Microwave-Assisted Synthesis, Biological Activity Evaluation, Molecular Docking, and ADMET Studies of Some Novel Pyrrolo [2,3-b] Pyrrole Derivatives

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Result and Discussion
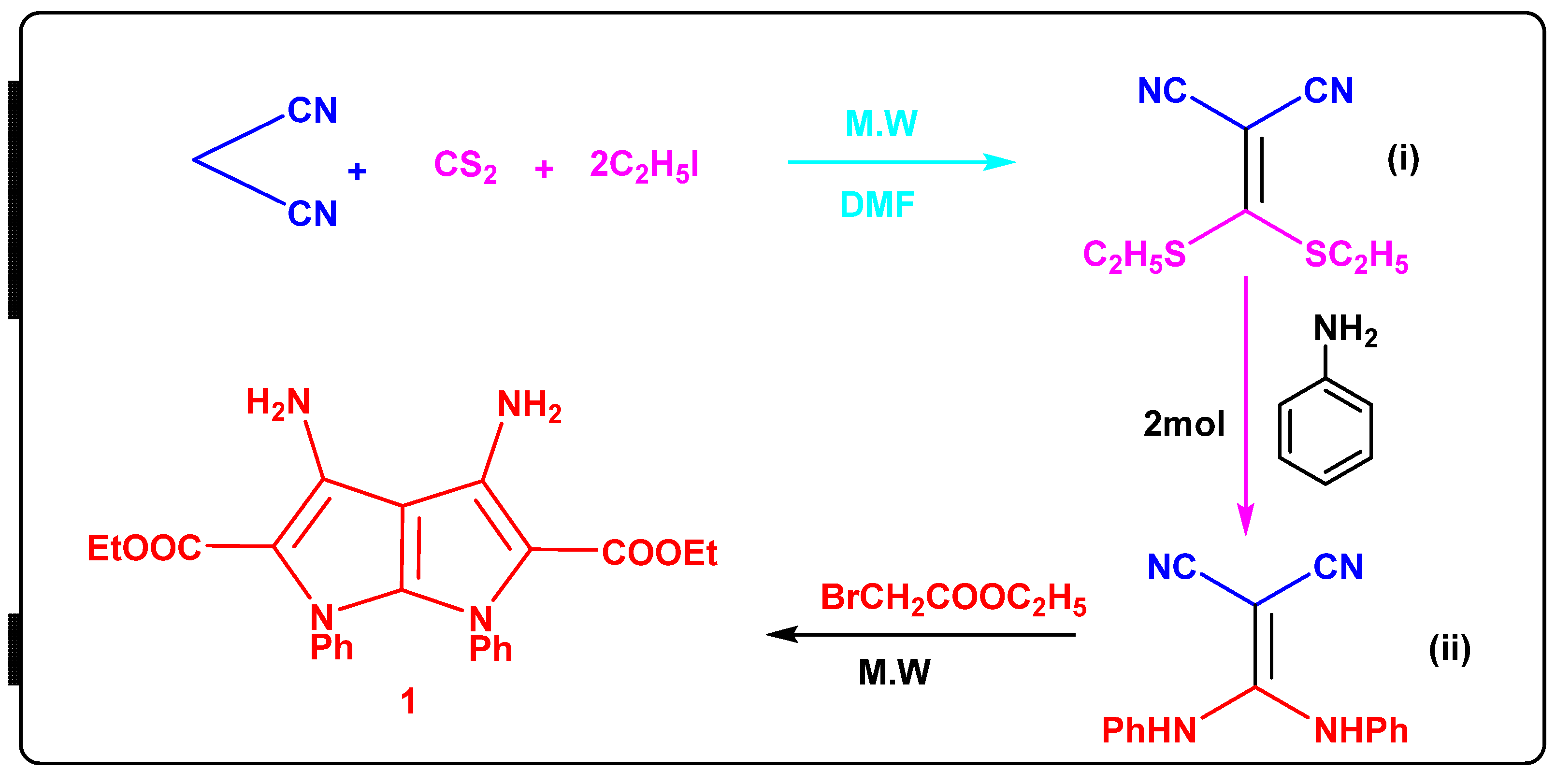
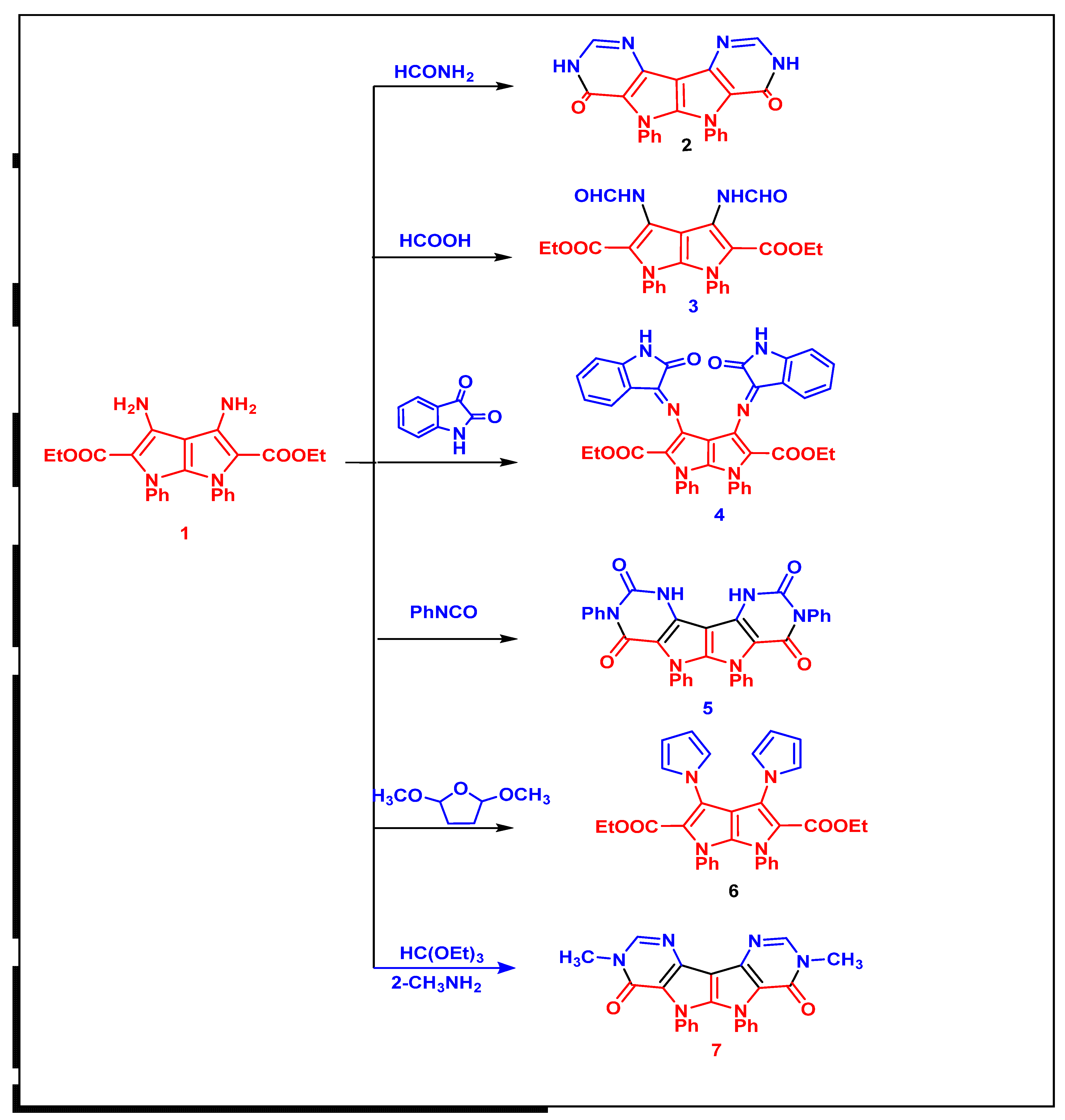
2.1. Chemistry
2.2. Biological Activities
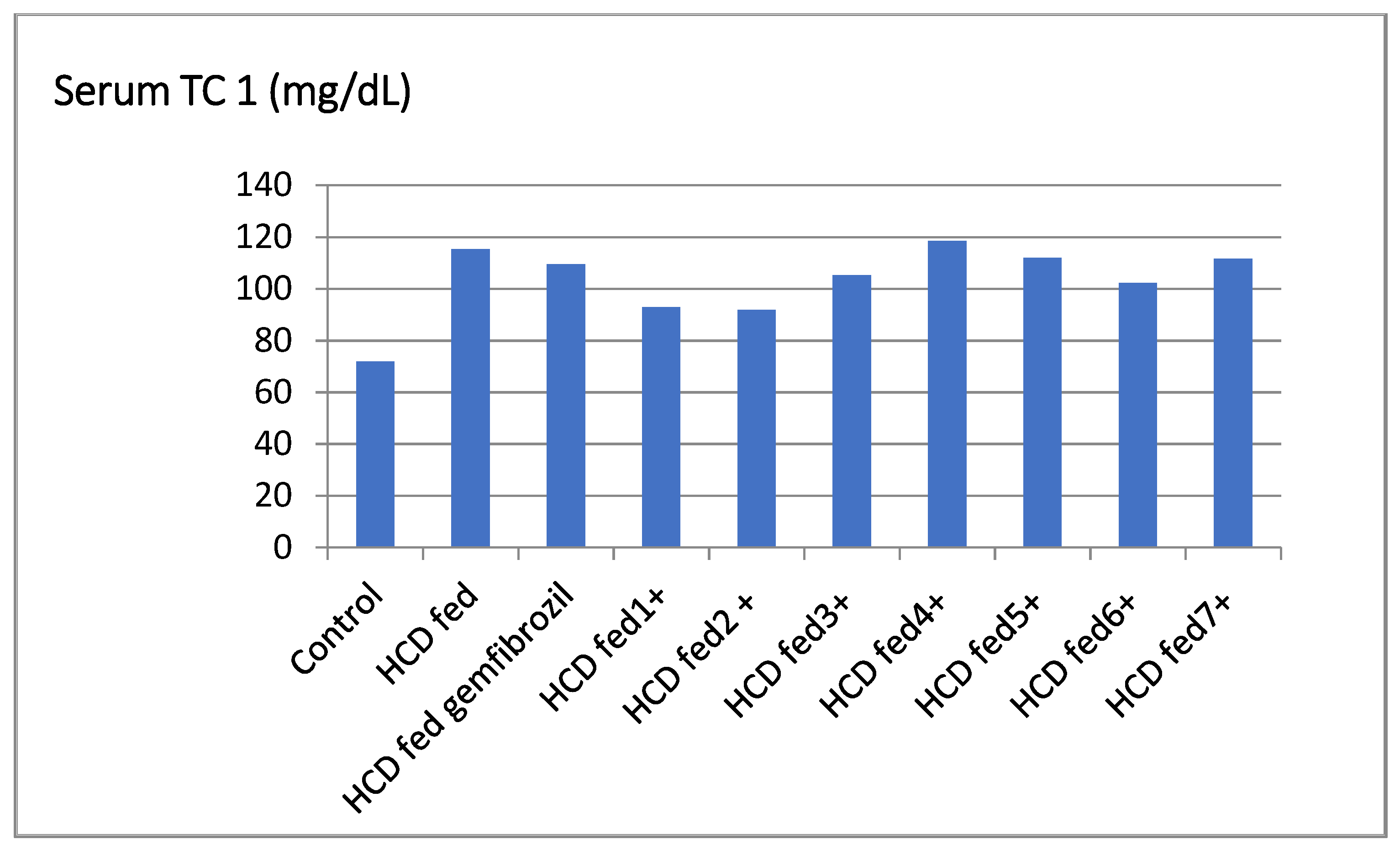
2.2.1. Hypolipidemic Activity
2.2.2. Screening of Antibacterial and Antifungal Activities
2.2.3. In Vitro Cytotoxic Activities
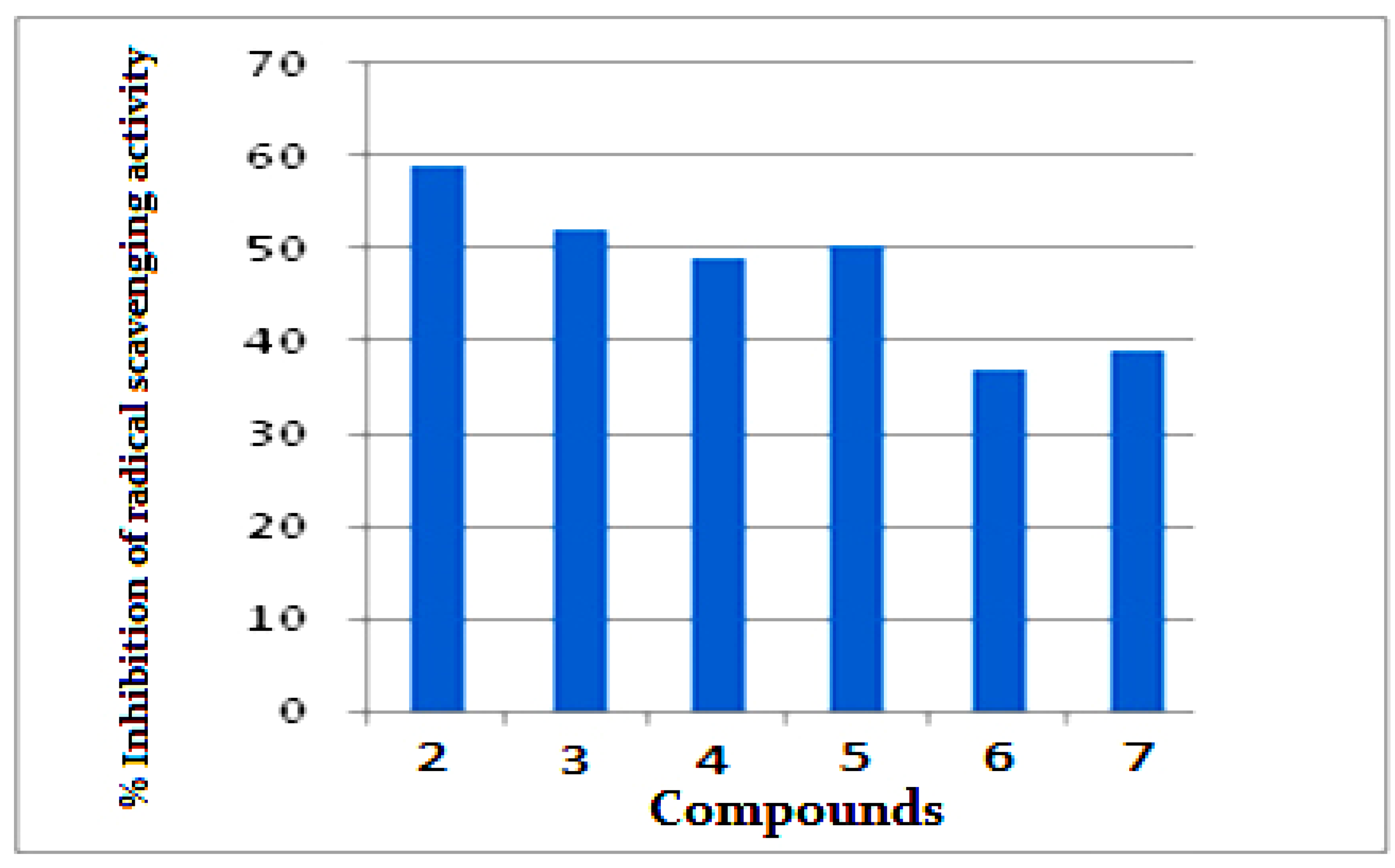
2.2.4. Antioxidant Activity by ABTS Method
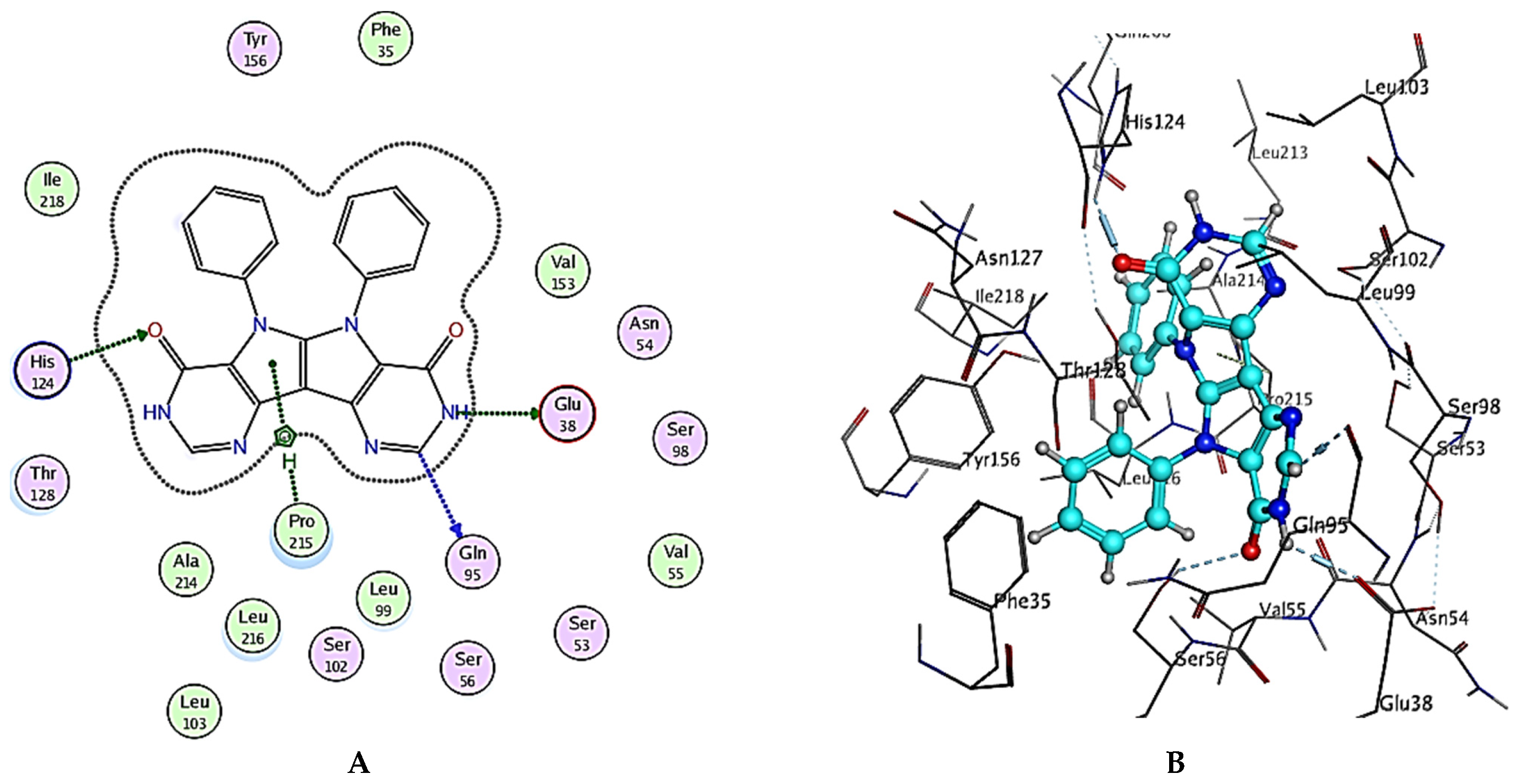
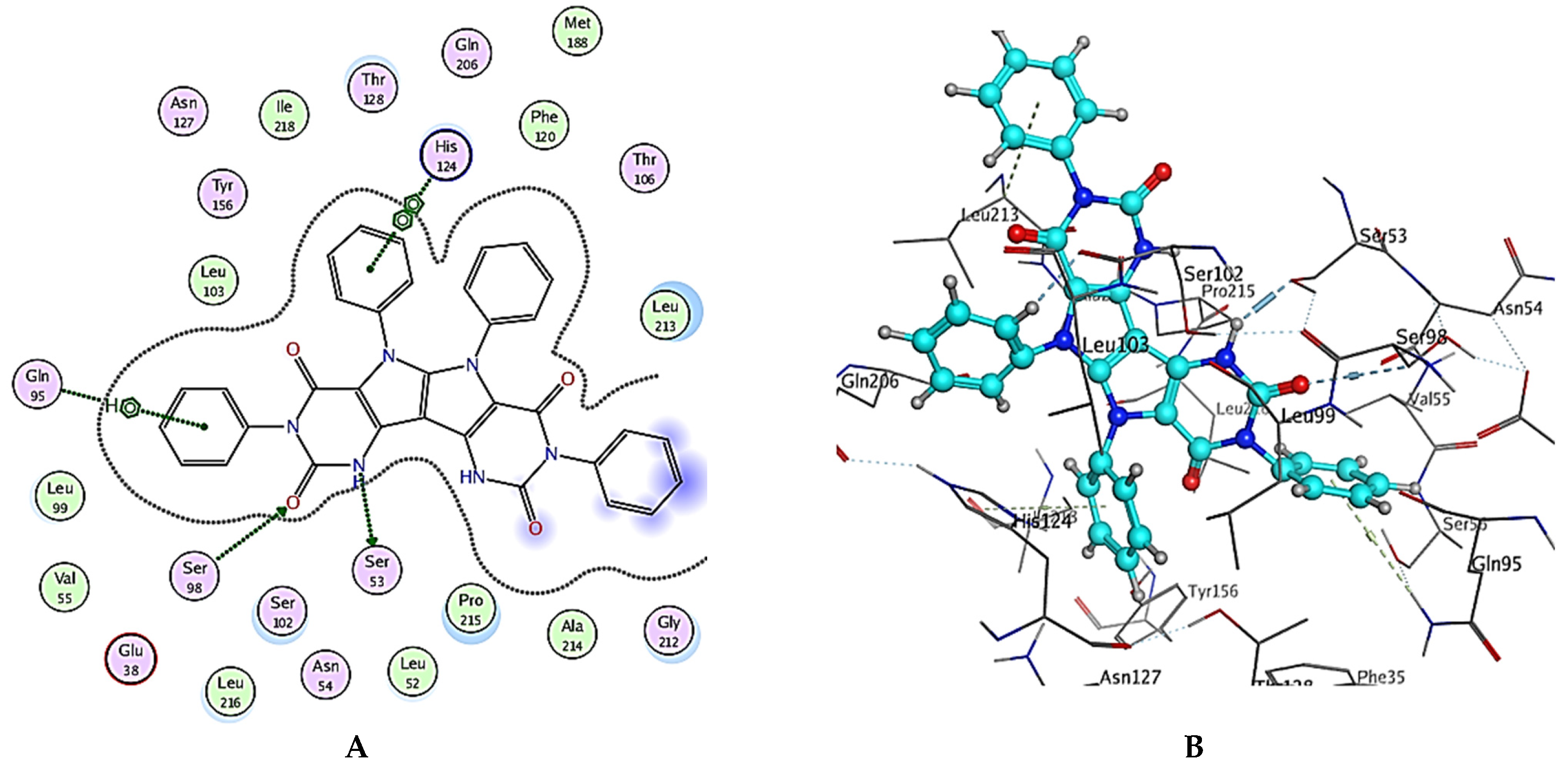
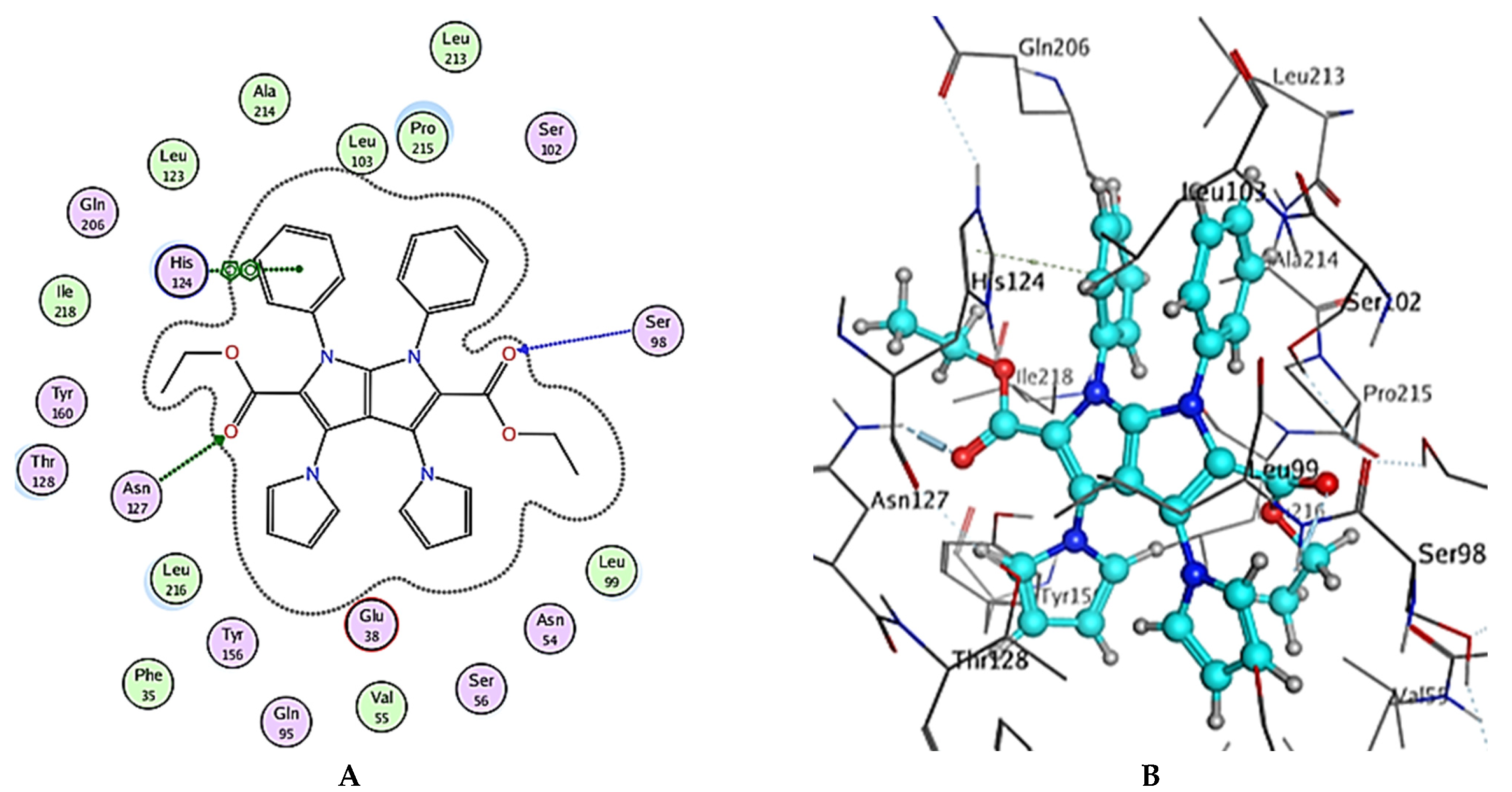
3. Molecular Docking Study
4. In Silico Pharmacokinetics and Toxicological Profile
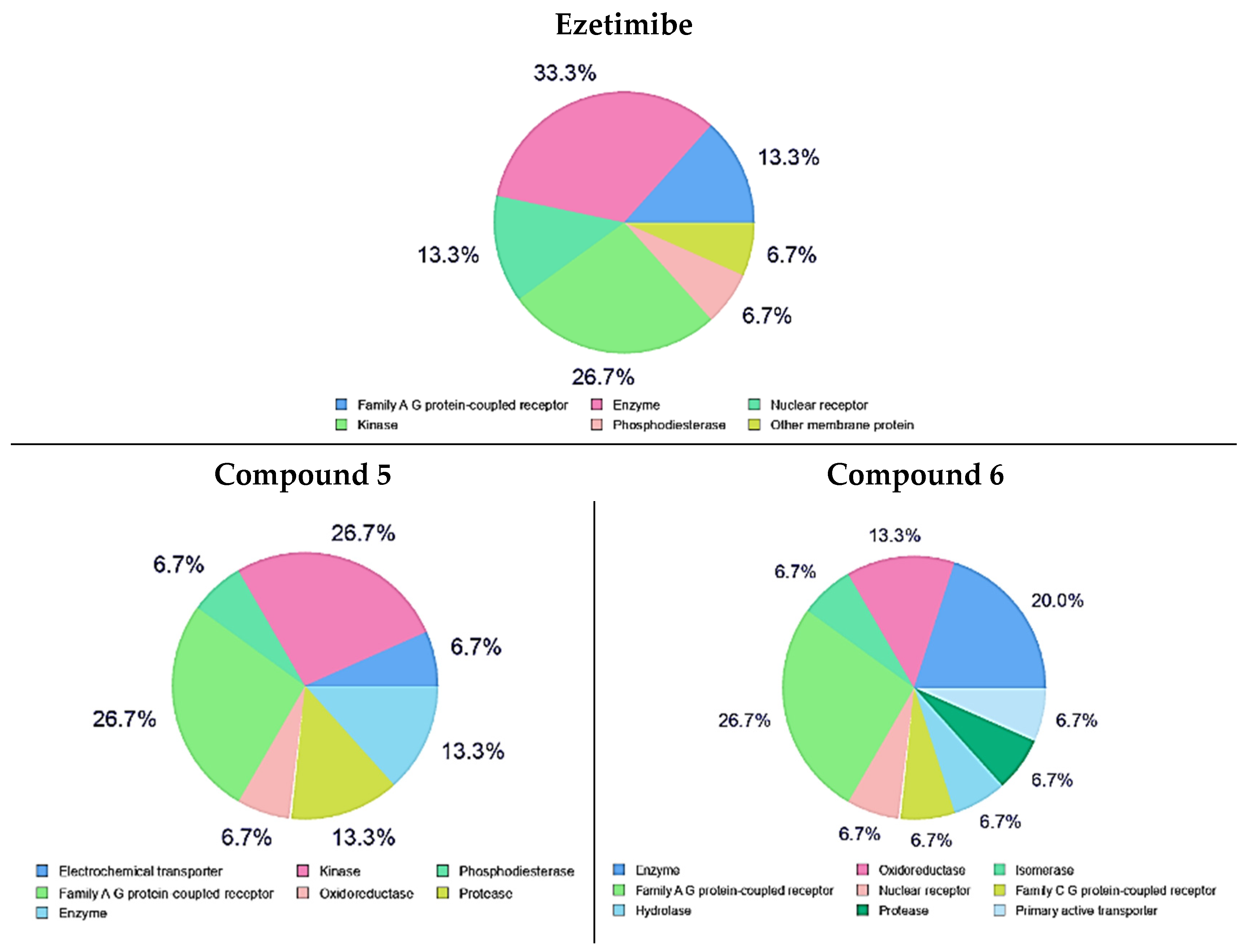
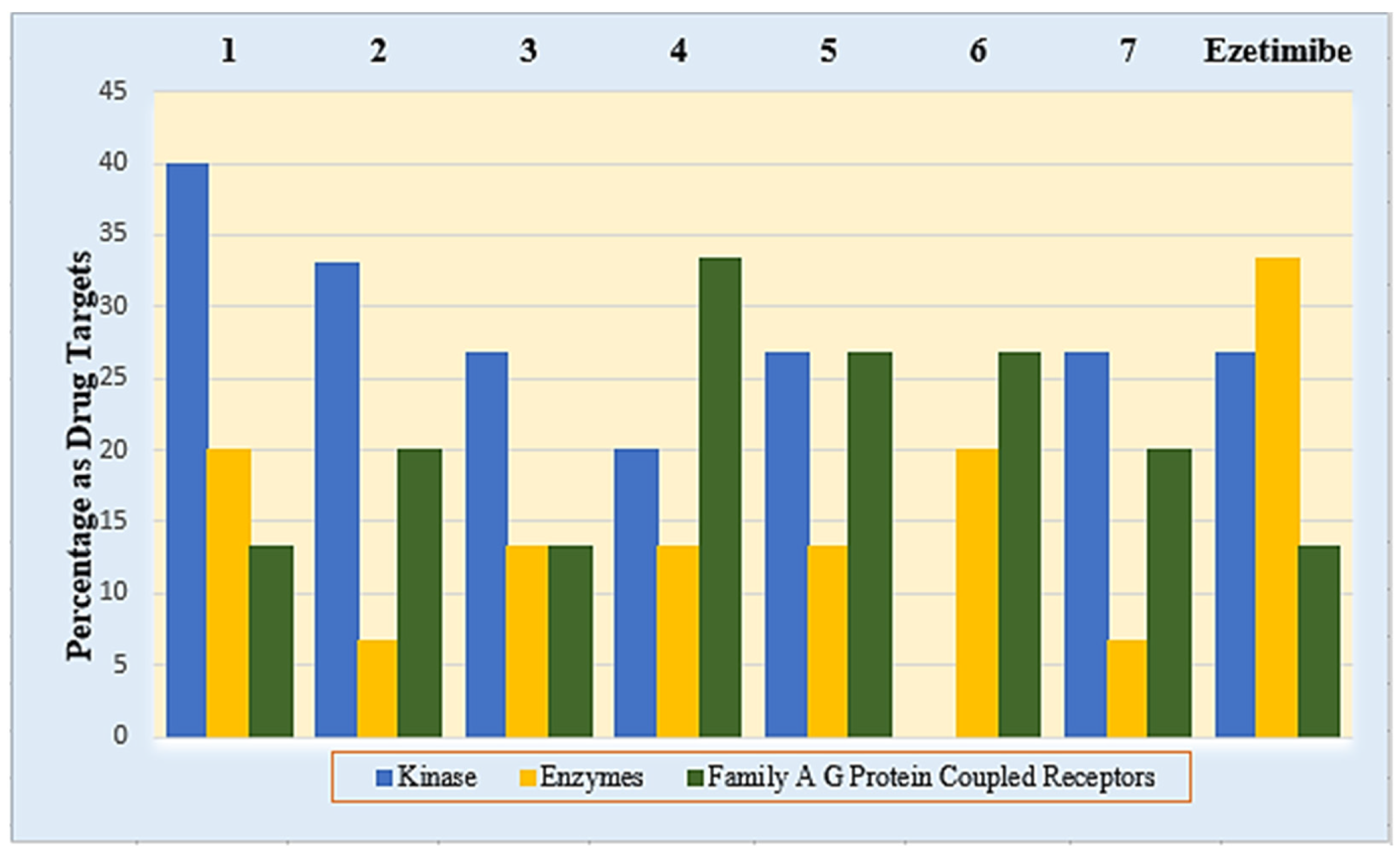
5. In Silico Target Prediction of Compounds 1–7 and Ezetimibe
6. Experimental Section
6.1. Chemistry
6.1.1. Materials and Methods
6.1.2. Synthesis of Diethyl 3,4-Diamino-1,6-diphenyl-1,6-dihydropyrrolo[2,3-b]pyrrole-2,5-dicarboxylate (1)
6.1.3. Synthesis of Compounds (2 and 3)
6.1.4. 5,6-Diphenyl-3,5,6,8-tetrahydropyrimido[4′′,3′′:4′,5′]pyrrolo[3′,2′:4,5]pyrrolo[3,2-d]pyrimidine-4,7-dione (2)
6.1.5. Diethyl 3,4-Diformamido-1,6-diphenyl-1,6-dihydropyrrolo[2,3-b]pyrrole-2,5-dicarboxylate (3)
6.1.6. Synthesis of Diethyl 3,4-bis(2-Oxoindolin-3-ylidene)amino)-1,6-diphenyl-1,6-dihydropyrrolo[2,3-b]pyrrole-2,5-dicarboxylate (4)
6.1.7. Synthesis of 3,5,6,8-Tetraphenyl-1,5,6,10-tetrahydropyrimido [4′′,3′′:4′,5′]pyrrolo[3′,2′:4,5]pyrrolo[3,2-d]pyrimidine-2,4,7,9(3H,8H)-tetraone (5)
6.1.8. Synthesis of Diethyl 1,6-diphenyl-3,4-di(1H-pyrrol-1-yl)-1,6-dihydro pyrrolo[2,3-b]pyrrole-2,5-dicarboxylate (6)
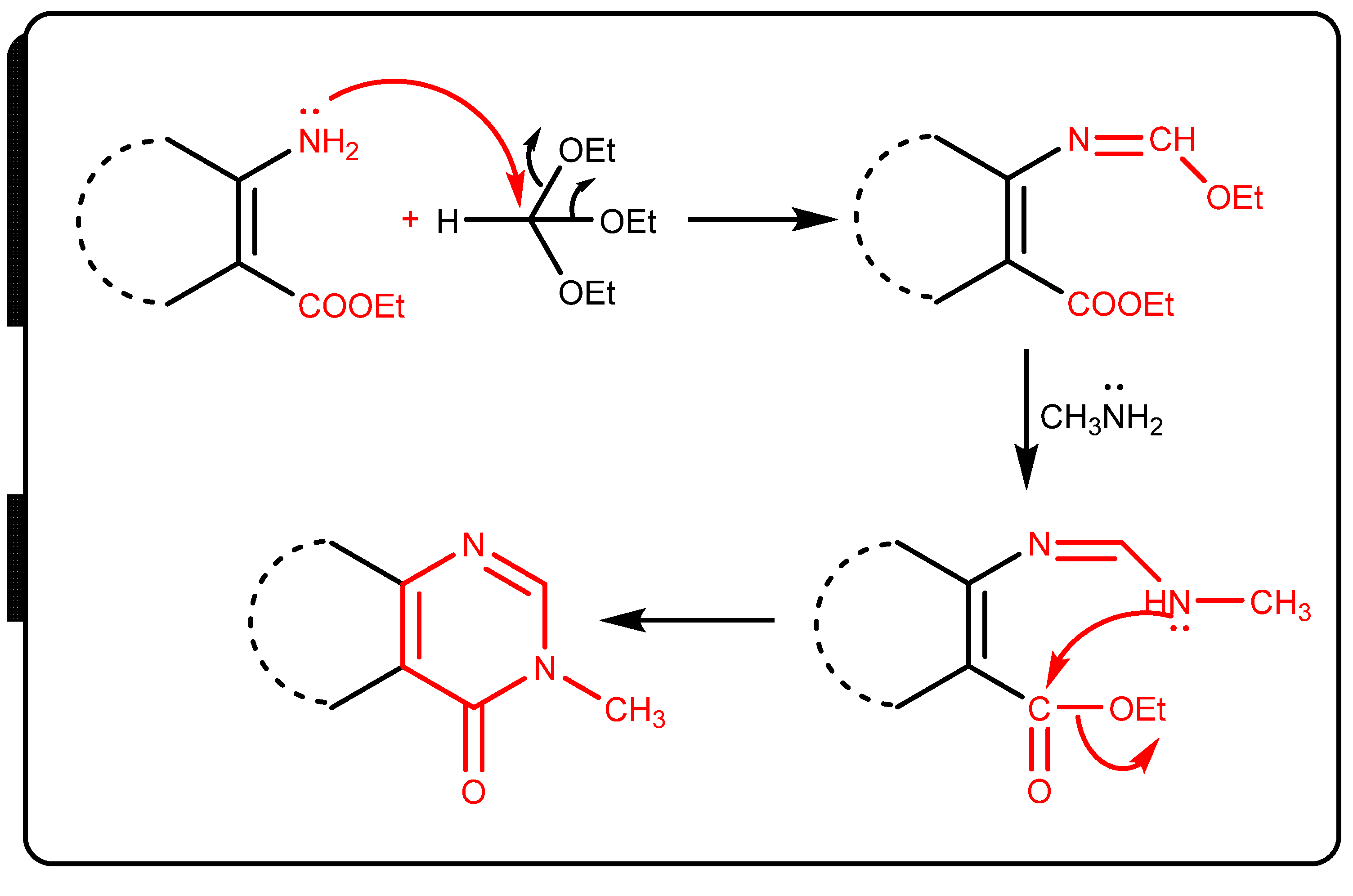
6.1.9. General Procedure of Synthesis 3,8-Dimethyl-5,6-diphenyl-3,5,6,8-tetrahydro-pyrimido [4′′,3′′:4′,5′]pyrrolo[3′,2′:4,5]pyrrolo[3,2-d]pyrimidine-4,7-dione (7)
6.2. Biological Activities
6.2.1. Hypolipidemic Activity
6.2.2. Antimicrobial and Antifungal Activity
6.2.3. In Vitro Cytotoxic Activity Screening
6.2.4. Antioxidant Activity by ABTS Method
7. Molecular Modeling
8. In Silico ADME/Tox Profile of the Synthesized Compounds (1–7) and Ezetimibe
9. In Silico Target Prediction of the Synthesized Compounds (1–7) and Ezetimibe
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rawat, P.; Singh, R. Assessment of conformational, spectral, antimicrobial activity, chemical reactivity and NLO application of Pyrrole-2, 5-dicarboxaldehyde bis (oxaloyldihydrazone). Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2015, 140, 344–355. [Google Scholar] [CrossRef]
- Pagadala, L.R.; Mukkara, L.D.; Singireddi, S.; Singh, A.; Thummaluru, V.R.; Jagarlamudi, P.S.; Guttala, R.S.; Perumal, Y.; Dharmarajan, S.; Upadhyayula, S.M. Design, synthesis and anti-mycobacterial activity of 1, 2, 3, 5-tetrasubstituted pyrrolyl-N-acetic acid derivatives. Eur. J. Med. Chem. 2014, 84, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, N.; Raghunathan, R.; Almansour, A.I.; Karama, U. An efficient synthesis of highly functionalized novel chromeno [4, 3-b] pyrroles and indolizino [6, 7-b] indoles as potent antimicrobial and antioxidant agents. Bioorganic Med. Chem. Lett. 2012, 22, 1375–1379. [Google Scholar] [CrossRef]
- Irfan, M.; Deng, R.; Sumra, I.; Zhu, X.-F.; Liu, T.; Zeng, Z. Stereoselective synthesis of E, E/E, Z isomers based on 1-(4-iodophenyl)-2, 5-divinyl-1H-pyrrole core skeleton: A configuration-controlled fluorescence characteristics and highly selective anti-cancer activity. Dye. Pigment. 2021, 184, 108733. [Google Scholar] [CrossRef]
- Xu, X.-T.; Mou, X.-Q.; Xi, Q.-M.; Liu, W.-T.; Liu, W.-F.; Sheng, Z.-J.; Zheng, X.; Zhang, K.; Du, Z.-Y.; Zhao, S.-Q. Anti-inflammatory activity effect of 2-substituted-1, 4, 5, 6-tetrahydrocyclopenta [b] pyrrole on TPA-induced skin inflammation in mice. Bioorganic Med. Chem. Lett. 2016, 26, 5334–5339. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.-B.; Qian, X.; Feng, D.; Fisher, M.; Crumley, T.; Darkin-Rattray, S.J.; Dulski, P.M.; Gurnett, A.; Leavitt, P.S.; Liberator, P.A. N-Alkyl-4-piperidinyl-2, 3-diarylpyrrole derivatives with heterocyclic substitutions as potent and broad spectrum anticoccidial agents. Bioorg. Med. Chem. Lett. 2008, 18, 2019–2022. [Google Scholar] [CrossRef] [PubMed]
- Boukouvala, M.C.; Kavallieratos, N.G.; Athanassiou, C.G.; Hadjiarapoglou, L.P. Insecticidal effect of two novel pyrrole derivatives against two major stored product insect species. Crop Prot. 2016, 84, 1–7. [Google Scholar] [CrossRef]
- N’guessan, R.; Boko, P.; Odjo, A.; Akogbeto, M.; Yates, A.; Rowland, M. Chlorfenapyr: A pyrrole insecticide for the control of pyrethroid or DDT resistant Anopheles gambiae (Diptera: Culicidae) mosquitoes. Acta Trop. 2007, 102, 69–78. [Google Scholar] [CrossRef]
- Ali El-Remaily, M.A.E.A.A.; El-Dabea, T.; Alsawat, M.; Mahmoud, M.H.; Alfi, A.A.; El-Metwaly, N.; Abu-Dief, A.M. Development of New Thiazole Complexes as Powerful Catalysts for Synthesis of Pyrazole-4-Carbonitrile Derivatives under Ultrasonic Irradiation Condition Supported by DFT Studies. ACS Omega 2021, 6, 21071–21086. [Google Scholar] [CrossRef]
- Shoman, M.E.; Aboelez, M.O.; Shaykhon, M.S.A.; Ahmed, S.A.; Abuo-Rahma, G.E.-D.A.; Elhady, O.M. New nicotinic acid-based 3, 5-diphenylpyrazoles: Design, synthesis and antihyperlipidemic activity with potential NPC1L1 inhibitory activity. Mol. Divers. 2021, 25, 673–686. [Google Scholar] [CrossRef]
- Shattat, G.F. A review article on hyperlipidemia: Types, treatments and new drug targets. Biomed. Pharmacol. J. 2014, 7, 399–409. [Google Scholar] [CrossRef]
- Jain, K.S.; Kathiravan, M.K.; Somani, R.S.; Shishoo, C.J. Thebiology and chemistry of hyperlipidemia. Bioorg. Med. Chem. 2007, 15, 4674–4699. [Google Scholar] [CrossRef] [PubMed]
- Luijten, J.; van Greevenbroek, M.M.J.; Schaper, N.C.; Meex, S.J.R.; vander Steen, C.; Meijer, L.J.; de Boer, D.; de Graaf, J.; Stehouwer, C.D.A.; Brouwers, M.C.G.J. Incidence of cardiovascular disease in familial combined hyperlipidemia: A 15-year follow-up study. Atherosclerosis 2019, 280, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bodor, E.T.; Offermanns, S. Nicotinic acid: An old drug with a promising future. Br. J. Pharmacol. 2008, 153, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Jia, L.; Betters, J.L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu. Rev. Phys. 2011, 73, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, L.M.R.; de Mejia, E.G. Bean peptides have higher in silico binding affinities than ezetimibe for the N-terminal domain of cholesterol receptor Niemann-Pick C1 Like-1. Peptides 2017, 90, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Shokr, E.K.; Kamel, M.S.; Abdel-Ghany, H.; Ali, M.A.E.A.A. Optical characterization and effects of iodine vapor & gaseous HCl adsorption investigation of Novel Synthesized Organic dye Based on Thieno [2, 3-b] thiophene. Optik 2021, 243, 167385. [Google Scholar]
- Shokr, E.K.; Kamel, M.S.; Abdel-Ghany, H.; Ali, M.A.E.A.A. Optoelectronic characteristics of as-deposited, annealed and I2–Treated thin films of newly synthesized organic dye based on pyrrolo [2, 3-b] pyrrole. Curr. Res. Green Sustain. Chem. 2021, 4, 100090. [Google Scholar] [CrossRef]
- Minhajuddin, M.; Beg, Z.H.; Iqbal, J. Hypolipidemic and antioxidant properties of tocotrienol rich fraction isolated from rice bran oil in experimentally induced hyperlipidemic rats. Food Chem. Toxicol. 2005, 43, 747–753. [Google Scholar] [CrossRef]
- Lorke, D. A New Approach to Practical Acute Toxicity Testing. Arch. Toxicol. 1983, 54, 275–287. [Google Scholar] [CrossRef]
- Maxwell, R.; Nawrocki, J.; Uhlendorf, P. Some comparative effects of gemfibrozil, clofibrate, bezafibrate, cholestyramine and compactin on sterol metabolism in rats. Atherosclerosis 1983, 48, 195–203. [Google Scholar] [CrossRef]
- Molecular Operating Enviroment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2013.
- Shoichet, B.K.; McGovern, S.L.; Wei, B. Lead discovery using molecular docking. Curr. Opin. Chem. Biol. 2002, 6, 439–446. [Google Scholar] [CrossRef]
- Durán-Iturbide, N.A.; Díaz-Eufracio, B.I.; Medina-Franco, J.L. In Silico ADME/Tox Profiling of Natural Products: A Focus on BIOFACQUIM. ACS Omega 2020, 5, 16076–16084. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.swissadme.ch/ (accessed on 25 January 2022).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug- likeness and medicinal chemistry friendliness of small molecules. Nat. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://biosig.unimelb.edu.au/pkcsm/prediction (accessed on 27 January 2022).
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Taylor, J.B.; Triggle, D.J. Comprehensive Medicinal Chemistry II, 5th ed.; Elsevier Ltd.: Amsterdam, The Netherlands; London, UK, 2007; pp. 669–697. [Google Scholar]
- Zerroug, A.; Belaidi, S.; BenBrahim, I.; Sinha, L.; Chtita, S. Virtual screening in drug-likeness and structure/activity relationship of pyridazine derivatives as Anti-Alzheimer drugs. J. King Saud Univ. Sci. 2019, 31, 595–601. [Google Scholar] [CrossRef]
- Watanabe, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Ohashi, R.; Mizuguchi, K. Predicting Fraction Unbound in Human Plasma from Chemical Structure: Improved Accuracy in the Low Value Ranges. Mol. Pharm. 2018, 15, 5302–5311. [Google Scholar] [CrossRef] [Green Version]
- Nagini, P.M. Siddavaram. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Ye, T.; Wang, R.; Zhang, C.; Zhang, X.; Sun, G.; Sun, X. An in silico model for predicting drug-induced hepatotoxicity. Int. J. Mol. Sci. 2019, 20, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedewald, W.; Levy, R.; Fredrickson, D. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499. [Google Scholar] [CrossRef] [PubMed]
- Ermondi, G.; Caron, G.; Lawrence, R.; Longo, D. Docking studies on NSAID/COX2 isozyme complexes using Contact Statistics analysis. J. Comput. Aided Mol. Des. 2004, 18, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.swisstargetprediction.ch/ (accessed on 28 January 2022).
- Mosmann, T. Rapidcolorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Thabrew, M.I.; Hughes, R.D.; McFarlane, I.G. Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay. J. Pharm. Pharmacol. 1997, 49, 1132–1135. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
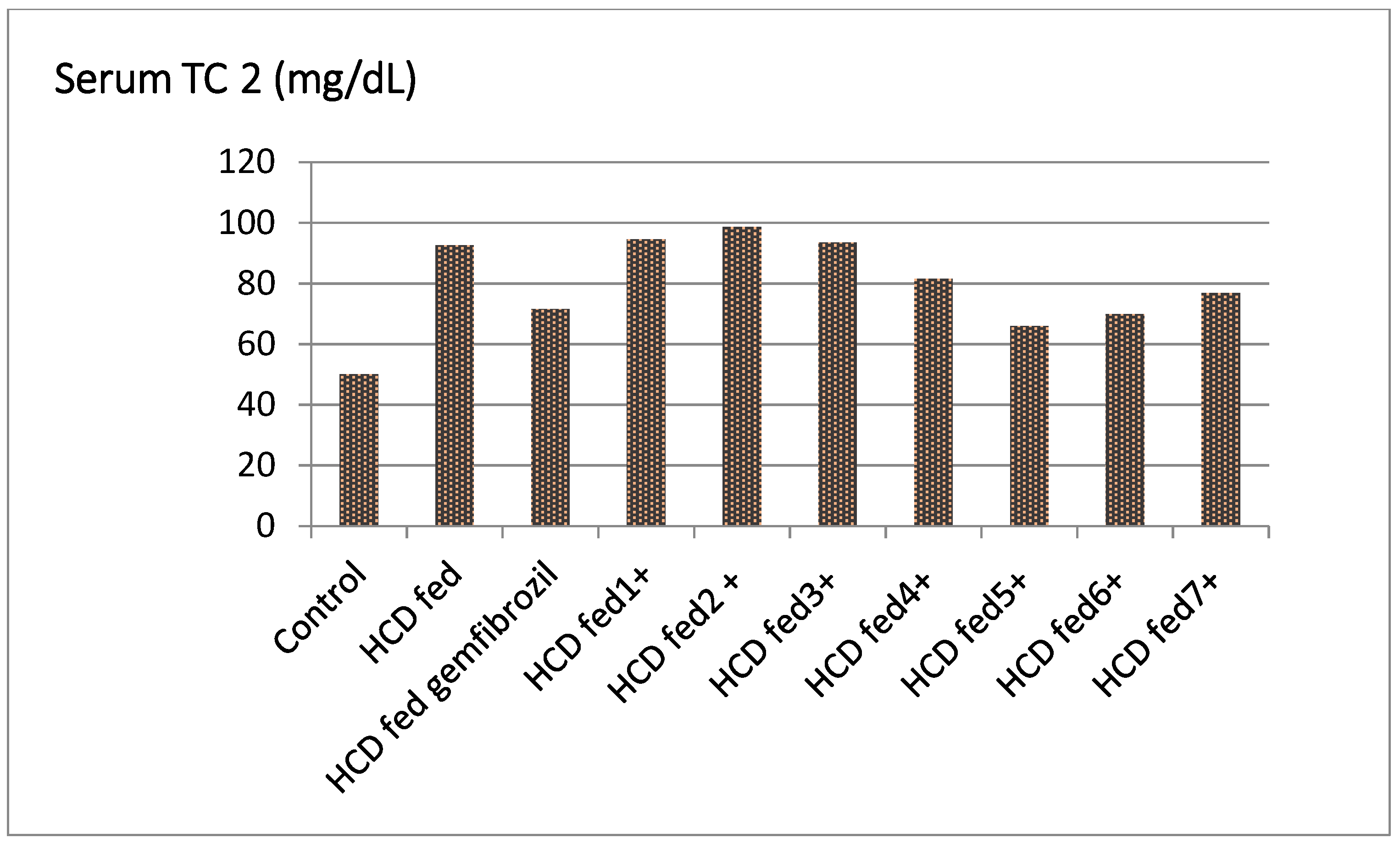
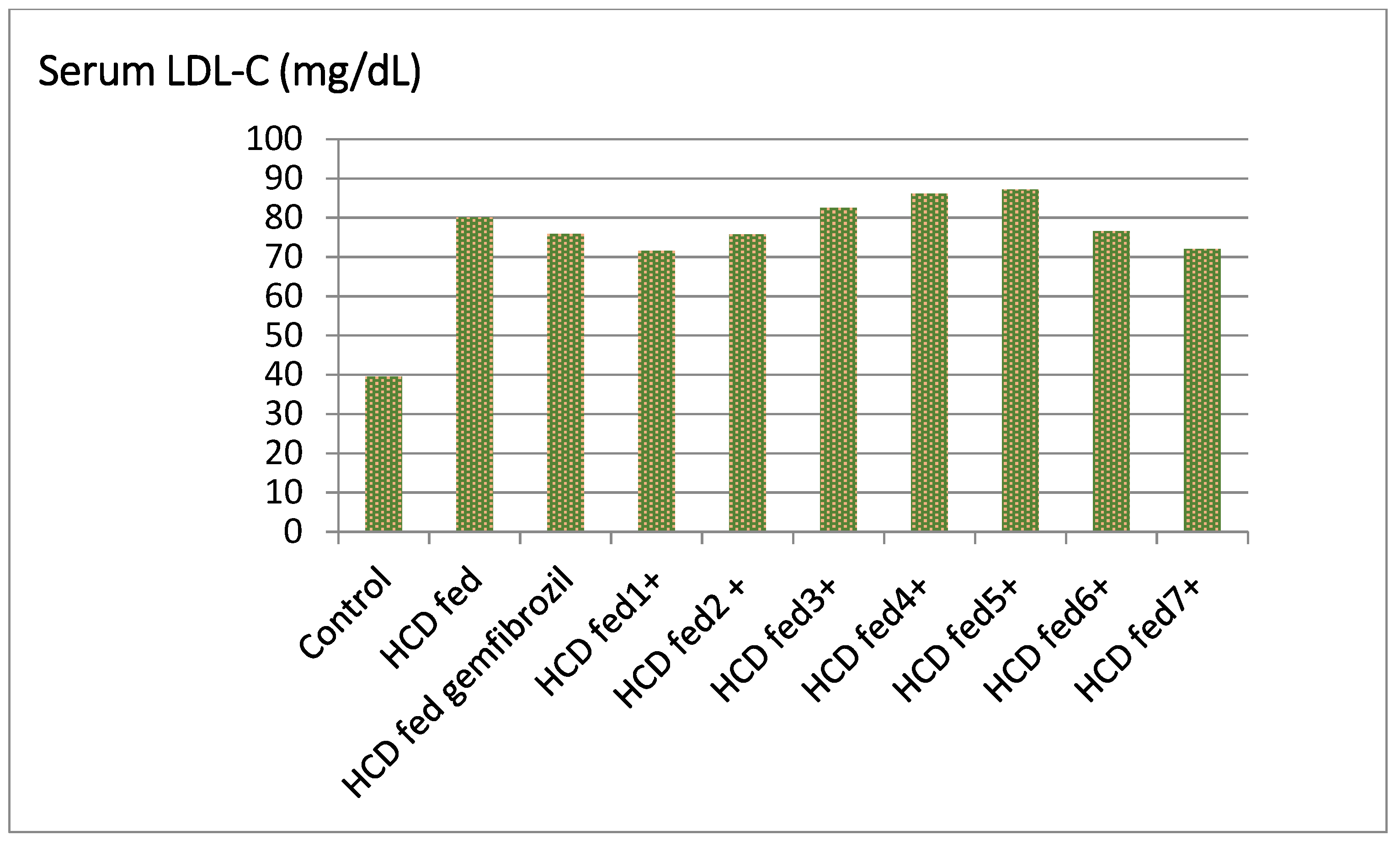
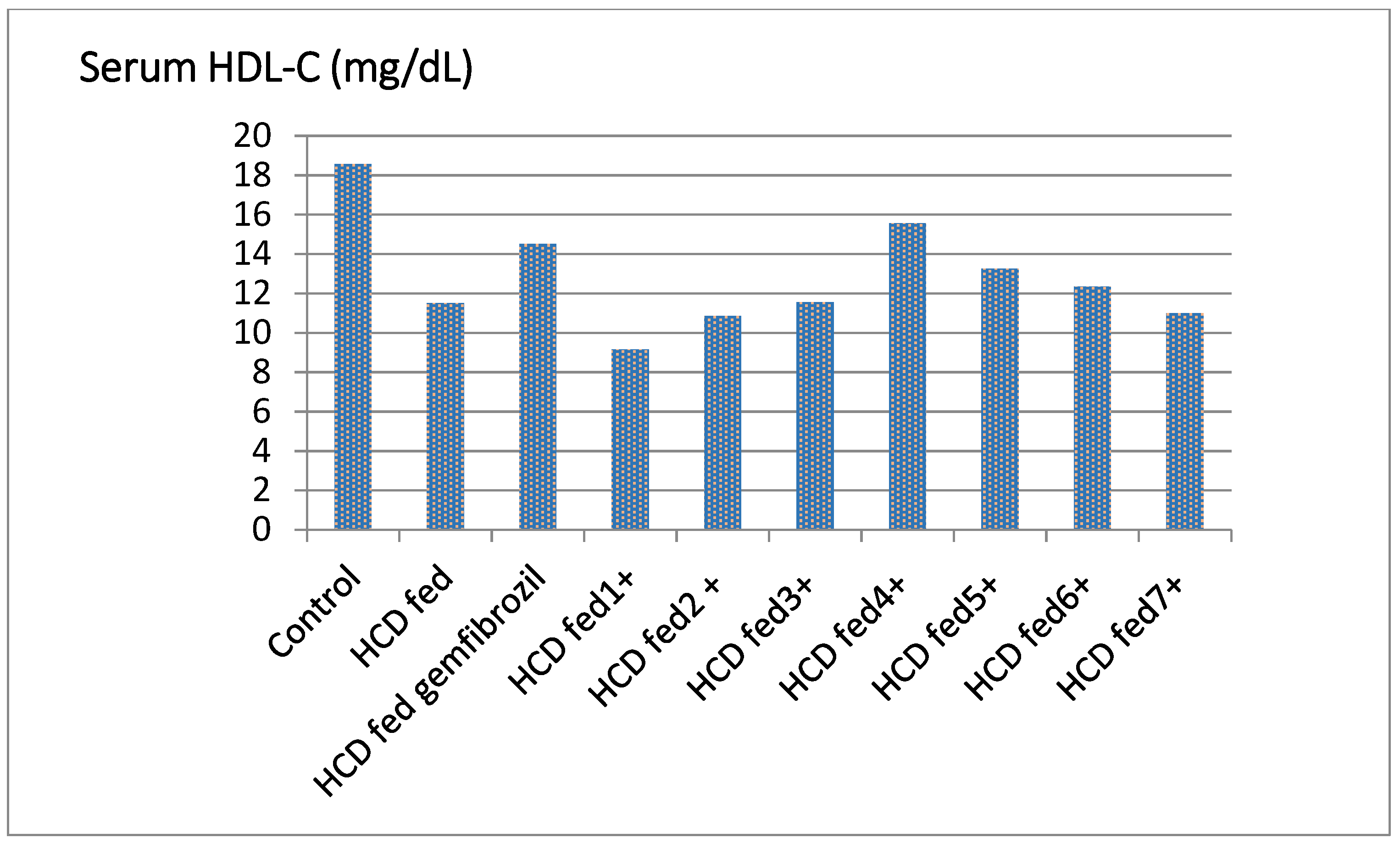
| Groups | TC ** (mg/dL) | % Fall | TG (mg/dL) | % Fall | HDL-C (mg/dL) | % Rise | LDL-C (mg/dL) | % Fall | LDL/HDL |
|---|---|---|---|---|---|---|---|---|---|
| Control | 71.83 ± 1.50 | - | 50.00 ± 1.14 | - | 18.56 ± 1.52 | - | 39.50 ± 2.71 | - | 2.13 |
| HCD fed | 115.33 ± 1.60 | - | 92.50 ± 0.71 | - | 11.50 ± 0.50 | - | 80.00 ± 1.91 | - | 6.69 |
| HCD fed gemfibrozil | 109.50 ± 6.27 | 4.65 | 71.52 ± 3.04 ** | 22.68 | 14.50 ± 0.52 ** | 25.54 | 75.89 ± 7.24 * | 5.13 | 5.23 |
| HCD fed ±1 | 92.89 ± 1.10 | −1.79 b | 94.51 ± 2.11 ** | −3.67 b | 9.15 ± 1.43 ** | −3.54 b | 71.50 ± 2.10 ** | −1.87 c | 7.44 |
| HCD fed ± 2 | 91.87± 1.32 ** | 2.88 | 98.65 ± 4.02 ** | 12.11 | 10.85 ± 2.63 ** | 20.43 | 75.77 ± 2.60 ** | 1.13 | 5.21 |
| HCD fed ±3 | 105.22 ± 1.04 | −1.04 b | 93.48 ± 2.67 | −1.55 b | 11.55 ± 0.52 | −8.65 b | 82.49 ± 3.30 | −2.18 c | 7.82 |
| HCD fed ±4 | 118.45 ± 4.46 ** | 5.46 | 81.57 ± 5.90 ** | 9.81 | 15.55 ± 1.02 ** | 16.30 | 86.05 ± 4.50 * | 4.43 | 5.21 |
| HCD fed ±5 | 112.00 ± 3.88 ** | 19.45 | 65.88 ± 3.64 ** | 18.77 | 13.25 ± 1.03 ** | 38.13 | 87.20 ± 2.24 | 15.11 | 4.24 |
| HCD fed ±6 | 102.33 ± 3.40 ** | 14.71 | 69.88 ± 2.53 ** | 17.45 | 12.35 ± 1.32 ** | 30.47 | 76.52 ± 2.60 * | 15.85 | 4.79 |
| HCD fed ±7 | 111.56 ± 5.42 ** | 9.17 | 76.82 ± 590 ** | 12.19 | 10.99 ± 0.50 ** | 22.11 | 71.99 ± 4.43 * | 3.91 | 5.02 |
| Microorganism | Escherichia coli IZ MIC | Pseudomonas aeruginosa IZ MIC | Staphylococcus aureus IZ MIC | Candida albicans IZ MIC |
|---|---|---|---|---|
| 18 25 | - - | 22 12.5 | - - | |
| 28 12.5 | 38 25 | 30 25 | - - | |
| - - | - - | - - | 40 12.5 | |
| 1 | 19 200 | 30 50 | 17 200 | >40 50 |
| 2 | 19 200 | 20 100 | 26 25 | 20 200 |
| 3 | 18 200 | 16 200 | 8 200 | 26 200 |
| 4 | 17 200 | 20 200 | 15 200 | 23 200 |
| 5 | 21 200 | 29 100 | 12 200 | 26 200 |
| 6 | 19 200 | 19 200 | >50 100 | 20 200 |
| 7 | 20 200 | 20 200 | 20 200 | 34 25 |
| Compounds | IC50 (µM) | ||
|---|---|---|---|
| MCF-7 | HCT-116 | A549 | |
| 1 | 9.23 ± 0.26 | 7.33 ± 0.19 | 5.13 ± 0.18 |
| 2 | 5.88 ± 0.21 | 5.43 ± 0.18 | 7.75 ± 0.11 |
| 3 | 9.17 ± 0.35 | 8.18 ± 0.37 | 5.44 ± 0.21 |
| 4 | 10.77 ± 0.32 | 8.28 ± 0.32 | 6.80 ± 0.18 |
| 5 | 18.44 ± 0.61 | 11.17 ± 0.30 | 8.76 ± 0.29 |
| 6 | 16.24 ± 0.61 | 14.24 ± 0.61 | 10.28 ± 0.27 |
| 7 | 18.14 ± 0.61 | 14.96 ± 0.46 | 9.33 ± 0.23 |
| Erlotinib | 22.46 ± 1.77 | 15.76 ± 1.60 | 18.55 ± 1.70 |
| Doxorubicin | 7.25 ± 0.46 | 5.12 ± 0.21 | NT |
| Compound No. | Binding Score (Kcal/mol) | Interactions | ||
|---|---|---|---|---|
| Receptor | Type | Length (Ǻ) | ||
| Ezetimibe | −8.43 | Glu38 | HB-donor | 2.66 |
| His124 | HB-acceptor | 2.82 | ||
| His124 | H-pi | 3.95 | ||
| Compound1 | −8.26 | Asn54 | HB-donor | 2.40 |
| Ser56 | HB-acceptor | 2.61 | ||
| His124 | H-pi | 4.01 | ||
| Compound2 | −8.48 | Glu38 | HB-donor | 2.68 |
| Gln95 | HB-donor | 2.74 | ||
| His124 | HB-acceptor | 2.94 | ||
| Pro215 | H-pi | 4.19 | ||
| Compound3 | −8.31 | Ser98 | HB-donor | 2.43 |
| Glu38 | HB-donor | 3.04 | ||
| Gln95 | HB-acceptor | 2.68 | ||
| His124 | H-pi | 4.11 | ||
| Compound4 | −8.36 | Leu99 | HB-acceptor | 3.06 |
| Ser53 | HB-acceptor | 2.68 | ||
| Compound5 | −8.51 | Ser53 | HB-donor | 2.84 |
| Ser98 | HB-acceptor | 3.23 | ||
| Gln95 | Pi-H | 4.92 | ||
| His124 | pi-pi | 3.81 | ||
| Compound6 | −8.73 | Ser98 | HB-acceptor | 2.45 |
| Asn127 | HB-acceptor | 2.61 | ||
| His124 | pi-pi | 3.44 | ||
| Compound7 | −8.41 | Asn54 | HB-donor | 3.10 |
| Gln95 | HB-acceptor | 3.27 | ||
| His124 | H-pi | 4.13 | ||
| Property | 1 | 2 | 3 | 4 | 5 | 6 | 7 | Ezetimibe |
|---|---|---|---|---|---|---|---|---|
| Absorption | ||||||||
| Bioavailability Score a | 0.55 | 0.55 | 0.55 | 0.17 | 0.17 | 0.55 | 0.55 | 0.55 |
| TPSA (Ų) a | 114.50 | 101.36 | 120.66 | 145.38 | 119.58 | 72.32 | 79.64 | 60.77 |
| Log S (ESOL), Class a | −5.76, Mod. Soluble | −4.5, Mod. Soluble | −5.63, Mod. Soluble | −8.18, Poor Soluble | −7.35, Poor Soluble | −7.01, Poor Soluble | −4.86, Mod. Soluble | −4.92, Mod. Soluble |
| Log Po/w (XLOGP3) a | 5.24 | 2.63 | 4.98 | 6.76 | 5.58 | 6.30 | 2.99 | 3.96 |
| % Human intestinal absorption b | 91.67 | 92.76 | 92.78 | 100 | 99.43 | 100 | 100 | 91.98 |
| Distribution | ||||||||
| BBB permeant a | No | No | No | No | No | No | No | Yes |
| P-gp substrate a | No | Yes | No | Yes | No | No | Yes | Yes |
| Fraction unbound (human) b | 0.155 | 0.249 | 0.063 | 0.266 | 0.362 | 0.387 | 0.375 | 0.078 |
| Metabolism | ||||||||
| CYP1A2 inhibitor a | No | No | No | No | No | No | No | No |
| CYP2C19 inhibitor a | Yes | No | Yes | No | No | Yes | No | Yes |
| CYP2C9 inhibitor a | Yes | No | Yes | Yes | No | Yes | Yes | No |
| CYP2D6 inhibitor a | No | No | No | No | No | No | No | Yes |
| CYP3A4 inhibitor a | No | No | Yes | Yes | No | Yes | No | Yes |
| Excretion | ||||||||
| Total clearance b (log ml/min/kg) | 0.505 | 0.641 | 0.242 | −0.041 | 0.983 | 1.198 | 0.924 | −0.269 |
| Renal OCT2 substrate b | No | No | No | No | No | No | No | No |
| Toxicity | ||||||||
| Oral rat acute toxicity (LD50) b (mol/kg) | 3.202 | 2.621 | 3.118 | 3.727 | 2.48 | 2.208 | 2.4 | 1.908 |
| Hepatotoxicity b | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamel, M.S.; Belal, A.; Aboelez, M.O.; Shokr, E.K.; Abdel-Ghany, H.; Mansour, H.S.; Shawky, A.M.; El-Remaily, M.A.E.A.A.A. Microwave-Assisted Synthesis, Biological Activity Evaluation, Molecular Docking, and ADMET Studies of Some Novel Pyrrolo [2,3-b] Pyrrole Derivatives. Molecules 2022, 27, 2061. https://doi.org/10.3390/molecules27072061
Kamel MS, Belal A, Aboelez MO, Shokr EK, Abdel-Ghany H, Mansour HS, Shawky AM, El-Remaily MAEAAA. Microwave-Assisted Synthesis, Biological Activity Evaluation, Molecular Docking, and ADMET Studies of Some Novel Pyrrolo [2,3-b] Pyrrole Derivatives. Molecules. 2022; 27(7):2061. https://doi.org/10.3390/molecules27072061
Chicago/Turabian StyleKamel, Moumen S., Amany Belal, Moustafa O. Aboelez, E. Kh. Shokr, H. Abdel-Ghany, Hany S. Mansour, Ahmed M. Shawky, and Mahmoud Abd El Aleem Ali Ali El-Remaily. 2022. "Microwave-Assisted Synthesis, Biological Activity Evaluation, Molecular Docking, and ADMET Studies of Some Novel Pyrrolo [2,3-b] Pyrrole Derivatives" Molecules 27, no. 7: 2061. https://doi.org/10.3390/molecules27072061
APA StyleKamel, M. S., Belal, A., Aboelez, M. O., Shokr, E. K., Abdel-Ghany, H., Mansour, H. S., Shawky, A. M., & El-Remaily, M. A. E. A. A. A. (2022). Microwave-Assisted Synthesis, Biological Activity Evaluation, Molecular Docking, and ADMET Studies of Some Novel Pyrrolo [2,3-b] Pyrrole Derivatives. Molecules, 27(7), 2061. https://doi.org/10.3390/molecules27072061