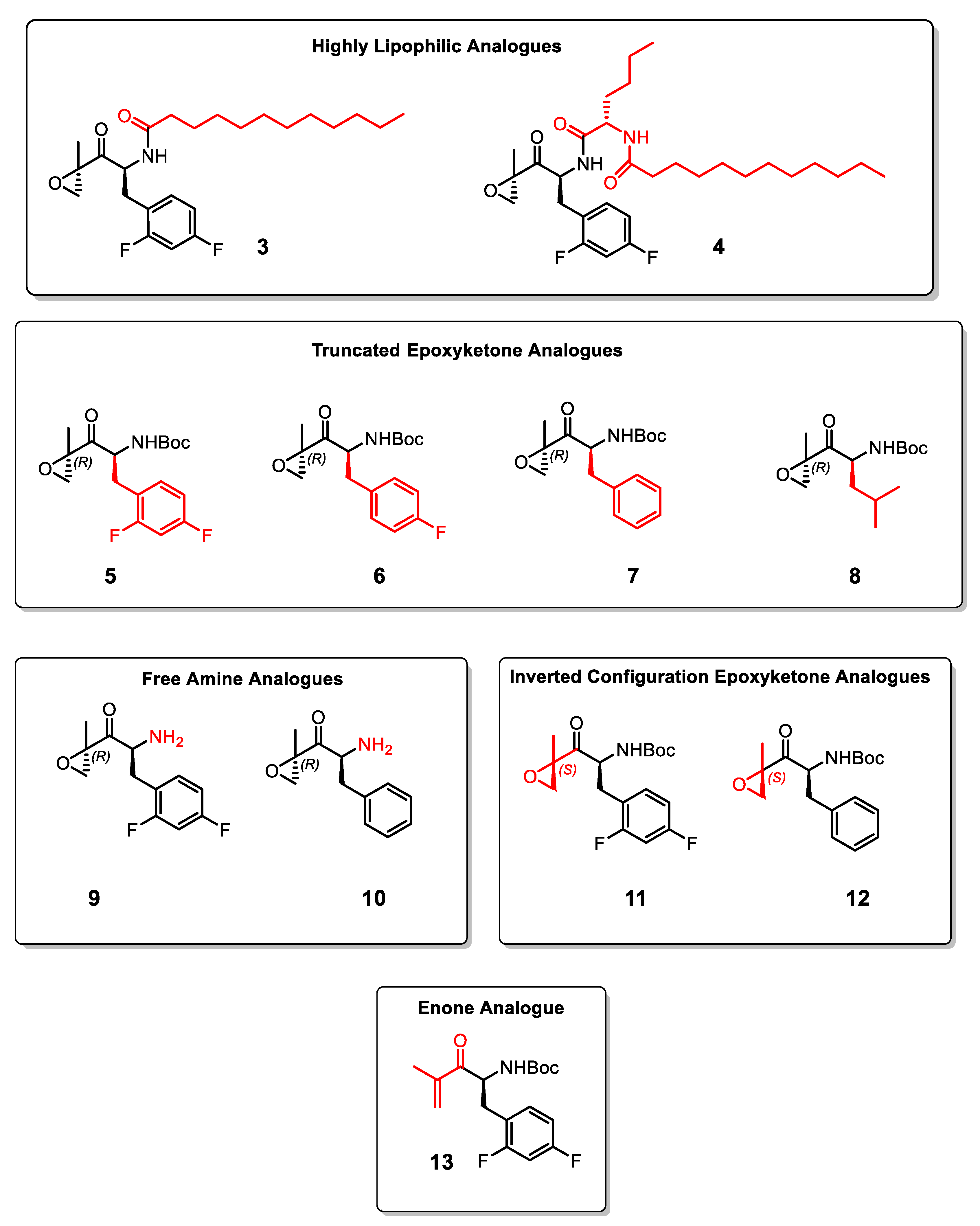
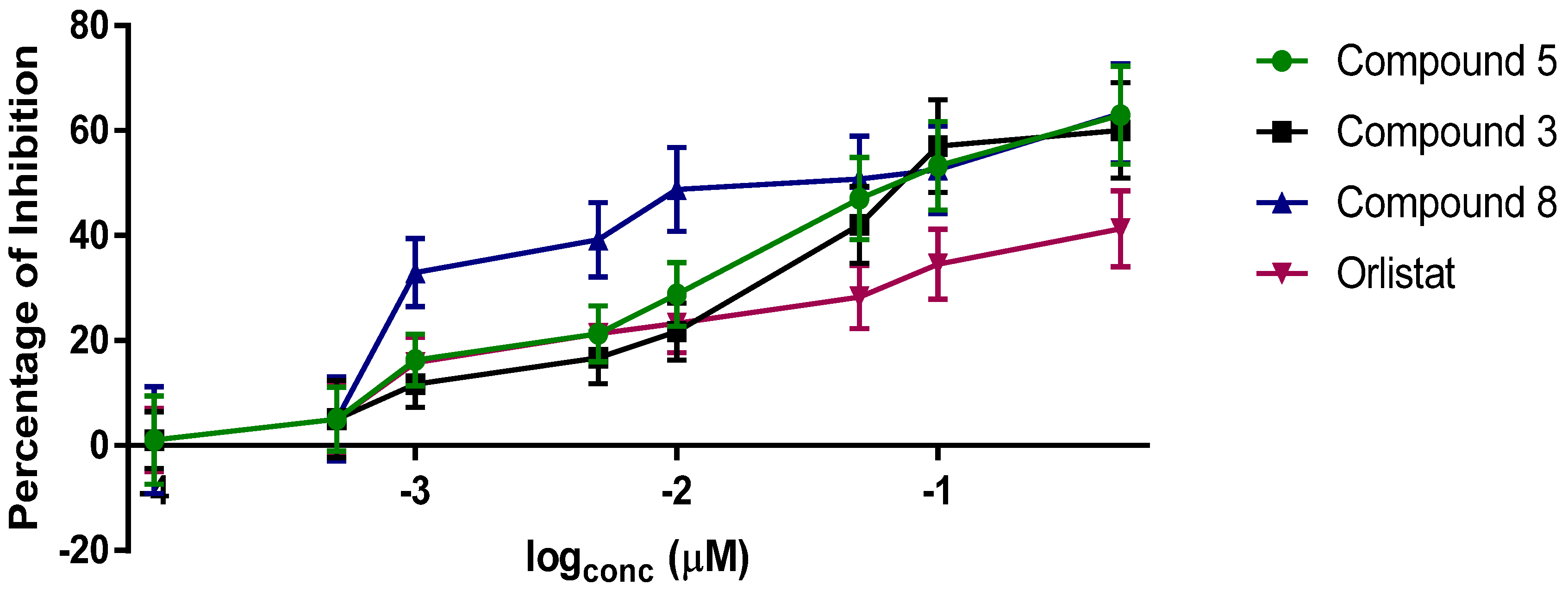
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Instrumentations
3.2. Synthesis
3.3. Anti-Lipase Activity
3.3.1. Preparation of the Candidate Inhibitors for Biological Testing
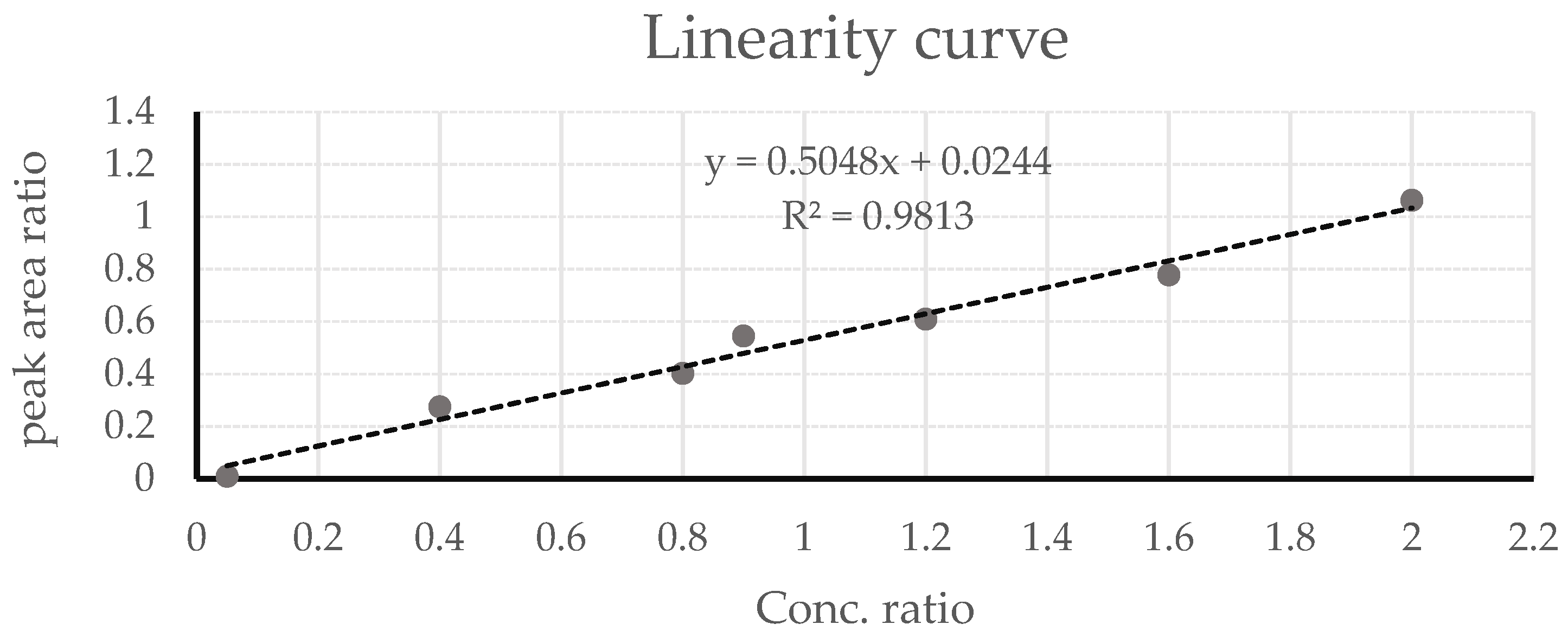
3.3.2. GC-FID Method Validation
3.4. Cytotoxic Activity
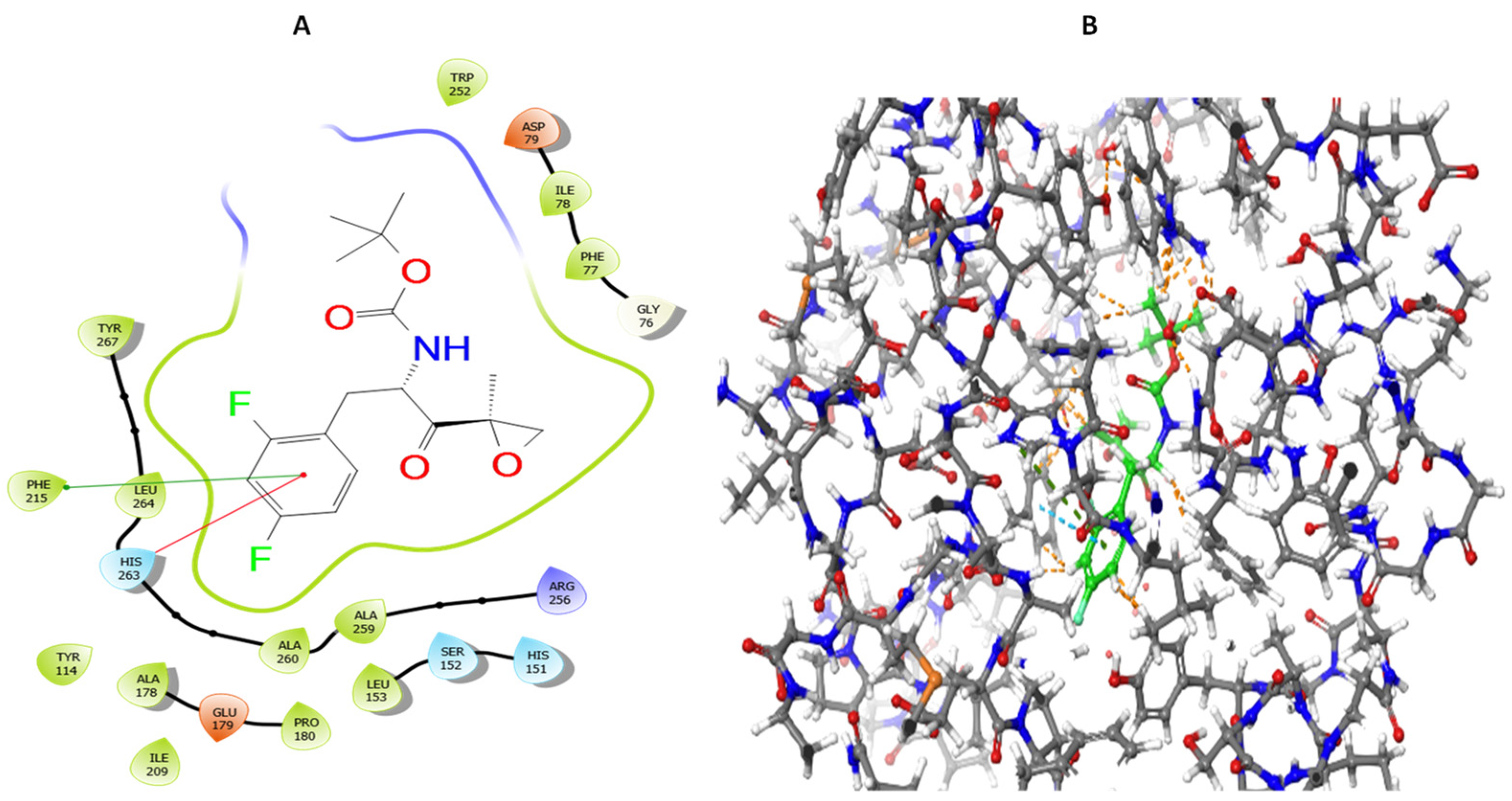
3.5. Molecular Docking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Blüher, M. Obesity: Global Epidemiology and Pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Fabricatore, A.N.; Wadden, T.A. Obesity. Annu. Rev. Clin. Psychol. 2006, 2, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Kyrou, I.; Randeva, H.S.; Tsigos, C.; Kaltsas, G.; Weickert, M.O. Clinical Problems Caused by Obesity. Endotext. 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK278973/ (accessed on 23 January 2022).
- Kompaniyets, L.; Goodman, A.B.; Belay, B.; Freedman, D.S.; Sucosky, M.S.; Lange, S.J.; Gundlapalli, A.V.; Boehmer, T.K.; Blanck, H.M. Body Mass Index and Risk for COVID-19–Related Hospitalization, Intensive Care Unit Admission, Invasive Mechanical Ventilation, and Death—United States, March–December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yang, Y.; Zhang, J. Obesity Is Associated with Severe Disease and Mortality in Patients with Coronavirus Disease 2019 (COVID-19): A Meta-Analysis. BMC Public Health 2021, 21, 1505. [Google Scholar] [CrossRef]
- Hall, K.D.; Kahan, S. Maintenance of Lost Weight and Long-Term Management of Obesity. Med. Clin. N. Am. 2018, 102, 183. [Google Scholar] [CrossRef]
- Chanoine, J.P.; Hampl, S.; Jensen, C.; Boldrin, M.; Hauptman, J. Effect of Orlistat on Weight and Body Composition in Obese Adolescents: A Randomized Controlled Trial. JAMA 2005, 293, 2873–2883. [Google Scholar] [CrossRef] [Green Version]
- Bray, G.A.; Heisel, W.E.; Afshin, A.; Jensen, M.D.; Dietz, W.H.; Long, M.; Kushner, R.F.; Daniels, S.R.; Wadden, T.A.; Tsai, A.G.; et al. The Science of Obesity Management: An Endocrine Society Scientific Statement. Endocr. Rev. 2018, 39, 79–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löffler, M.C.; Betz, M.J.; Blondin, D.P.; Augustin, R.; Sharma, A.K.; Tseng, Y.H.; Scheele, C.; Zimdahl, H.; Mark, M.; Hennige, A.M.; et al. Challenges in Tackling Energy Expenditure as Obesity Therapy: From Preclinical Models to Clinical Application. Mol. Metab. 2021, 51, 101237. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Eckel, R.H. Lipoprotein Lipase: From Gene to Obesity. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 271–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrelli, G.M.; Trono, D. Recombinant Lipases and Phospholipases and Their Use as Biocatalysts for Industrial Applications. Int. J. Mol. Sci. 2015, 16, 20774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.T.; Liu, X.T.; Chen, Q.X.; Shi, Y. Lipase Inhibitors for Obesity: A Review. Biomed. Pharmacother. 2020, 128, 110314. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.V.S.; Dias, M.M.G.; Brilhante, A.J.V.C.; Terra, M.F.; García-Arévalo, M.; Figueira, A.C.M. Multifactorial Basis and Therapeutic Strategies in Metabolism-Related Diseases. Nutrients 2021, 13, 2830. [Google Scholar] [CrossRef]
- Eichmann, T.O.; Lass, A. DAG Tales: The Multiple Faces of Diacylglycerol—Stereochemistry, Metabolism, and Signaling. Cell. Mol. Life Sci. 2015, 72, 3931–3952. [Google Scholar] [CrossRef] [Green Version]
- Heck, A.M.; Yanovski, J.A.; Calis, K.A. Orlistat, a New Lipase Inhibitor for the Management of Obesity. Pharmacotherapy 2000, 20, 270. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Bhattarai, D.; Yoo, J.; Miller, Z.; Park, J.E.; Lee, S.; Lee, W.; Driscoll, J.J.; Kim, K.B. Development of Novel Epoxyketone-Based Proteasome Inhibitors as a Strategy to Overcome Cancer Resistance to Carfilzomib and Bortezomib. J. Med. Chem. 2019, 62, 4444–4455. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dixit, V.M. Drugging the Undruggables: Exploring the Ubiquitin System for Drug Development. Cell Res. 2016, 26, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Almaliti, J.; Miller, B.; Pietraszkiewicz, H.; Glukhov, E.; Naman, C.B.; Kline, T.; Hanson, J.; Li, X.; Zhou, S.; Valeriote, F.A.; et al. Exploration of the Carmaphycins as Payloads in Antibody Drug Conjugate Anticancer Agents. Eur. J. Med. Chem. 2019, 161, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome Inhibitors-Molecular Basis and Current Perspectives in Multiple Myeloma. J. Cell. Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Matsukawa, T.; Murata, K.; Nishitani, R.; Yamagami, M.; Tomohiro, N.; Kajiyama, I.; Fumuro, M.; Iijima, M.; Shigeoka, S.; et al. Pancreatic Lipase Inhibitory Activity of Citrus Unshiu Leaf Extract. Nat. Prod. Commun. 2019, 14, 1–5. [Google Scholar] [CrossRef]
- Pereira, A.R.; Kale, A.J.; Fenley, A.T.; Byrum, T.; Debonsi, H.M.; Gilson, M.K.; Valeriote, F.A.; Moore, B.S.; Gerwick, W.H. The Carmaphycins: New Proteasome Inhibitors Exhibiting an α,β-Epoxyketone Warhead from a Marine Cyanobacterium. ChemBioChem 2012, 13, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Sankar, V.; Maida Engels, S. Synthesis, Biological Evaluation, Molecular Docking and in Silico ADME Studies of Phenacyl Esters of N-Phthaloyl Amino Acids as Pancreatic Lipase Inhibitors. Future J. Pharm. Sci. 2018, 4, 276–283. [Google Scholar] [CrossRef]
- Nguyen, P.T.V.; Huynh, H.A.; van Truong, D.; Tran, T.D.; Vo, C.V.T. Exploring Aurone Derivatives as Potential Human Pancreatic Lipase Inhibitors through Molecular Docking and Molecular Dynamics Simulations. Molecules 2020, 25, 4657. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Van Der Linden, W.A.; Overkleeft, H.S. Proteasome Inhibitors: An Expanding Army Attacking a Unique Target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaMonte, G.M.; Almaliti, J.; Bibo-Verdugo, B.; Keller, L.; Zou, B.Y.; Yang, J.; Antonova-Koch, Y.; Orjuela-Sanchez, P.; Boyle, C.A.; Vigil, E.; et al. Development of a Potent Inhibitor of the Plasmodium Proteasome with Reduced Mammalian Toxicity. J. Med. Chem. 2017, 60, 6721–6732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaliti, J.; Fajtová, P.; O’Donoghue, A.J.; AlHindy, M.; Gerwick, W.H. Improved Scalable Synthesis of Clinical Candidate KZR-616, a Selective Immunoproteasome Inhibitor. ChemistrySelect 2021, 6, 12461–12465. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analogue | IC50 (nM) against PL |
|---|---|
| orlistat (1) | 100.8 ± 13.2 |
| carmaphycin B (2) | 25.0 ± 1.1 |
| 3 | 29.3 ± 2.4 |
| 4 | 9.3 ± 2.5 |
| 5 | 29.4 ± 4.8 |
| 6 | 46.4 ± 3.7 |
| 7 | 49.1 ± 11.4 |
| 8 | 120 ± 15.2 |
| 9 | 41.5 ± 7.1 |
| 10 | 73.1 ± 9.8 |
| 11 | 29.0 ± 4.2 |
| 12 | 49.2 ± 9.3 |
| 13 | >10,000 |
| Compound Code | CCD-1064Sk Fibroblasts IC50 (nM) | HepG2 Cells IC50 (nM) |
|---|---|---|
| 3 | >15,000 | 2598 |
| 4 | >15,000 | 2325 |
| 5 | >15,000 | >15,000 |
| 6 | >15,000 | >15,000 |
| 7 | >15,000 | >15,000 |
| 8 | >15,000 | >15,000 |
| 9 | >15,000 | >15,000 |
| 10 | >15,000 | >15,000 |
| 11 | >15,000 | >15,000 |
| 12 | >15,000 | >15,000 |
| 13 | >15,000 | >15,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almaliti, J.; Alzweiri, M.; Alhindy, M.; Al-Helo, T.; Daoud, I.; Deknash, R.; Naman, C.B.; Abu-Irmaileh, B.; Bustanji, Y.; Hamad, I. Discovery of Novel Epoxyketone Peptides as Lipase Inhibitors. Molecules 2022, 27, 2261. https://doi.org/10.3390/molecules27072261
Almaliti J, Alzweiri M, Alhindy M, Al-Helo T, Daoud I, Deknash R, Naman CB, Abu-Irmaileh B, Bustanji Y, Hamad I. Discovery of Novel Epoxyketone Peptides as Lipase Inhibitors. Molecules. 2022; 27(7):2261. https://doi.org/10.3390/molecules27072261
Chicago/Turabian StyleAlmaliti, Jehad, Muhammed Alzweiri, Momen Alhindy, Tamam Al-Helo, Ibrahim Daoud, Raghad Deknash, C. Benjamin Naman, Bashaer Abu-Irmaileh, Yasser Bustanji, and Islam Hamad. 2022. "Discovery of Novel Epoxyketone Peptides as Lipase Inhibitors" Molecules 27, no. 7: 2261. https://doi.org/10.3390/molecules27072261
APA StyleAlmaliti, J., Alzweiri, M., Alhindy, M., Al-Helo, T., Daoud, I., Deknash, R., Naman, C. B., Abu-Irmaileh, B., Bustanji, Y., & Hamad, I. (2022). Discovery of Novel Epoxyketone Peptides as Lipase Inhibitors. Molecules, 27(7), 2261. https://doi.org/10.3390/molecules27072261