
Selective Formation of Intramolecular Hydrogen-Bonding Palladium(II) Complexes with Nucleosides Using Unsymmetrical Tridentate Ligands


Abstract
: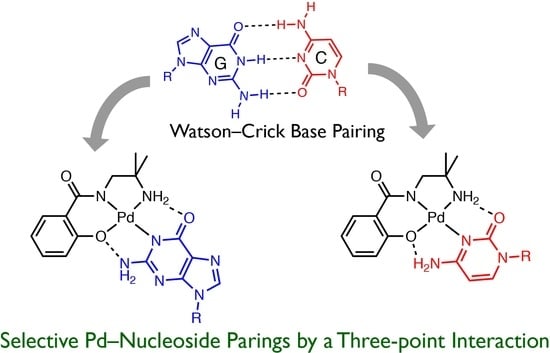
1. Introduction
2. Results and Discussion
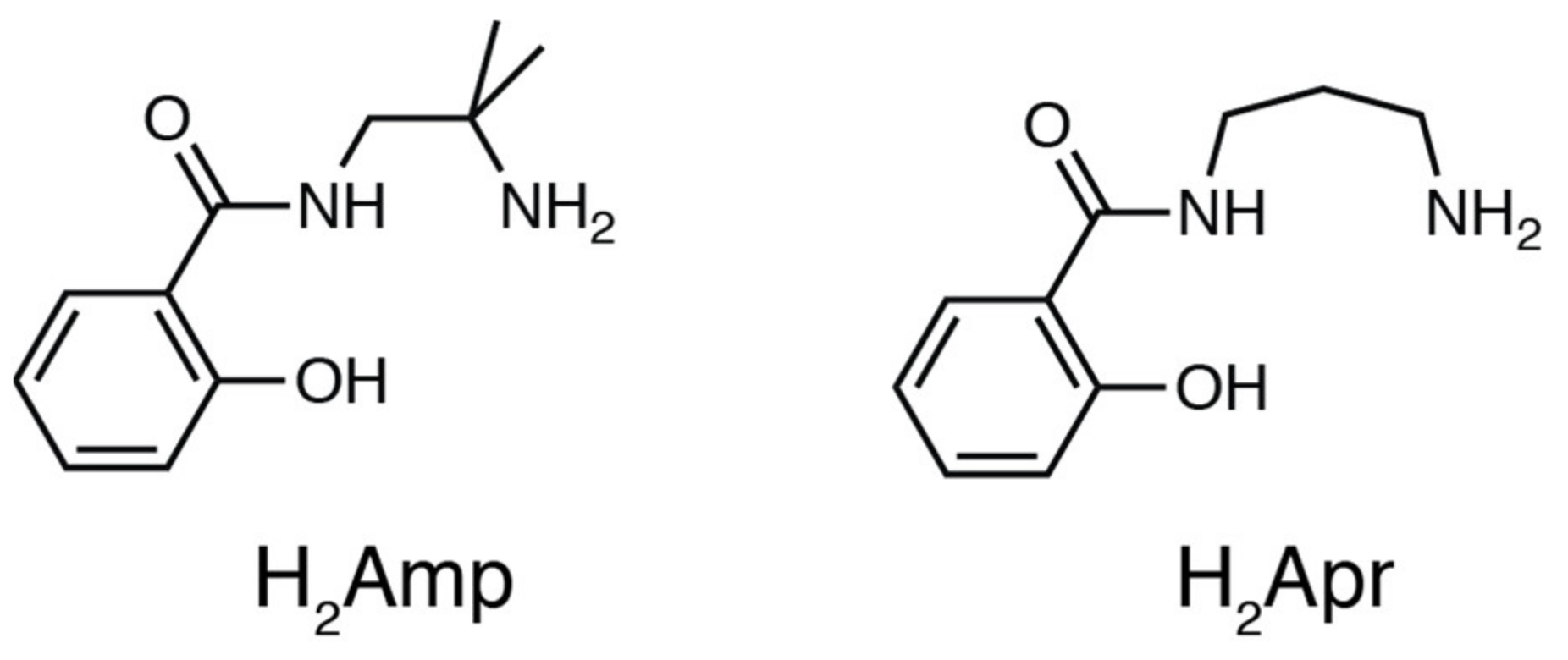
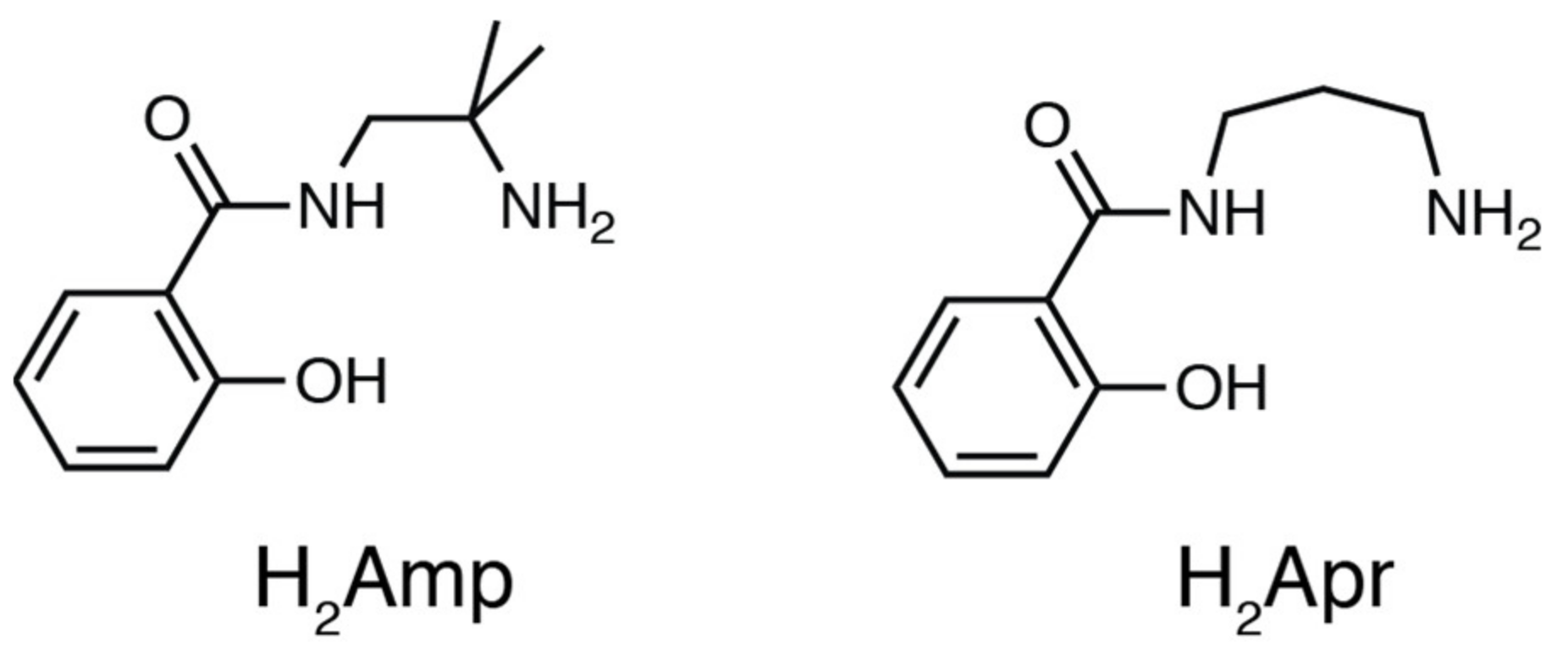
2.1. Preparation
Preparation of PdII Complexes
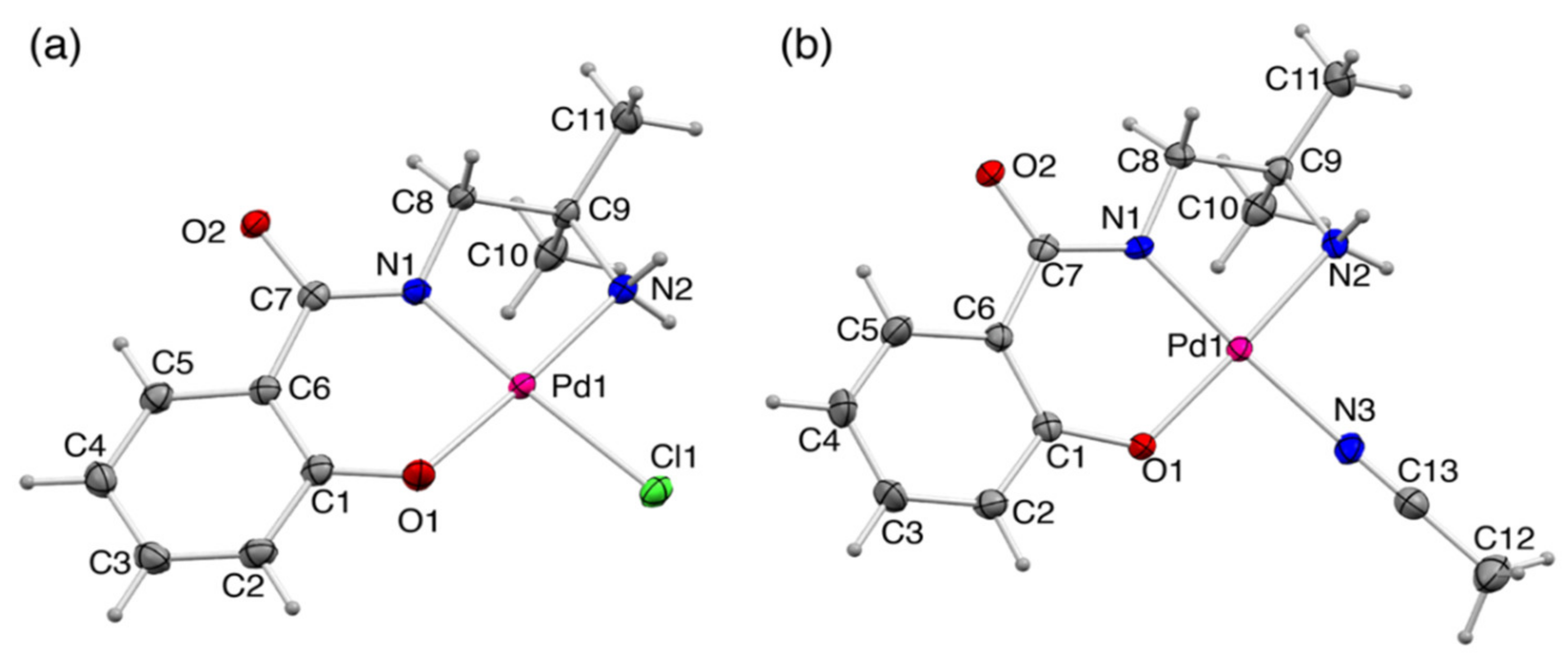
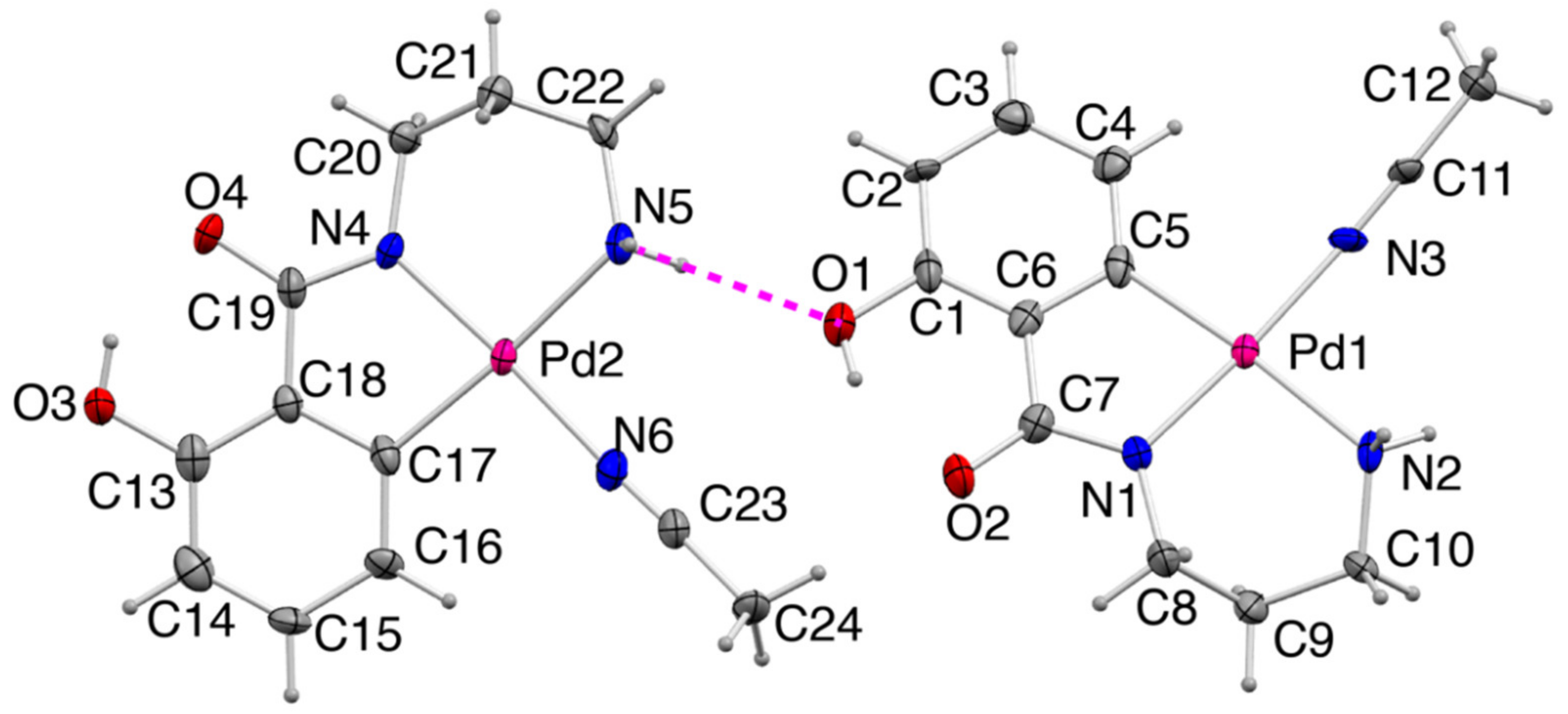
2.2. Crystal Strucre of PdII Complexes
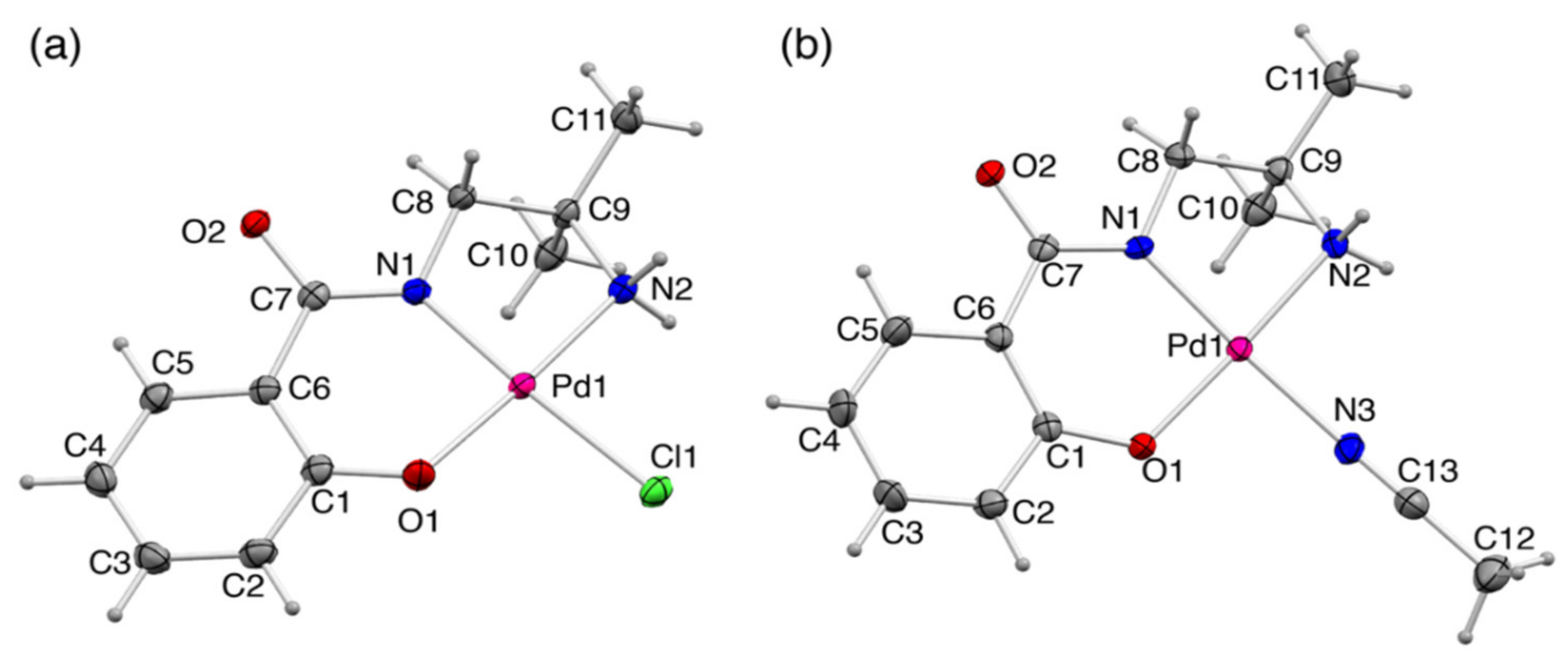
2.2.1. Crystal Structures of 1 and 1′
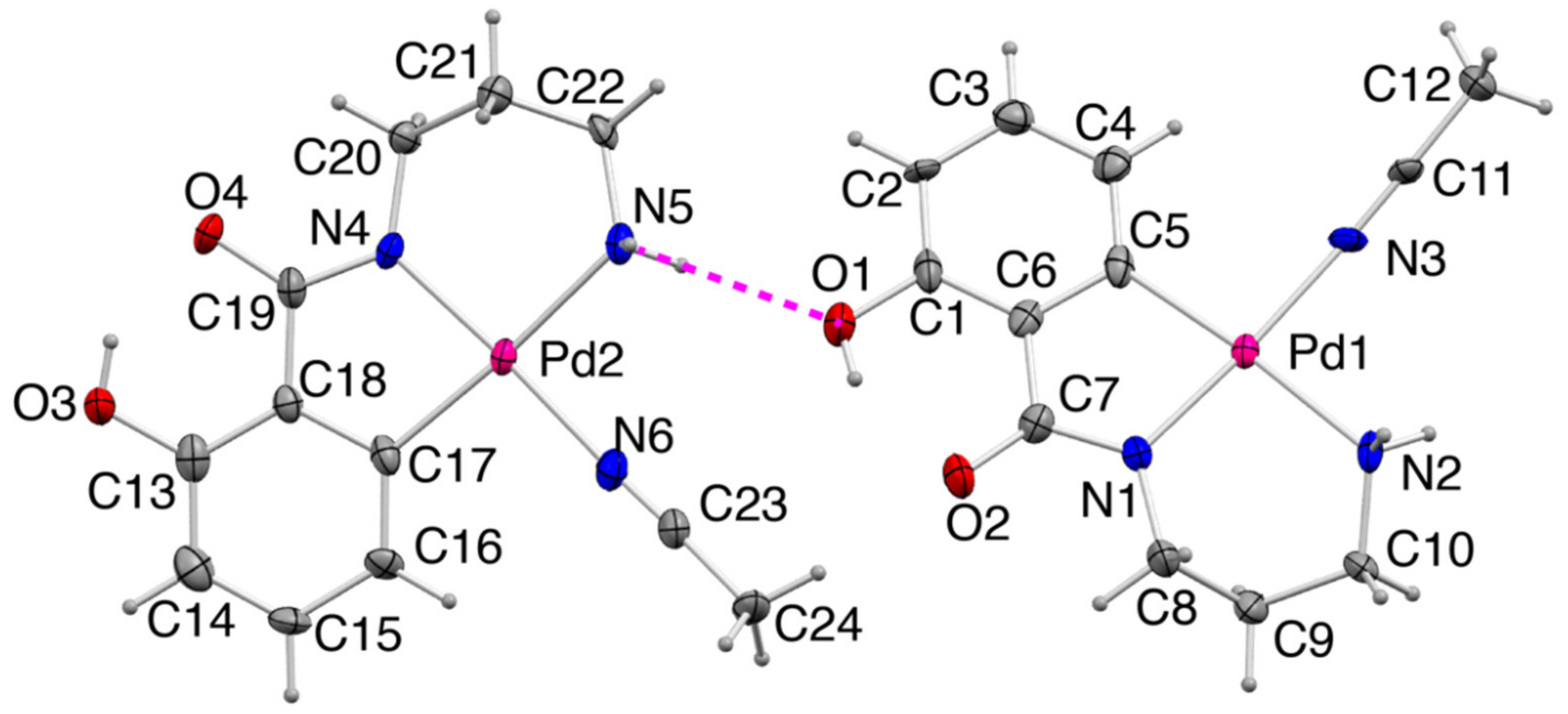
2.2.2. Crystal Structure of 2
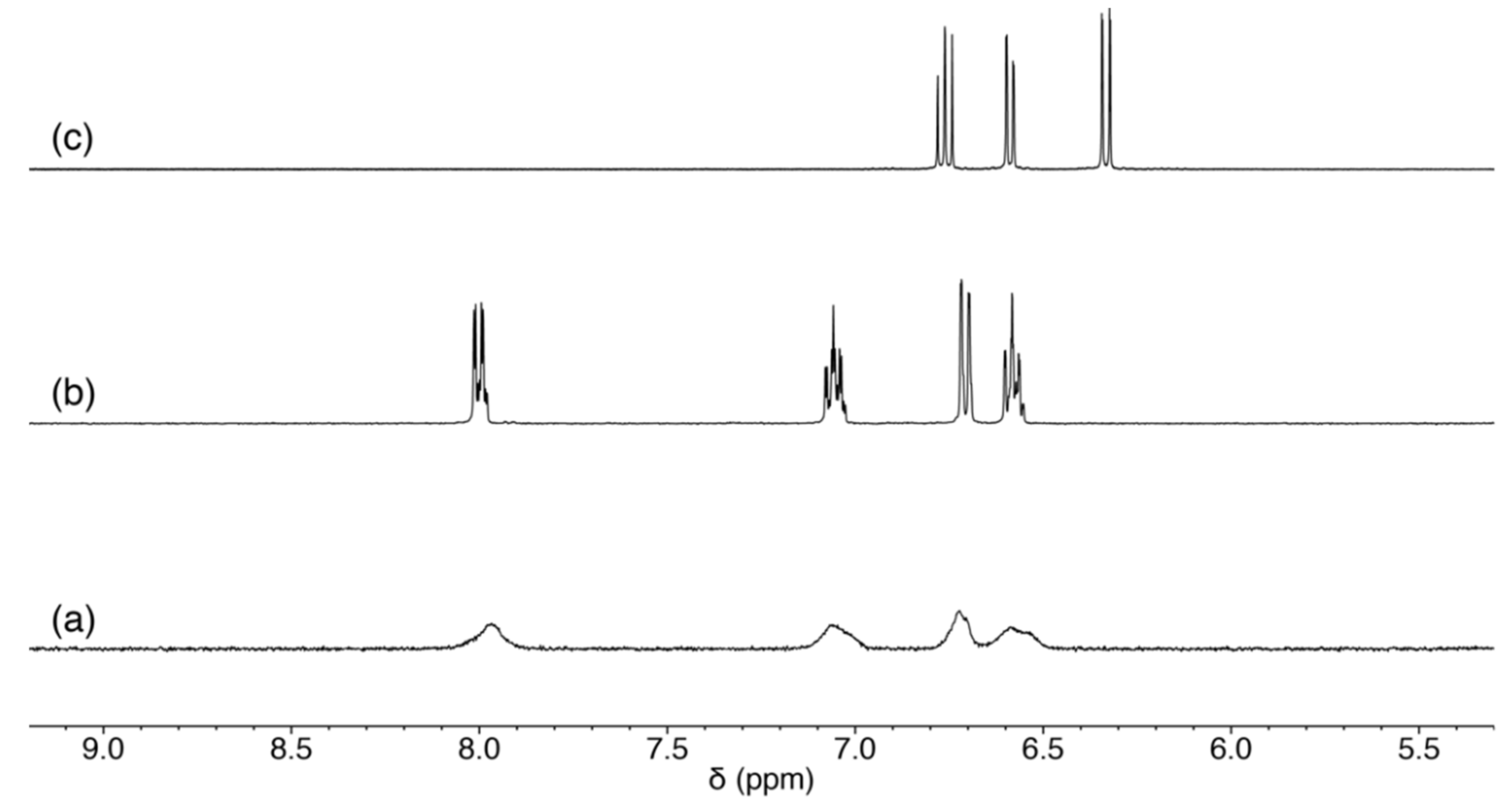
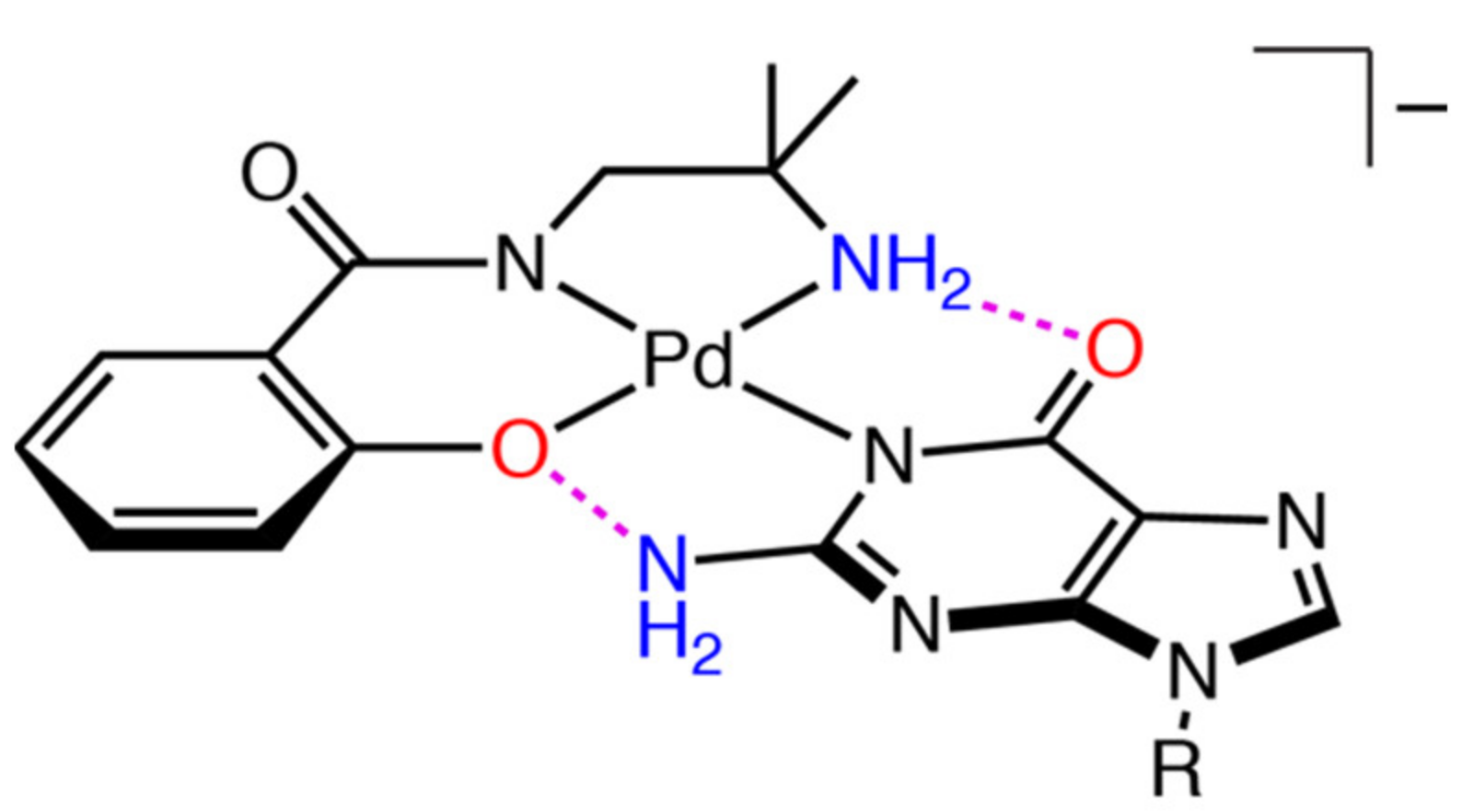
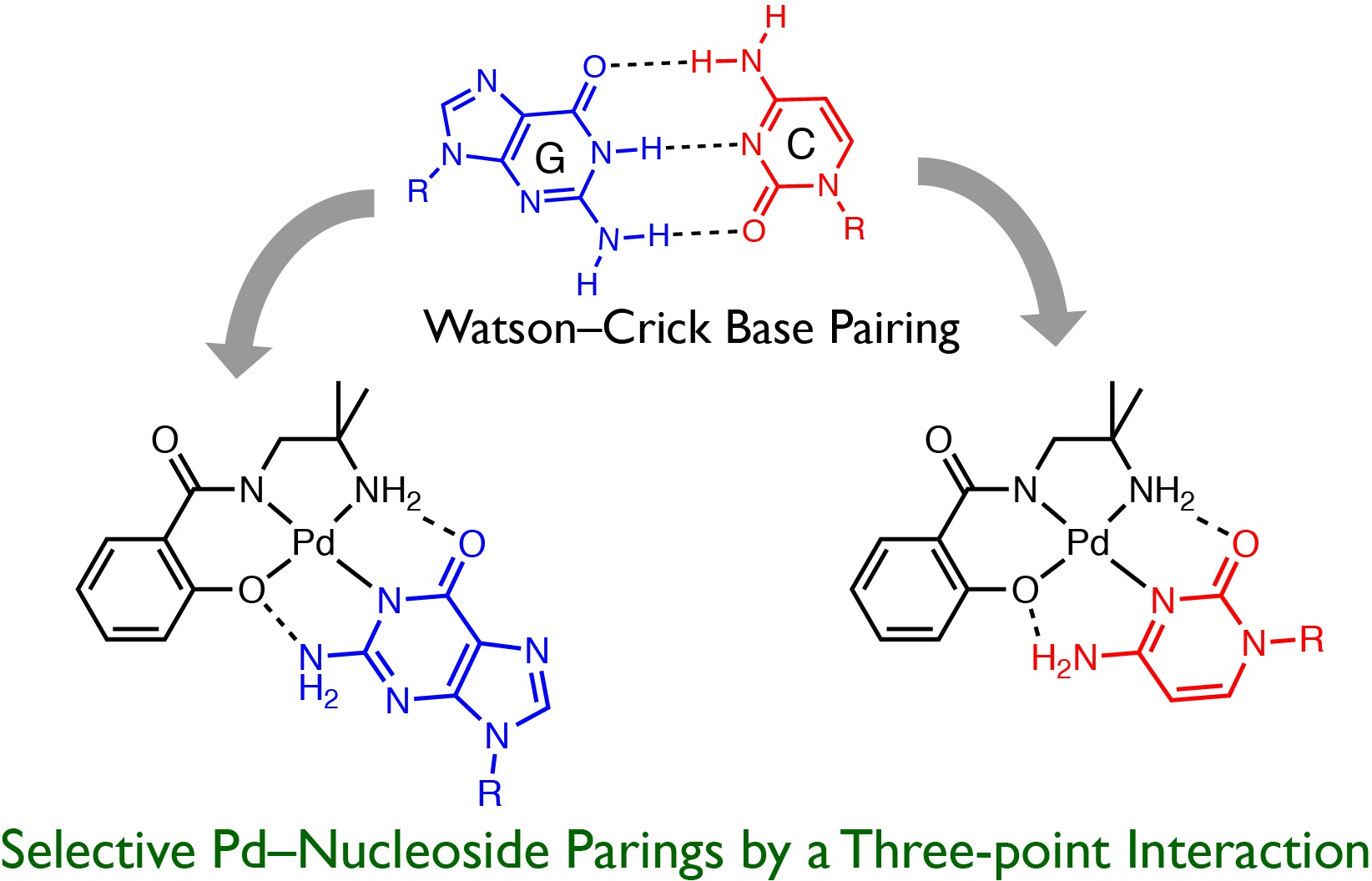
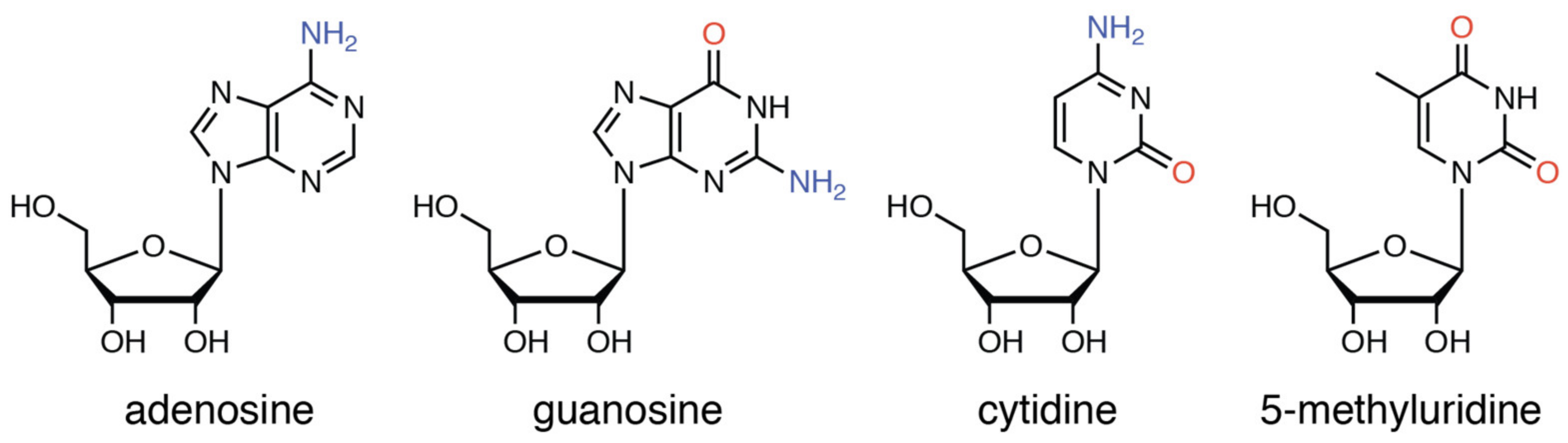
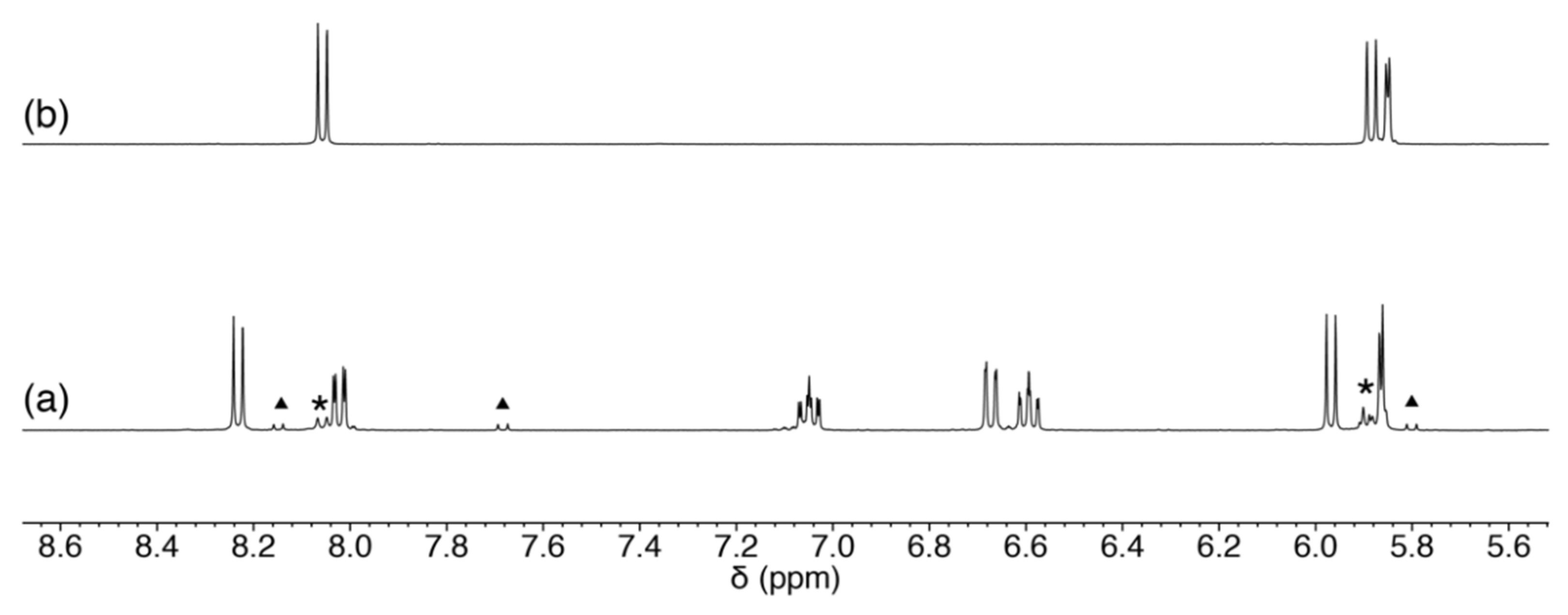
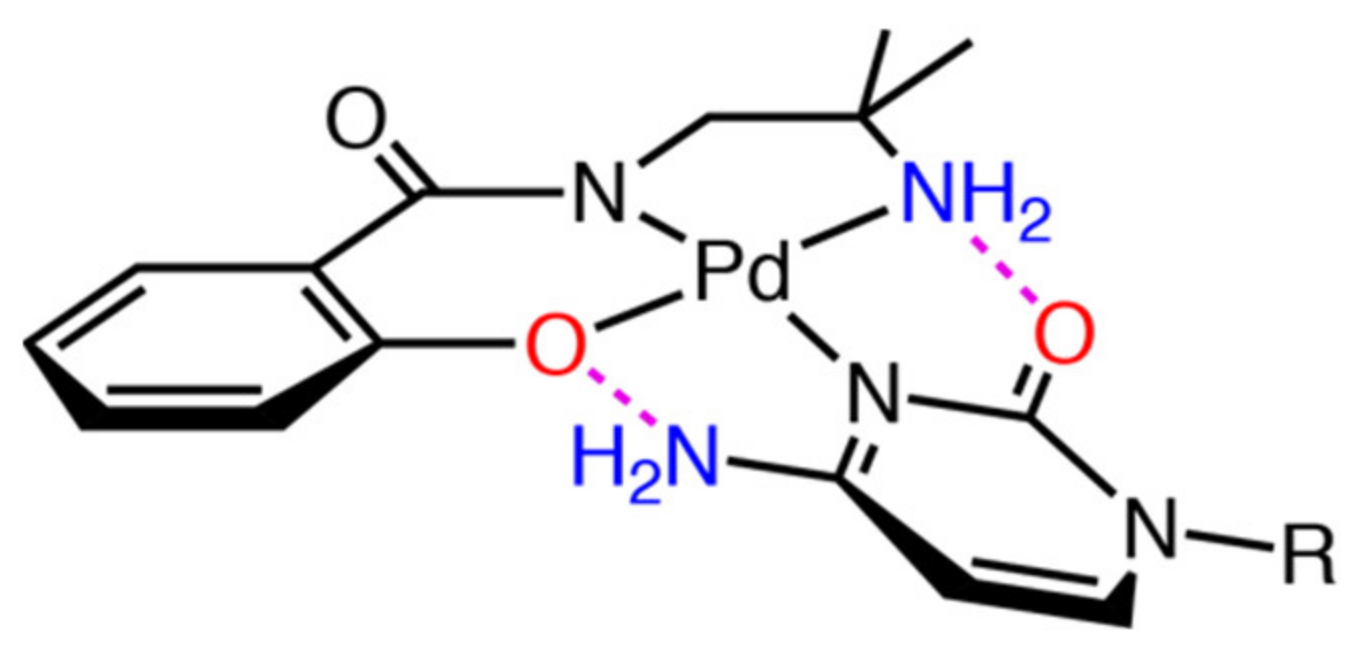
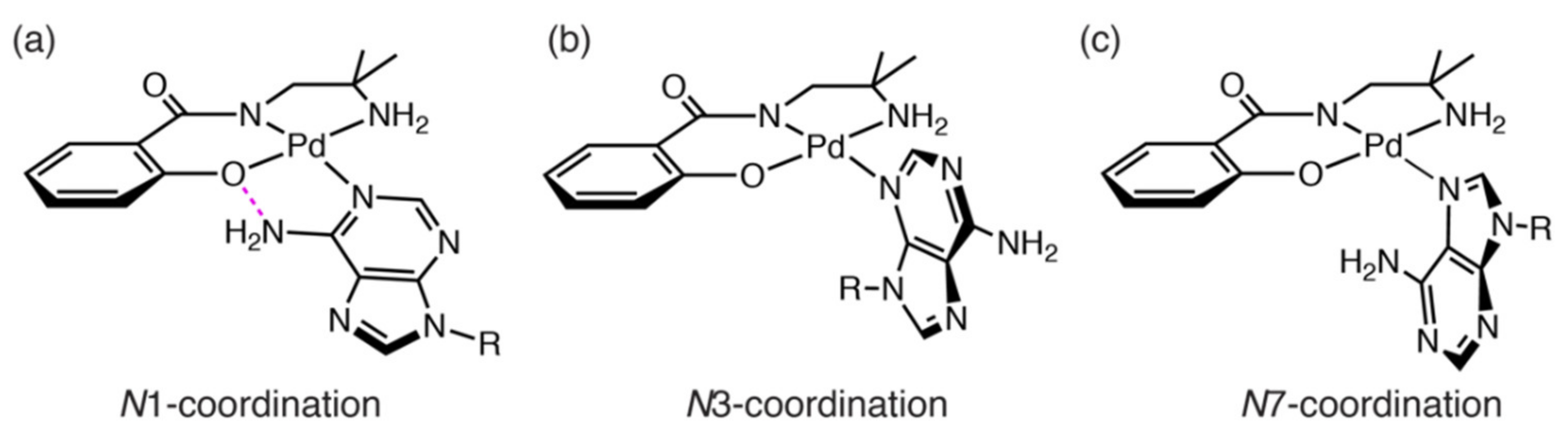
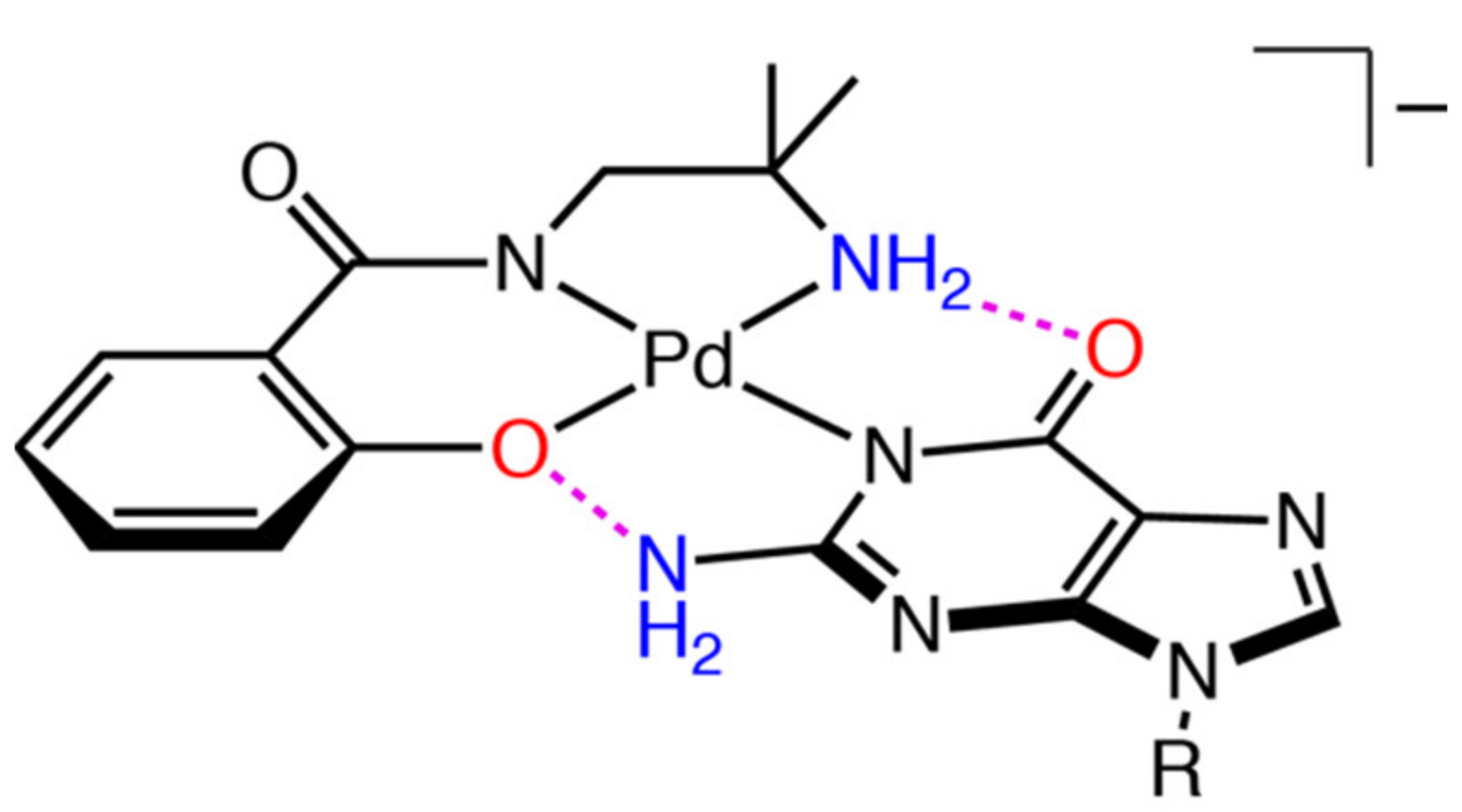
2.3. Reaction of 1 with Nucleosides
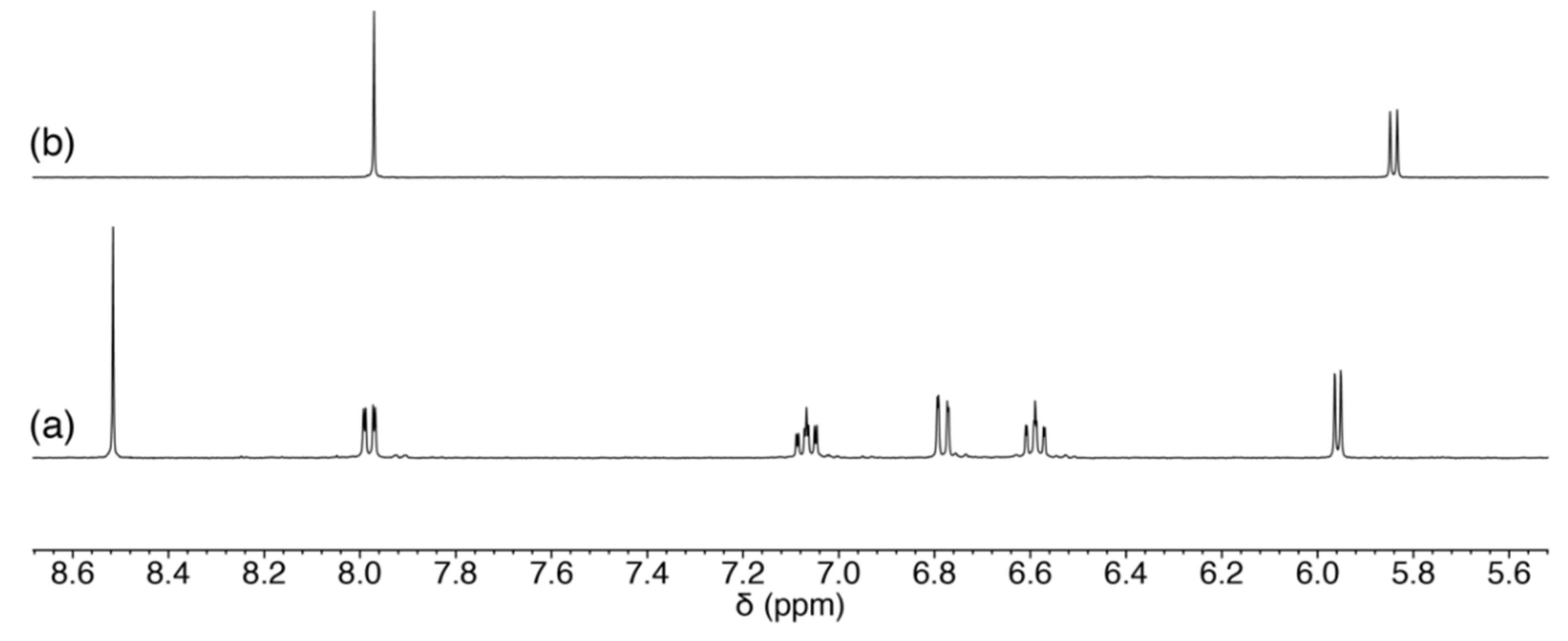
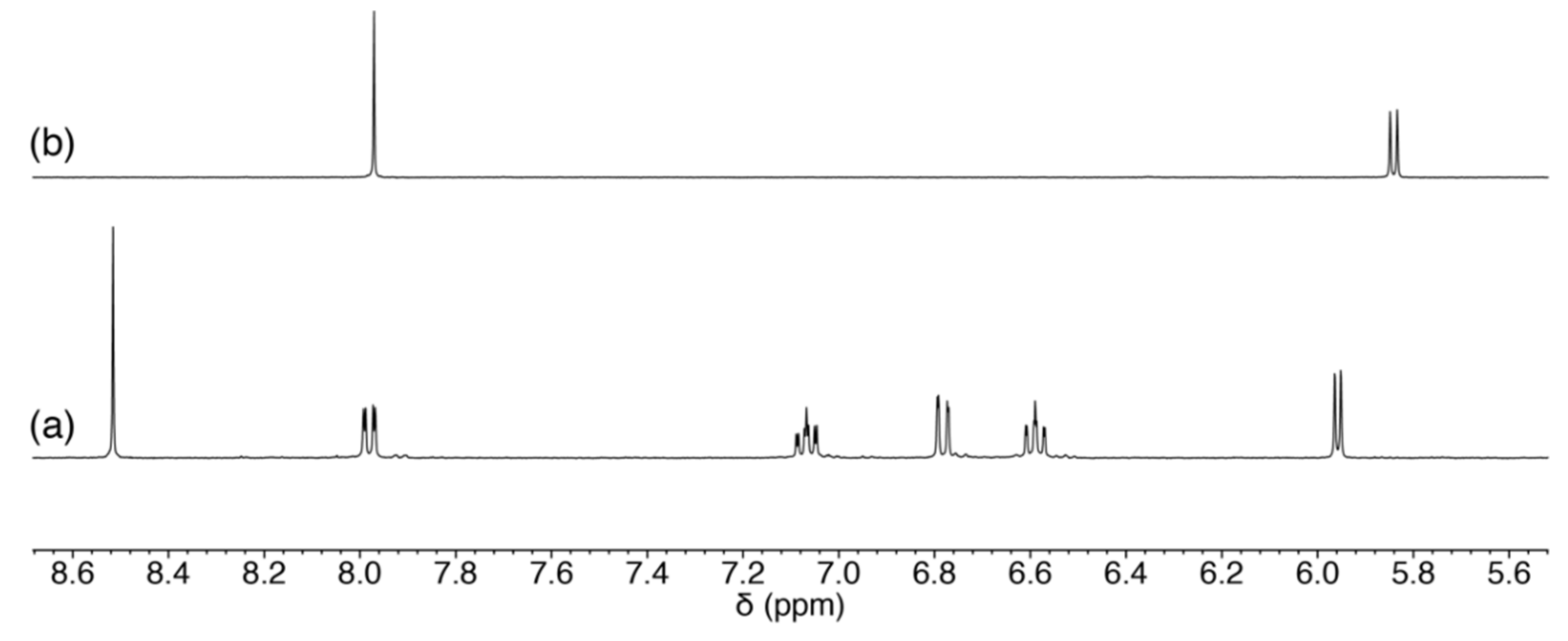
2.3.1. Reaction of 1 with Pyrimidine-Nucleosides
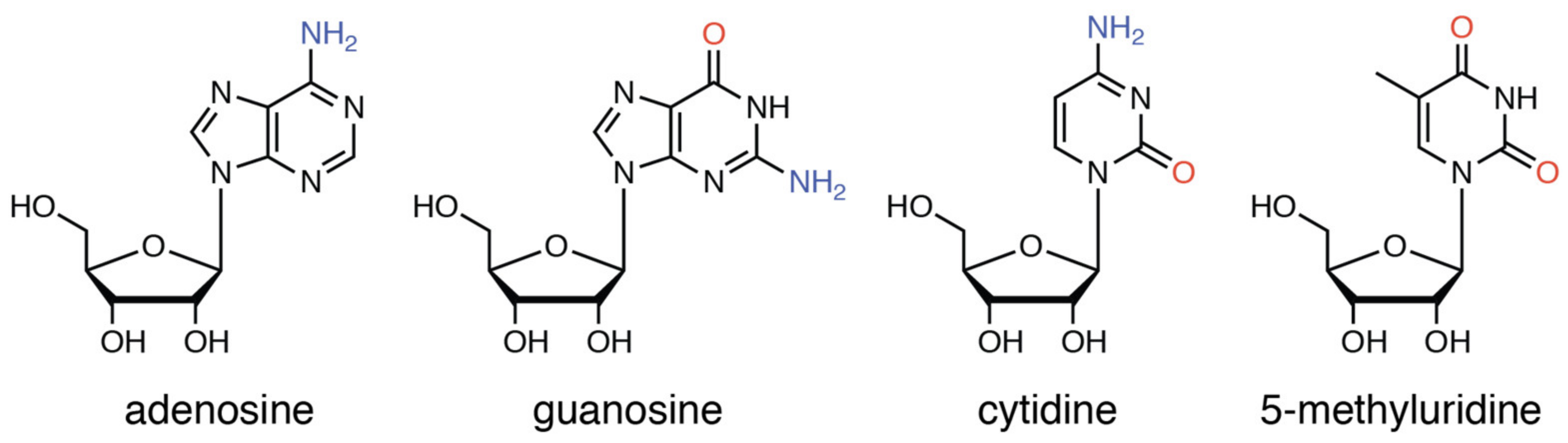
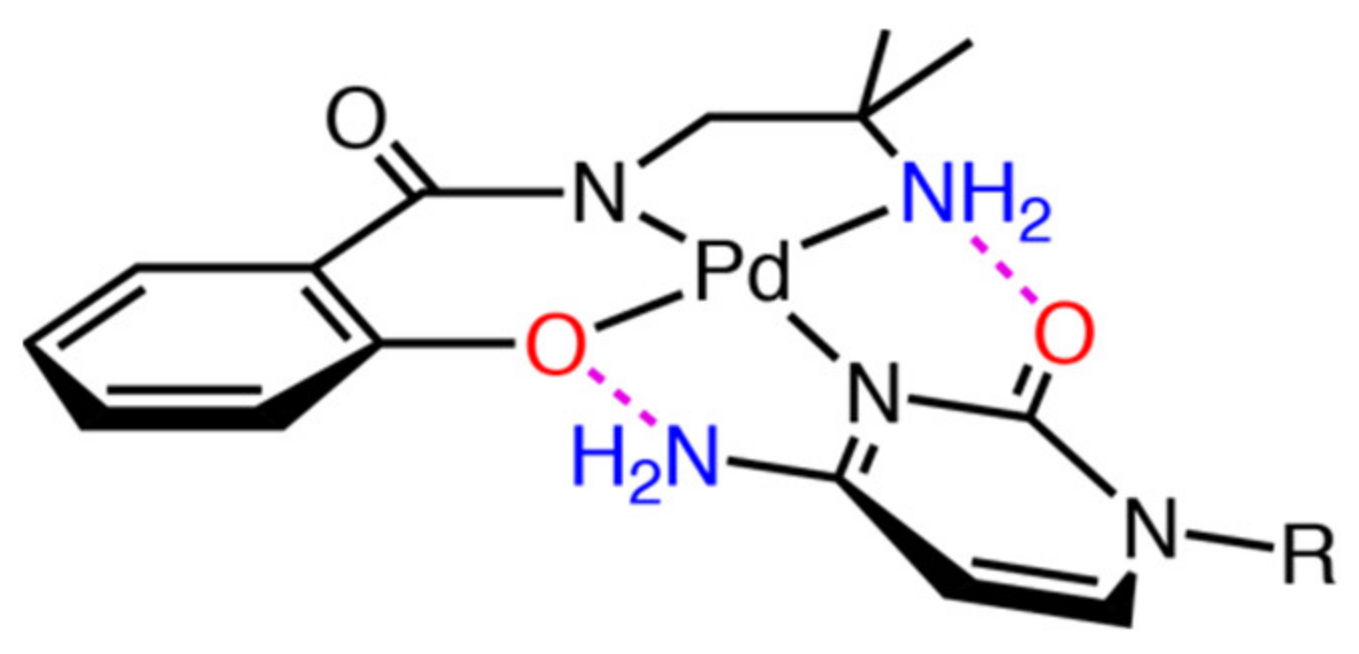
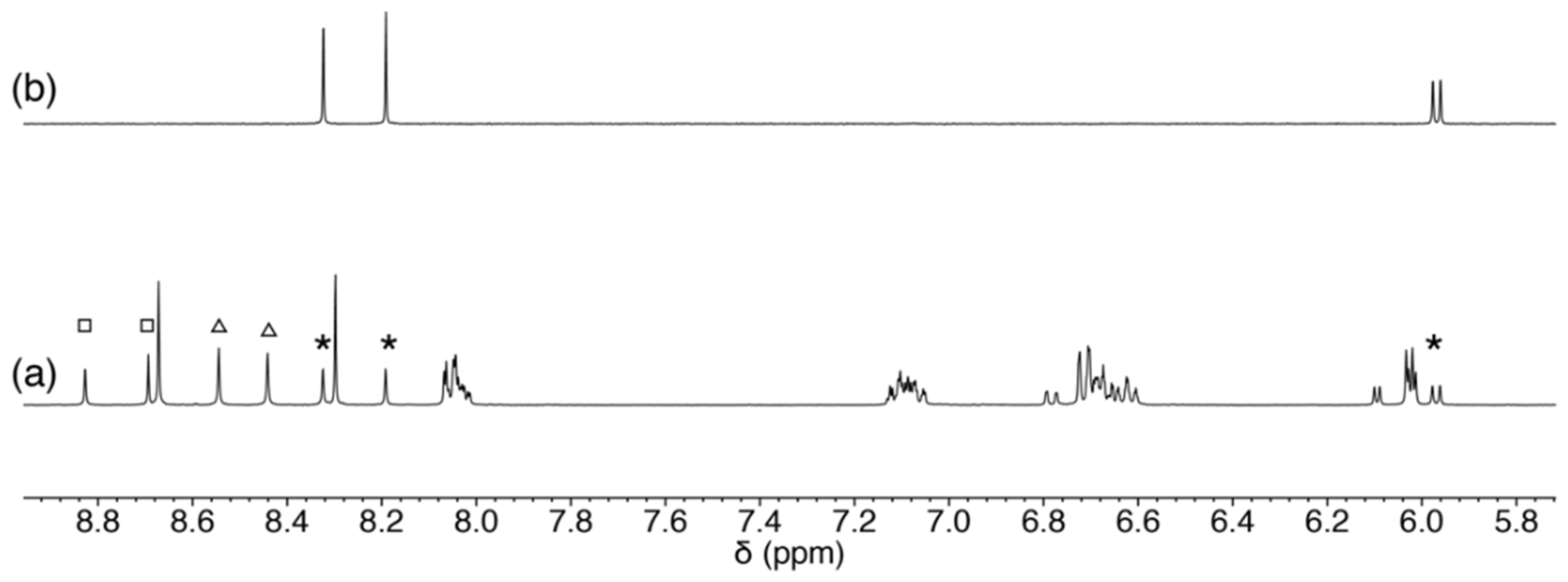
2.3.2. Reaction of 1 with Purine-Nucleosides
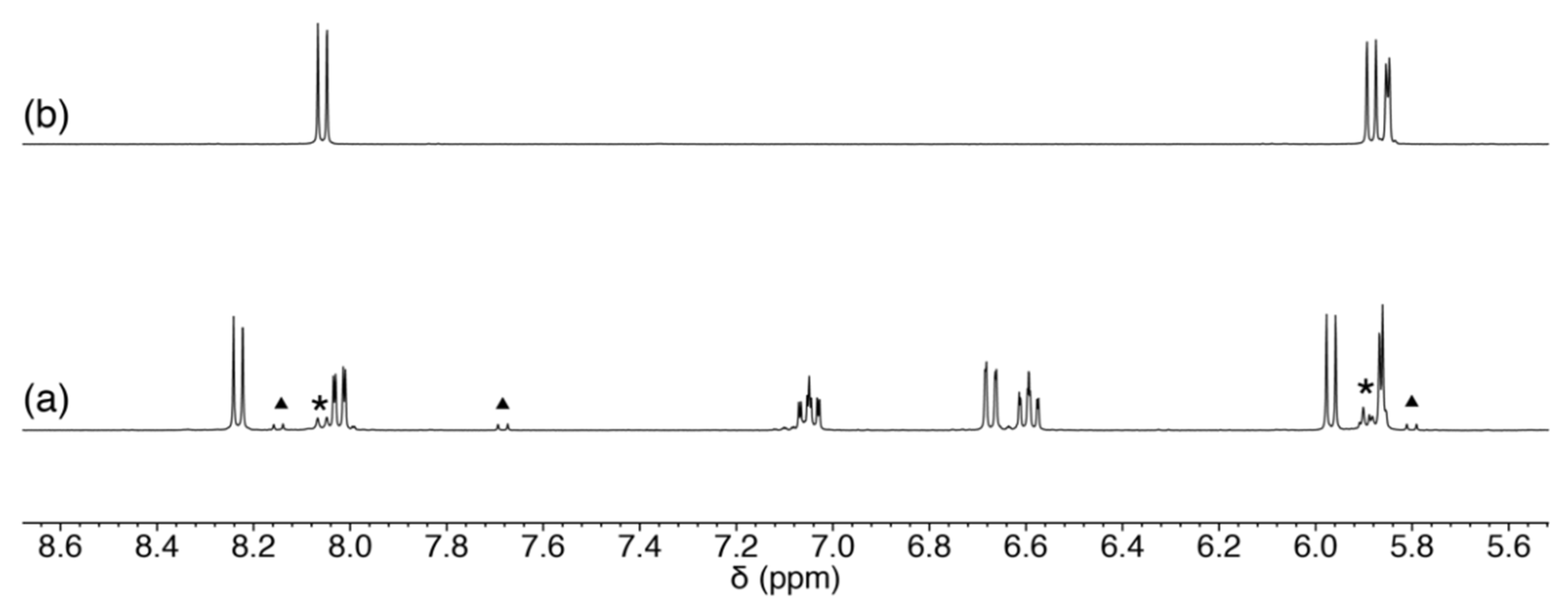
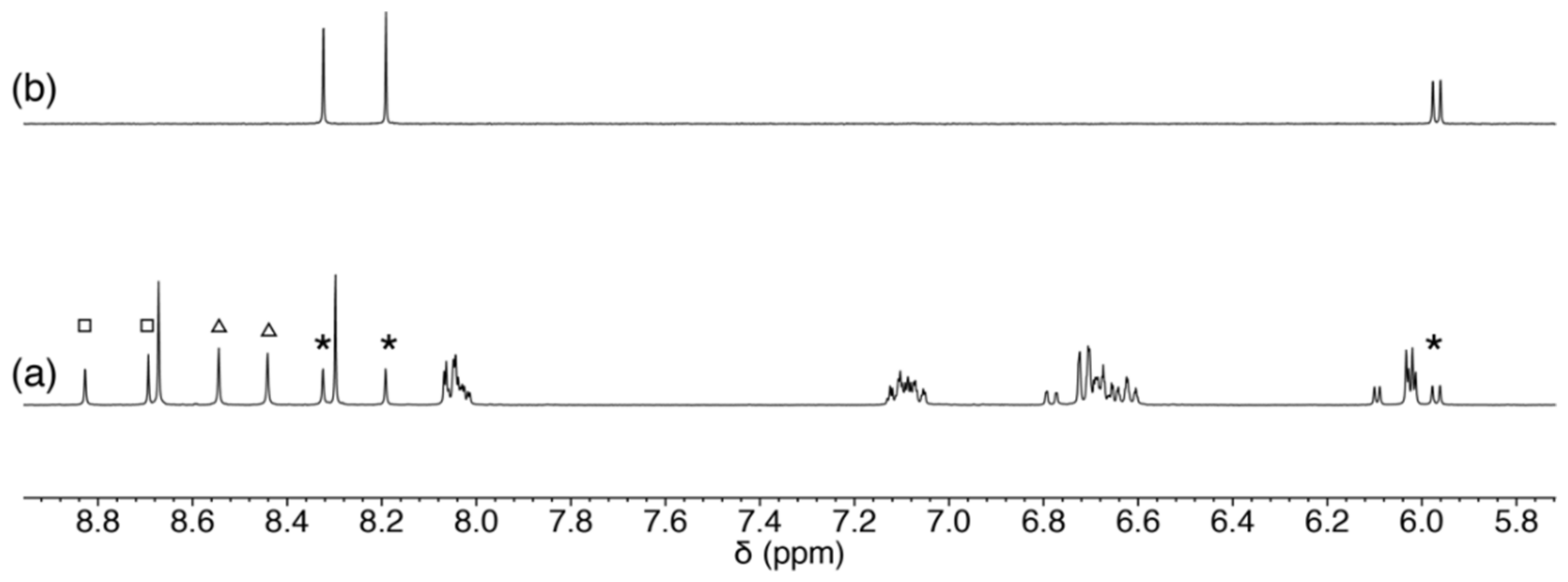
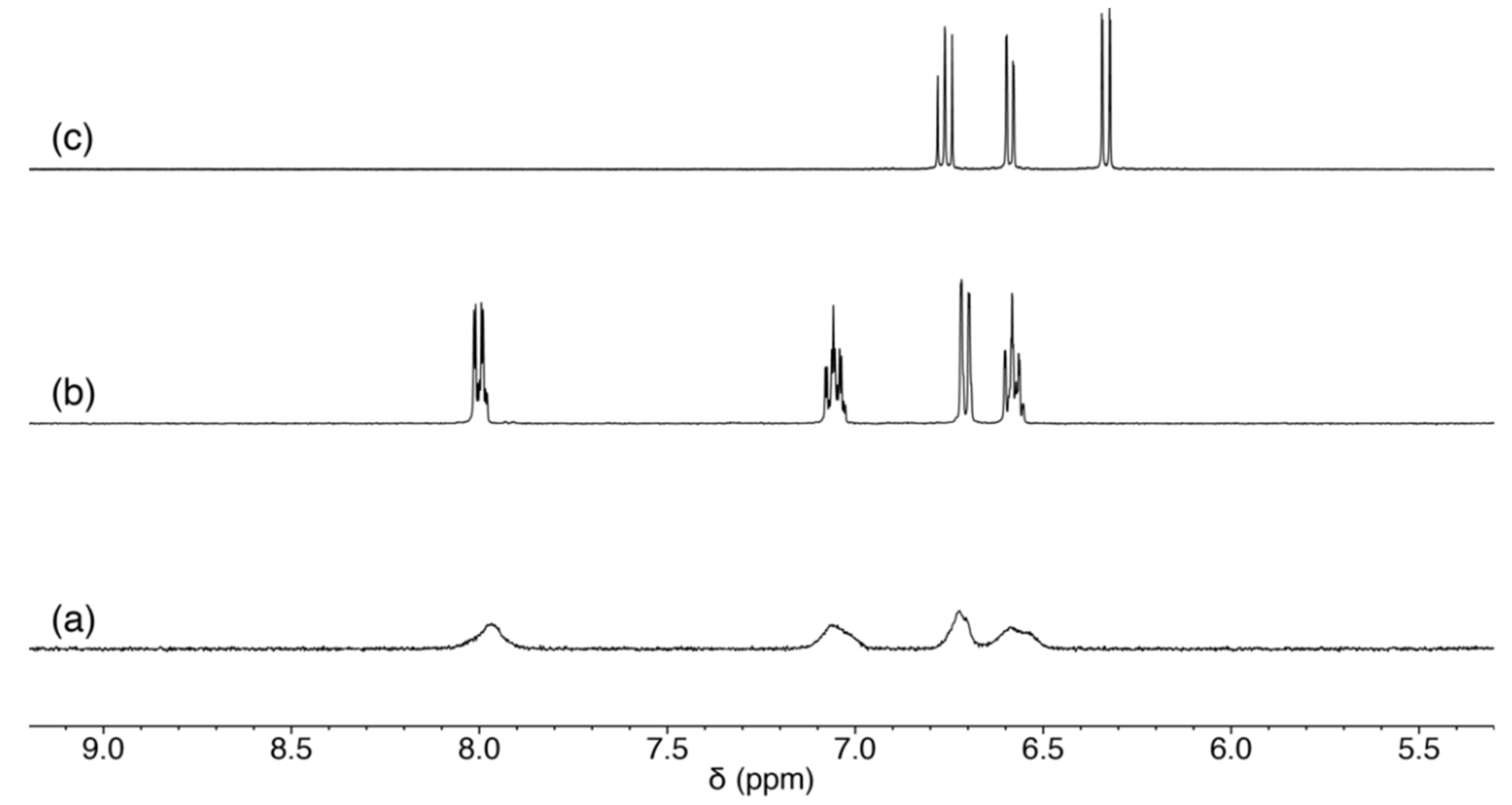
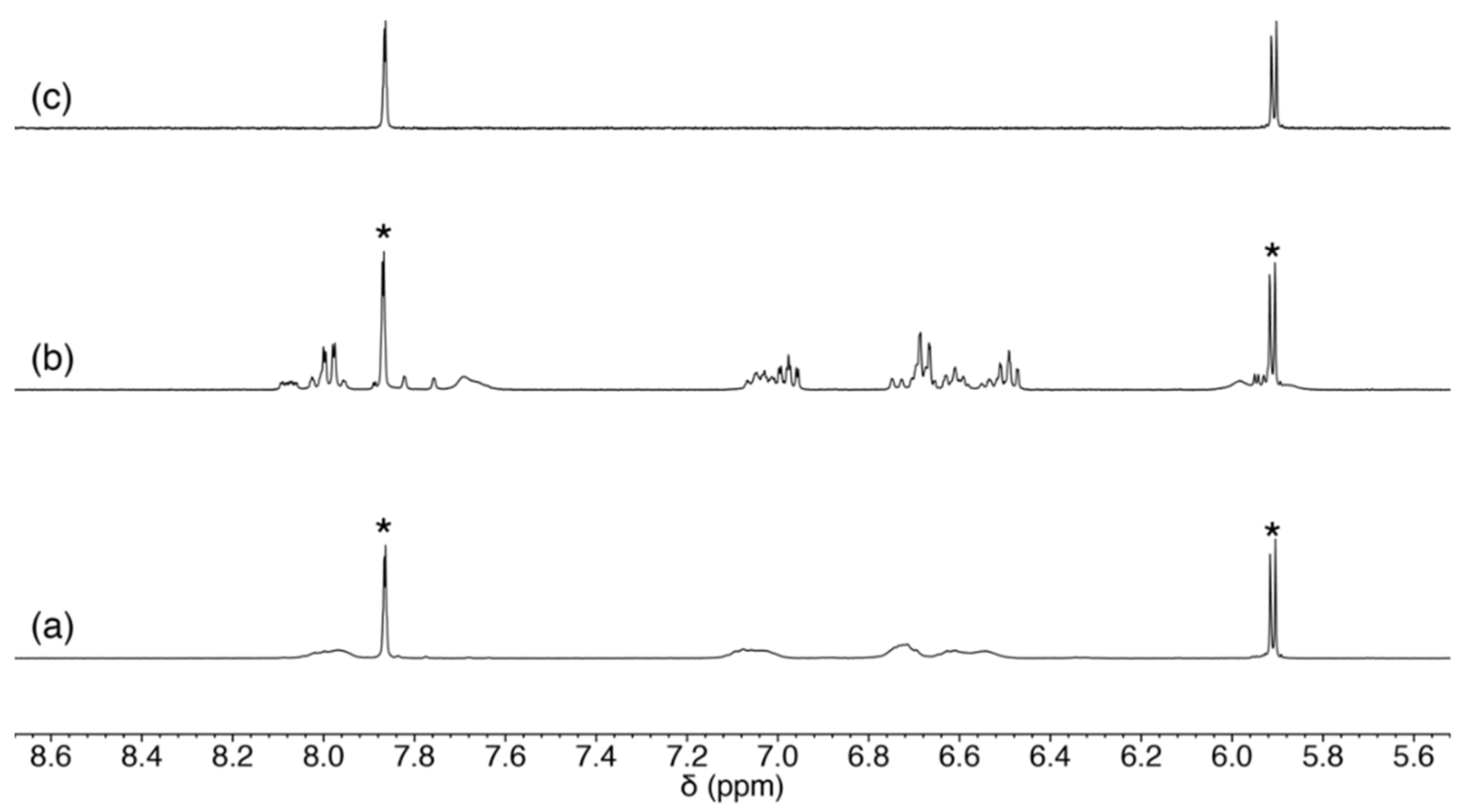
2.3.3. Scrambling Reaction of 1 with Two Nucleosides
3. Materials and Methods
3.1. Measurements
3.2. Materials
3.3. Preparations
3.4. Crystallography
3.5. Reaction of 1 with Nucleosides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 2001. [Google Scholar] [CrossRef]
- Gilli, G.; Gilli, P. The Nature of Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory; Oxford University Press: Oxford, UK, 2009. [Google Scholar] [CrossRef] [Green Version]
- Burrows, A.D. Crystal Engineering Using Multiple Hydrogen Bonds. In Supramolecular Assembly via Hydrogen Bond I Structure and Bondings; Mingos, D.M.P., Ed.; Springer: Amsterdam, The Netherlands, 2004; Volume 108, pp. 55–96. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Polito, M.; Grepioni, F. Crystal Engineering Using Multiple Hydrogen Bonds. In Supramolecular Assembly via Hydrogen Bond II Hydrogen Bonding Interactions Between Ions: A Powerful Took in Molecular Crystal Engineering; Mingos, D.M.P., Ed.; Springer: Amsterdam, The Netherlands, 2004; Volume 111, pp. 1–32. [Google Scholar] [CrossRef]
- Mitsuhashi, R.; Suzuki, T.; Hosoya, S.; Mikuriya, M. Hydrogen-Bonded Supramolecular Structures of Cobalt(III) Complexes with Unsymmetrical Bidentate Ligands: Mer/fac Interconversion Induced by Hydrogen-Bonding Interactions. Cryst. Growth Des. 2017, 17, 207–213. [Google Scholar] [CrossRef]
- Mitsuhashi, R.; Pedersen, K.S.; Ueda, T.; Suzuki, T.; Bendix, J.; Mikuriya, M. Field-induced single-molecule magnet behavior in ideal trigonal antiprismatic cobalt(II) complexes: Precise geometrical control by a hydrogen-bonded rigid metalloligand. Chem. Commun. 2018, 54, 8869–8872. [Google Scholar] [CrossRef]
- Mitsuhashi, R.; Hosoya, S.; Suzuki, T.; Sunatsuki, Y.; Sakiyama, H.; Mikuriya, M. Hydrogen-bonding interactions and magnetic relaxation dynamics in tetracoordinated cobalt(II) single-ion magnets. Dalton Trans. 2019, 48, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, R.; Hosoya, S.; Suzuki, T.; Sunatsuki, Y.; Sakiyama, H.; Mikuriya, M. Zero-field slow relaxation of magnetization in cobalt(II) single-ion magnets: Suppression of quantum tunneling of magnetization by tailoring the intermolecular magnetic coupling. RSC Adv. 2020, 10, 43472–43479. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, E.N.; Kim, E.; Wise, A.A.; O’Brien, P.J.; Andricioaei, I.; Al-Hashimi, H.M. Transient Hoogsteen base pairs in canonical duplex DNA. Nature 2011, 470, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, B.; Camp, L.V.; Krigas, T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1969, 205, 698–699. [Google Scholar] [CrossRef]
- Rosenberg, B.; Camp, L.V.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, K.S.; Todd, R.C.; Zhang, S.; McCormick, M.S.; D’Aquino, J.A.; Reardon, J.T.; Sancar, A.; Giacomini, K.M.; Lippard, S.J. cis-Diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: Uptake, structure, function, and prospects. Proc. Natl. Acad. Sci. USA 2008, 105, 8902–8907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, R.C.; Lippard, S.J. Structure of duplex DNA containing the cisplatin 1,2-{Pt(NH3)2}2+-d(GpG) cross-link at 1.77Å resolution. J. Inorg. Biochem. 2010, 104, 902–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuhashi, R.; Suzuki, T.; Sunatsuki, Y.; Kojima, M. Hydrogen-bonding interactions, geometrical selectivity and spectroscopic properties of cobalt(III) complexes with unsymmetrical tridentate amine-amidato-phenolato type ligands. Inorg. Chim. Acta 2013, 399, 131–137. [Google Scholar] [CrossRef]
- Mitsuhashi, R.; Ogawa, R.; Ishikawa, R.; Suzuki, T.; Sunatsuki, Y.; Kawata, S. Preparation, structures and properties of manganese complexes containing amine–(amido or amidato)–phenolato type ligands. Inorg. Chim. Acta 2016, 447, 113–120. [Google Scholar] [CrossRef]
- Schlosser, M. (Ed.) Organometallics in Synthesis: A Manual, 2nd ed.; Wiley: Hoboken, NJ, USA, 2004; 1126p. [Google Scholar]
- Hofmann, A.; Dahlenburg, L.; van Eldik, R. Cyclometalated analogues of platinum terpyridine complexes: Kinetic study of the strong sigma-donor cis and trans effects of carbon in the presence of a pi-acceptor ligand backbone. Inorg. Chem. 2003, 42, 6528–6538. [Google Scholar] [CrossRef] [PubMed]
- Odoko, M.; Okabe, N. (Adeninato-N9)[N-(2-aminoethyl)salicylideneiminato]palladium(II) 3.5-hydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, m2670–m2673. [Google Scholar] [CrossRef] [Green Version]
- Sherman, S.E.; Lippard, S.J. Structural aspects of platinum anticancer drug interactions with DNA. Chem. Rev. 1987, 87, 1153–1181. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
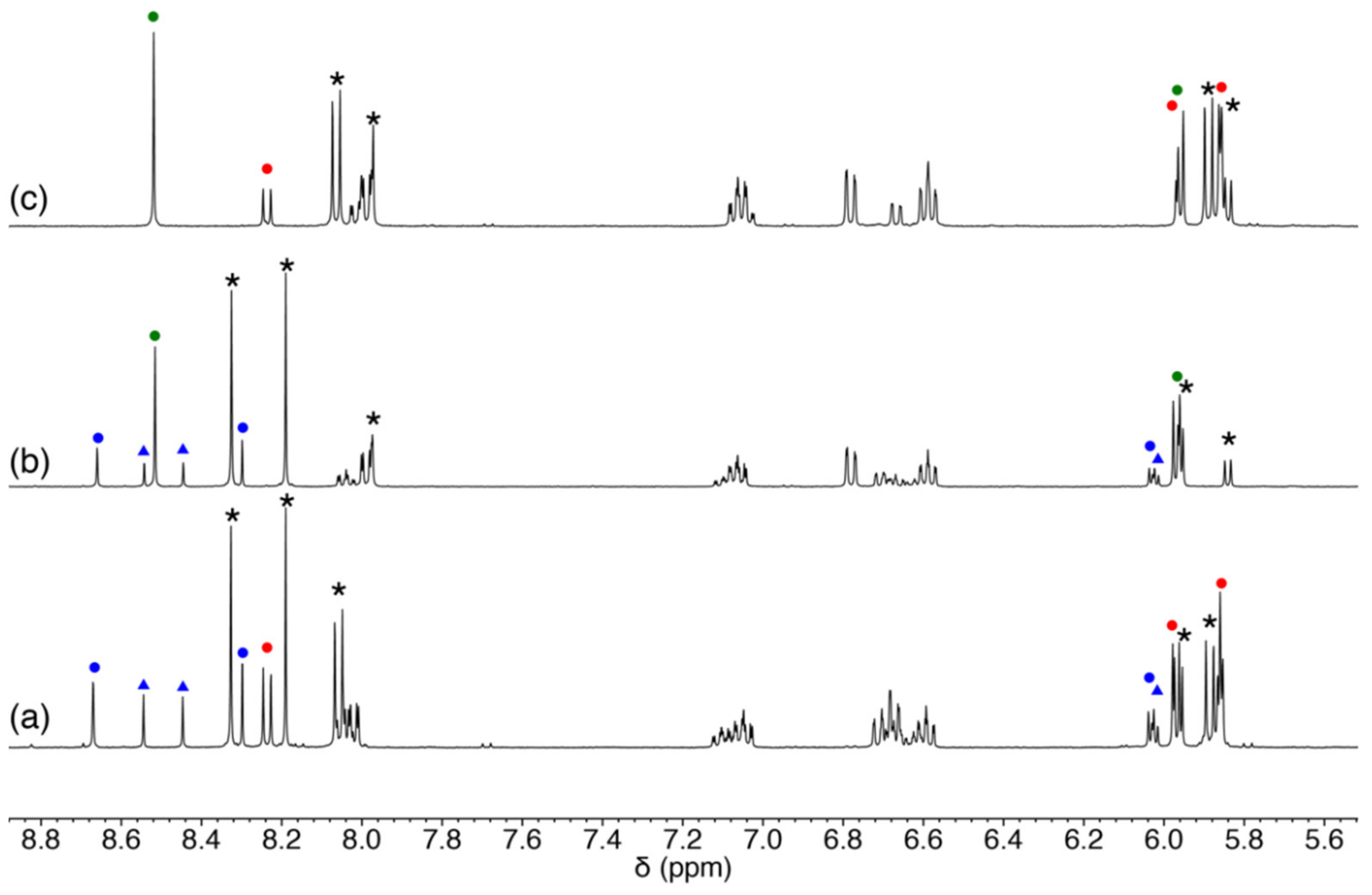{kind=link}
| Atom–Atom | Length/Å | Atom–Atom–Atom | Angle/° |
|---|---|---|---|
| Pd(1)–O(1) | 1.9877 (13) | N(1)–Pd(1)–O(1) | 93.70 (6) |
| Pd(1)–N(1) | 1.9647 (14) | N(1)–Pd(1)–N(2) | 83.87 (6) |
| Pd(1)–N(2) | 2.0264 (16) | O(1)–Pd(1)–N(2) | 177.15 (5) |
| Pd(1)–Cl(1) | 2.3389 (4) | N(1)–Pd(1)–Cl(1) | 176.50 (4) |
| O(1)–Pd(1)–Cl(1) | 89.79 (4) | ||
| N(2)–Pd(1)–Cl(1) | 92.64 (4) |
| Atom–Atom | Length/Å | Atom–Atom–Atom | Angle/° |
|---|---|---|---|
| Pd(1)–O(1) | 1.9771 (19) | N(1)–Pd(1)–O(1) | 94.73 (8) |
| Pd(1)–N(1) | 1.956 (2) | N(1)–Pd(1)–N(2) | 82.77 (9) |
| Pd(1)–N(2) | 2.025 (2) | O(1)–Pd(1)–N(2) | 176.79 (8) |
| Pd(1)–N(3) | 2.027 (2) | N(1)–Pd(1)–N(3) | 176.99 (8) |
| O(1)–Pd(1)–N(3) | 88.02 (8) | ||
| N(2)–Pd(1)–N(3) | 94.44 (9) |
| Atom–Atom | Length/Å | Atom–Atom–Atom | Angle/° |
|---|---|---|---|
| Pd(1)–C(5) | 1.982 (5) | N(1)–Pd(1)–C(5) | 81.8 (2) |
| Pd(1)–N(1) | 1.974 (5) | N(1)–Pd(1)–N(2) | 92.2 (2) |
| Pd(1)–N(2) | 2.144 (4) | C(5)–Pd(1)–N(2) | 173.2 (3) |
| Pd(1)–N(3) | 1.986 (5) | N(1)–Pd(1)–N(3) | 175.65 (17) |
| Pd(2)–C(17) | 1.981 (6) | C(5)–Pd(1)–N(3) | 95.1 (2) |
| Pd(1)–N(4) | 1.998 (4) | N(2)–Pd(1)–N(3) | 90.72 (19) |
| Pd(1)–N(5) | 2.121 (5) | N(4)–Pd(2)–C(17) | 81.8 (2) |
| Pd(1)–N(6) | 2.013 (5) | N(4)–Pd(2)–N(5) | 94.33 (18) |
| C(17)–Pd(2)–N(5) | 173.84 (19) | ||
| N(4)–Pd(2)–N(6) | 176.3 (2) | ||
| C(17)–Pd(2)–N(6) | 95.3 (2) | ||
| N(5)–Pd(2)–N(6) | 88.41 (19) |
| Complex | 1 | 1′ | 2 |
|---|---|---|---|
| Empirical formula | C15H28ClN3O3Pd | C15H20N4O2Pd | C12H15N3O2Pd |
| Formula weight | 440.25 | 394.75 | 339.67 |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Crystal dimensions/mm | 0.19 × 0.12 × 0.06 | 0.21 × 0.15 × 0.09 | 0.11 × 0.07 × 0.04 |
| Space group | P21/c | P21/c | P−1 |
| a/Å | 11.8363 (3) | 13.3692 (5) | 8.7885 (4) |
| b/Å | 8.7436 (2) | 10.9488 (4) | 10.8746 (5) |
| c/Å | 18.3451 (5) | 11.1827 (4) | 13.2005 (5) |
| α/° | 96.688 (4) | ||
| β/° | 104.732 (3) | 94.084 (3) | 90.840 (3) |
| γ/° | 104.073 (4) | ||
| V/Å3 | 1836.15 (8) | 1632.73 (10) | 1214.19 (9) |
| Z | 4 | 4 | 4 |
| T/K | 100 (2) | 100 (2) | 100 (2) |
| ρcalcd/g·cm−3 | 1.593 | 1.606 | 1.858 |
| µ/mm−1 | 1.173 | 1.149 | 1.526 |
| F(000) | 904 | 800 | 680 |
| 2θmax/◦ | 55 | 55 | 55 |
| No. of reflections measured | 17,469 | 11,616 | 17,415 |
| No. of independent reflections | 4207 (Rint = 0.0244) | 3737 (Rint = 0.0390) | 5543 (Rint = 0.1488) |
| Data/restraints/parameters | 4207/2/222 | 3737/0/203 | 5543/18/329 |
| R1 1 [I > 2.00 σ(I)] | 0.0217 | 0.0320 | 0.0555 |
| wR2 2 (all reflections) | 0.0558 | 0.0803 | 0.1348 |
| Goodness of fit indicator | 1.076 | 1.088 | 0.906 |
| Highest peak, deepest hole/e Å−3 | 0.445, −0.439 | 0.627, −1.156 | 1.646, −1.379 |
| CCDC deposition number | 2153005 | 2153006 | 2153007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitsuhashi, R.; Imai, Y.; Suzuki, T.; Hayashi, Y. Selective Formation of Intramolecular Hydrogen-Bonding Palladium(II) Complexes with Nucleosides Using Unsymmetrical Tridentate Ligands. Molecules 2022, 27, 2098. https://doi.org/10.3390/molecules27072098
Mitsuhashi R, Imai Y, Suzuki T, Hayashi Y. Selective Formation of Intramolecular Hydrogen-Bonding Palladium(II) Complexes with Nucleosides Using Unsymmetrical Tridentate Ligands. Molecules. 2022; 27(7):2098. https://doi.org/10.3390/molecules27072098
Chicago/Turabian StyleMitsuhashi, Ryoji, Yuya Imai, Takayoshi Suzuki, and Yoshihito Hayashi. 2022. "Selective Formation of Intramolecular Hydrogen-Bonding Palladium(II) Complexes with Nucleosides Using Unsymmetrical Tridentate Ligands" Molecules 27, no. 7: 2098. https://doi.org/10.3390/molecules27072098
APA StyleMitsuhashi, R., Imai, Y., Suzuki, T., & Hayashi, Y. (2022). Selective Formation of Intramolecular Hydrogen-Bonding Palladium(II) Complexes with Nucleosides Using Unsymmetrical Tridentate Ligands. Molecules, 27(7), 2098. https://doi.org/10.3390/molecules27072098