
Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases


 ,
,  , , and
, , and 
Abstract
1. Introduction
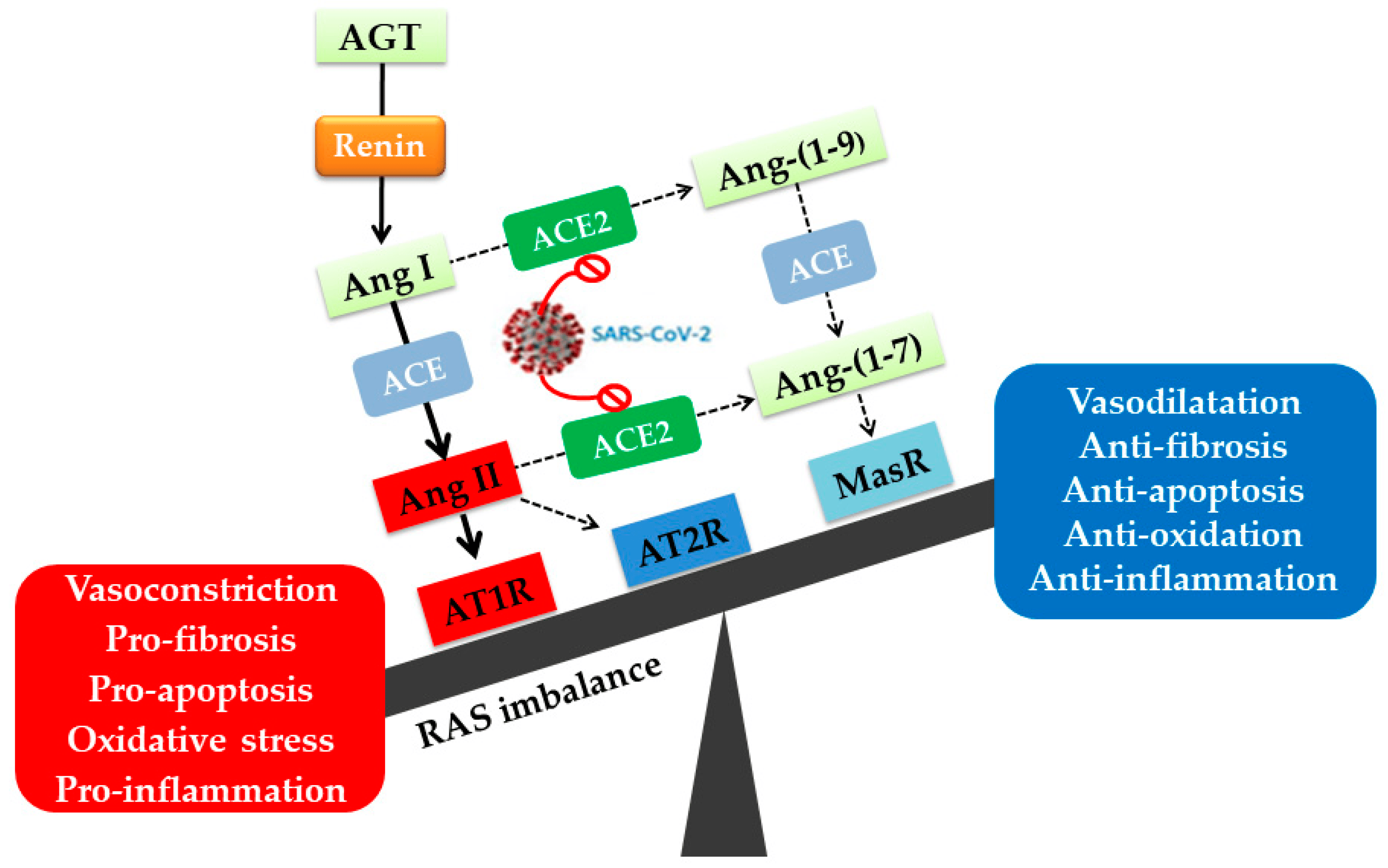
2. Angiotensin II Type I Receptor (AT1R) and COVID-19
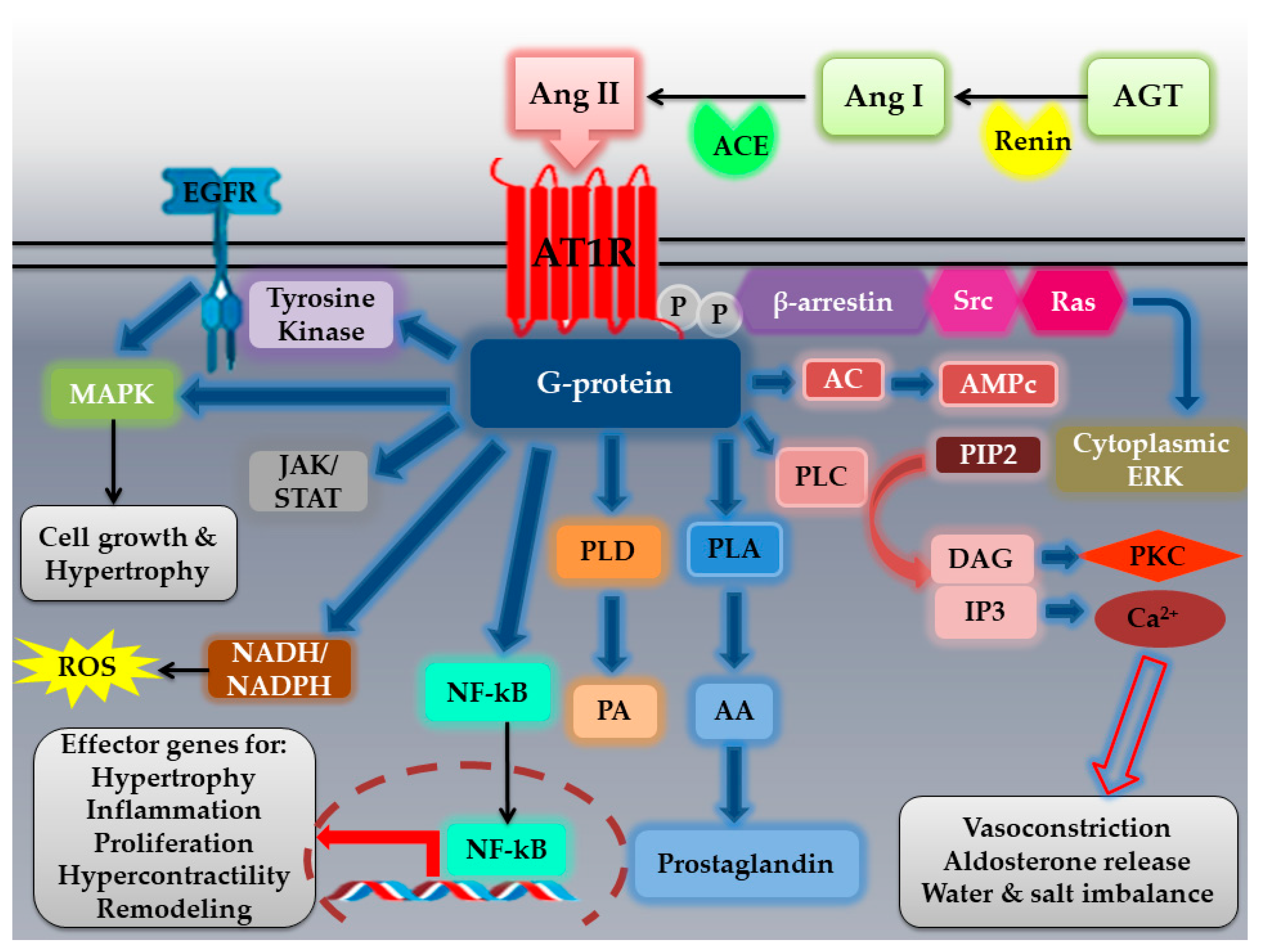
2.1. The AT1R: Structural Characteristics, Polymorphisms, Localization, Activation, and Signaling Pathways
2.1.1. Structural Characteristics
2.1.2. AT1R Polymorphisms and Associated Effects
2.1.3. AT1R Localization, Activation, and Signaling Pathways
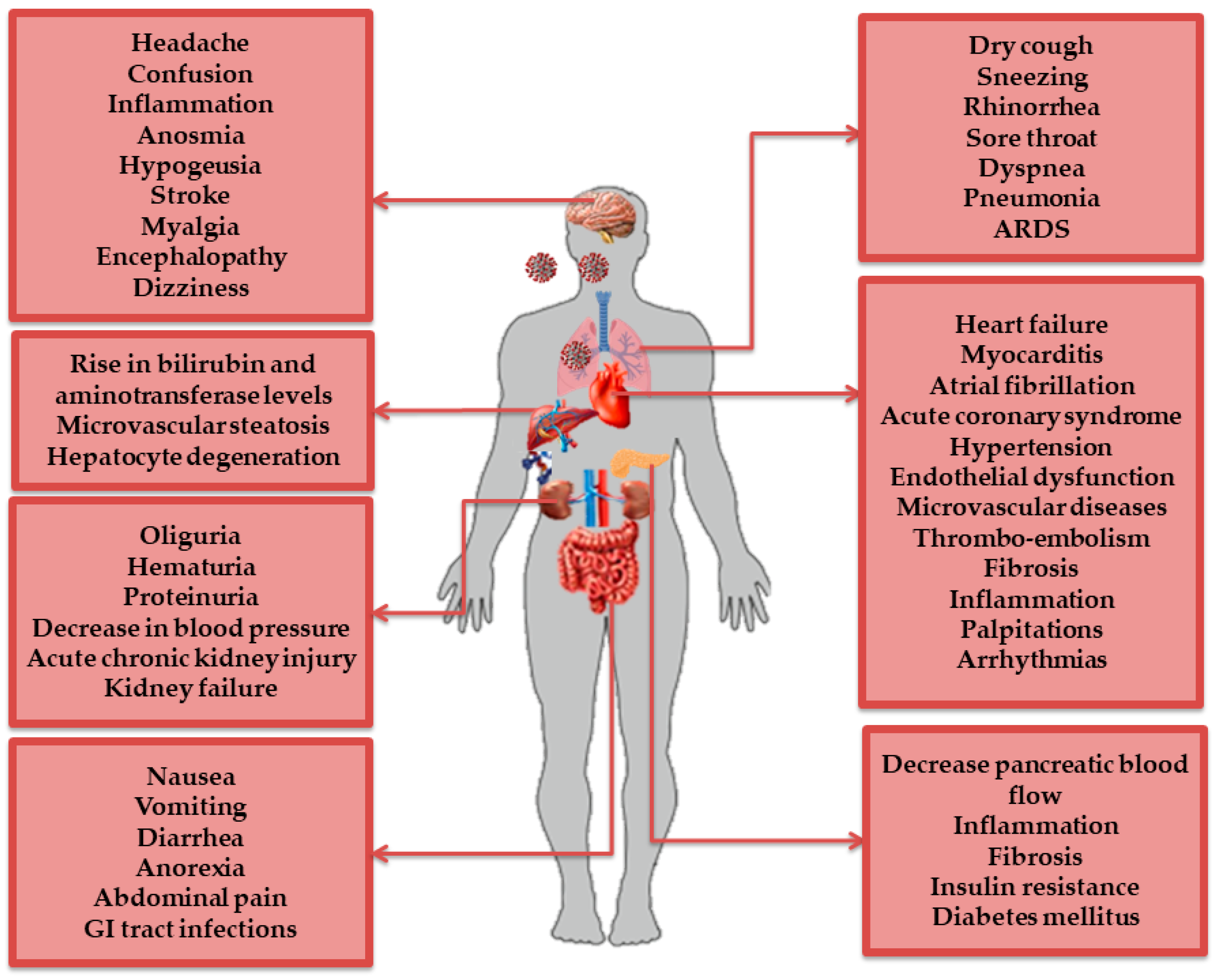
2.2. AT1R Role in COVID-19 Diseases
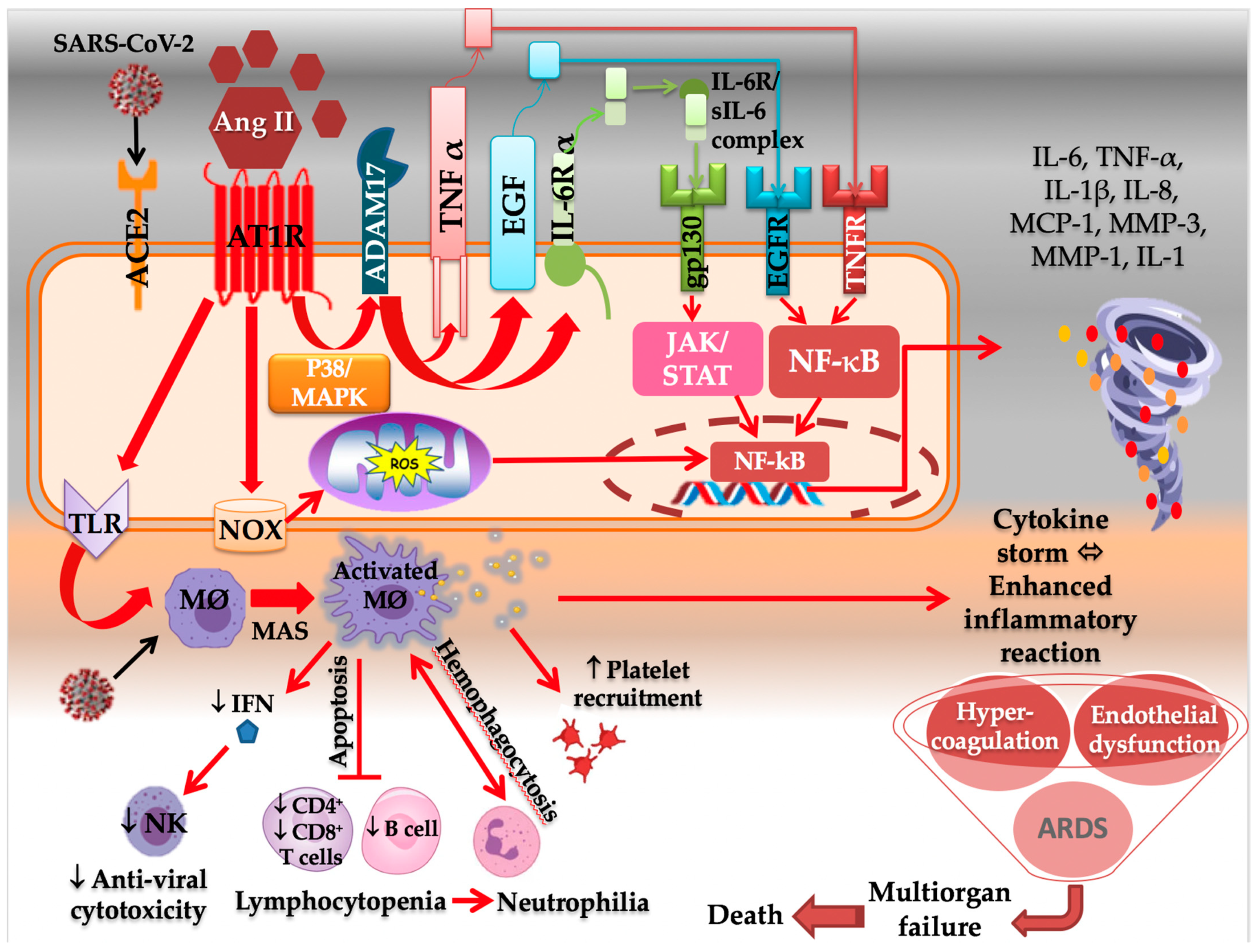
2.2.1. AT1R and the Immune System
2.2.2. AT1R and the Respiratory System
2.2.3. AT1R and the Cardiovascular System
2.2.4. AT1R and the Nervous System
2.2.5. AT1R and the Digestive System
2.2.6. AT1R and the Renal System
3. AT1R Blockers (ARBs): A Potential Treatment Strategy in COVID-19?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1282, pp. 1–23. [Google Scholar] [CrossRef]
- Nejat, R.; Sadr, A.S. Are losartan and imatinib effective against SARS-CoV-2 pathogenesis? A pathophysiologic-based in silico study. In Silico Pharmacol. 2021, 9, 1. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2014, 2, 88. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Nazerian, Y.; Vakili, K.; Ebrahimi, A.; Niknejad, H. Developing Cytokine Storm-Sensitive Therapeutic Strategy in COVID-19 Using 8P9R Chimeric Peptide and Soluble ACE2. Front. Cell Dev. Biol. 2021, 9, 717587. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 19 December 2021).
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Kariyanna, P.T.; Sutarjono, B.; Grewal, E.; Singh, K.P.; Aurora, L.; Smith, L.; Chandrakumar, H.P.; Jayarangaiah, A.; Goldman, S.A.; Salifu, M.O.; et al. A Systematic Review of COVID-19 and Myocarditis. Am. J. Med. Case Rep. 2020, 8, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.B.; Perez, L.G.; Palmeira, V.A.; Macedo e Cordeiro, T.; Ribeiro, V.T.; Lanza, K.; Simões e Silva, A.C. Insights on SARS-CoV-2 Molecular Interactions with the Renin-Angiotensin System. Front. Cell Dev. Biol. 2020, 8, 559841. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. Enzym. Catal. Regul. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Gerbino, A.; Lionetti, V.; Miragoli, M.; Munaron, L.M.; Pagliaro, P.; Pasqua, T.; Penna, C.; Rocca, C.; Samaja, M.; et al. COVID-19-associated cardiovascular morbidity in older adults: A position paper from the Italian Society of Cardiovascular Researches. GeroScience 2020, 42, 1021–1049. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Won Han, G.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Simões, S.C.; Balico-Silva, A.L.; Parreiras-e-Silva, L.T.; Bitencourt, A.L.B.; Bouvier, M.; Costa-Neto, C.M. Signal Transduction Profiling of Angiotensin II Type 1 Receptor with Mutations Associated to Atrial Fibrillation in Humans. Front. Pharmacol. 2020, 11, 600132. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Quan, X.; Li, X.; Su, X. Correlation between gene polymorphism in angiotensin II type 1 receptor and type 2 diabetes mellitus complicated by hypertension in a population of Inner Mongolia. BMC Med. Genet. 2020, 21, 83. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical Renin-Angiotensin System in Kidney Physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Pende, A.; Mach, F. The Renin-Angiotensin System Modulates Inflammatory Processes in Atherosclerosis: Evidence from Basic Research and Clinical Studies. Mediat. Inflamm. 2009, 2009, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Hernandez, W.; Ansari, R.A.; Ferder, L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 2015, 6, 209–217. [Google Scholar] [CrossRef]
- Wang, D.; Chai, X.; Magnussen, C.G.; Zosky, G.R.; Shu, S.H.; Wei, X.; Hu, S.S. Renin-angiotensin-system, a potential pharmacological candidate, in acute respiratory distress syndrome during mechanical ventilation. Pulm. Pharmacol. Ther. 1019, 58, 101833. [Google Scholar] [CrossRef]
- Tan, W.S.D.; Liao, W.; Zhou, S.; Mei, D.; Wong, W.S.F. Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr. Opin. Pharmacol. 2018, 40, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.L.; Du, Y.; Zhang, C.; Cheng, C.; Yang, H.Y.; Jin, Y.F.; Duan, G.C.; Chen, S.Y. Role of Renin-Angiotensin System in Acute Lung Injury Caused by Viral Infection. Infect. Drug Resist. 2020, 13, 3715–3725. [Google Scholar] [CrossRef] [PubMed]
- Papola, F.; Biancofiore, V.; Angeletti, C.; Grimaldi, A.; Carucci, A.C.; Cofini, V.; Necozione, S.; Rosciano, A.; Marinangeli, F.; Cervelli, C. Anti-AT1R autoantibodies and prediction of the severity of COVID-19. Hum. Immunol. 2021, 83, 130–133. [Google Scholar] [CrossRef]
- Su, C.; Xue, J.; Ye, C.; Chen, A. Role of the central renin-angiotensin system in hypertension. Int. J. Mol. Med. 2021, 47, 1–16. [Google Scholar] [CrossRef]
- Fu, G.D.; Sun, Y.L.; Hamet, P.; Inagami, T. The angiotensin II type 1 receptor and receptor-associated proteins. Cell Res. 2001, 11, 165–180. [Google Scholar] [CrossRef]
- Takezako, T.; Unal, H.; Node, K. The non-biphenyl-tetrazole AT1R antagonist eprosartan is a unique and robust inverse agonist of the active state of AT1R. J. Pharmacol. 2018, 175, 2454–2469. [Google Scholar] [CrossRef]
- Izmailova, O.; Shlykova, O.; Vatsenko, A.; Ivashchenko, D.; Dudchenko, M.; Koval, T.; Kaidashev, I. Allele C (rs5186) of at1r is associated with the severity of COVID-19 in the Ukrainian population. Infect. Genet. Evol. 2022, 98, 105227. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, T.; Mohamed, R.H.; Rezk, N.A. Association of angiotensin II type I and type II receptor genes polymorphisms with the presence of premature coronary disease and metabolic syndrome. Mol. Biol. Rep. 2014, 41, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Shahin, D.S.; Irshaid, Y.M.; Abu Saleh, A. The A1166C polymorphism of the AT1R gene is associated with an early onset of hypertension and high waist circumference in Jordanian males attending the Jordan University Hospital. Clin. Exp. Hypertens. 2014, 36, 333–339. [Google Scholar] [CrossRef]
- Chandra, S.; Narang, R.; Sreenivas, V.; Bhatia, J.; Saluja, D.; Srivastava, K. Association of Angiotensin II Type 1 Receptor (A1166C) Gene Polymorphism and Its Increased Expression in Essential Hypertension: A Case-Control Study. PLoS ONE 2014, 9, 101502. [Google Scholar] [CrossRef]
- Abdollahi, M.R.; Gaunt, T.R.; Syddall, H.E.; Cooper, C.; Phillips, D.I.; Ye, S.; Day, I.N.M. Angiotensin II type I receptor gene polymorphism: Anthropometric and metabolic syndrome traits. J. Med. Genet. 2005, 42, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Alavi-Shahri, J.; Behravan, J.; Hassany, M.; Tatari, F.; Kasaian, J.; Ganjali, R.; Tavallaie, S.; Sabouri, S.; Sahebkar, A.; Oladi, M.; et al. Association Between Angiotensin II Type 1 Receptor Gene Polymorphism and Metabolic Syndrome in a Young Female Iranian Population. Arch. Med. Res. 2010, 41, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Amir, O.; Amir, R.E.; Paz, H.; Attias, E.; Sagiv, M.; Lewis, B.S. Relation between AT1R Gene Polymorphism and Long-Term Outcome in Patients with Heart Failure. Cardiology 2009, 112, 151–157. [Google Scholar] [CrossRef]
- Oro, C.; Qian, H.; Thomas, W.G. Type 1 angiotensin receptor pharmacology: Signaling beyond G proteins. Pharmacol. Ther. 2007, 113, 210–226. [Google Scholar] [CrossRef]
- He, L.; Du, J.; Chen, Y.; Liu, C.; Zhou, M.; Adhikari, S.; Rubin, D.T.; Pekow, J.; Li, Y.C. Renin-angiotensin system promotes colonic inflammation by inducing TH17 activation via JAK2/STAT pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, 774–784. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Saito, J.; Zhao, H.; Sakamoto, A.; Hirota, K.; Ma, D. Inflammation Triggered by SARS-CoV-2 and ACE2 Augment Drives Multiple Organ Failure of Severe COVID-19: Molecular Mechanisms and Implications. Inflammation 2021, 44, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Wiese, O.J.; Allwood, B.W.; Zemlin, A.E. COVID-19 and the renin-angiotensin system (RAS): A spark that sets the forest alight? Med. Hypothes. 2020, 144, 110231. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.Y.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef] [PubMed]
- Silva-Filho, J.L.; Caruso-Neves, C.; Sá Pinheiro, A.A. Angiotensin II type-1 receptor (AT1R) regulates expansion, differentiation, and functional capacity of antigen-specific CD8+ T cells. Sci. Rep. 2016, 6, 35997. [Google Scholar] [CrossRef]
- Tawinwung, S.; Petpiroon, N.; Chanvorachotte, P. Blocking of Type 1 Angiotensin II Receptor Inhibits T-lymphocyte Activation and IL-2 Production. In Vivo 2018, 32, 1353–1359. [Google Scholar] [CrossRef]
- Mehrabadi, M.E.; Hemmati, R.; Tashakor, A.; Homaei, A.; Yousefzadeh, M.; Hemati, K.; Hosseinkhani, S. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed. Pharmacother. 2021, 137, 111363. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Almutlaq, M.; Alamro, A.A.; Alroqi, F.; Barhoumi, T. Classical and Counter-regulatory Renin-angiotensin System: Potential key roles in COVID-19 pathophysiology. Can. J. Cardiol. 2021, 3, 1060–1074. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Boukhris, M.; Hillani, A.; Moroni, F.; Abbate, A.; Vilca, L.M.; Azzalini, L. Cardiovascular Implications of the COVID-19 Pandemic: A Global Perspective. Can. J. Cardiol. 2020, 36, 1068–1080. [Google Scholar] [CrossRef]
- Garg, S.; Garg, M.; Prabhakar, N.; Malhotra, P.; Agarwal, R. Unraveling the mystery of COVID-19 cytokine storm: From skin to organ systems. Dermatol. Ther. 2020, 33, 13859. [Google Scholar] [CrossRef]
- Rello, J.; Storti, E.; Belliato, M.; Serrano, R. Clinical phenotypes of SARS-CoV-2: Implications for clinicians and researchers. Eur. Respir. J. 2020, 55, 2001028. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, L.F.; Kroft, L.J.M.; Van Der Wal, L.I.; Cannegieter, S.C.; Eikenboom, J.; De Jonge, E.; Huisman, M.V.; Klok, F.A. Clinical and computed tomography characteristics of COVID-19 associated acute pulmonary emboliam: A different phenotypes of thrombotic diseases. Thromb. Res. 2020, 193, 86–89. [Google Scholar] [CrossRef]
- Robba, C.; Battaglini, D.; Ball, L.; Patroniti, N.; Loconte, M.; Brunetti, L.; Vena, A.; Giacobbe, D.R.; Bassetti, M.; Rocco, P.R.M.; et al. Distinct phenotypes require distinct respiratory managemnet strategies in severe COVID-19. Respir. Physiol. Neurobiol. 2020, 279, 103455. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, 438–440. [Google Scholar] [CrossRef]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417. [Google Scholar] [CrossRef] [PubMed]
- Dielis, A.W.J.H.; Smid, M.; Spronk, H.M.H.; Hamulyak, K.; Kroon, A.A.; ten Cate, H.; de Leeuw, P.W. The Prothrombotic Paradox of Hypertension. Hypertension 2005, 46, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Lumpuy-Castillo, J.; Lorenzo-Almoros, A.; Pello-Lazaro, A.M.; Sanchez-Ferrer, C.; Egido, J.; Tunon, J.; Peiro, C.; Lorenzo, O. Cardiovascular damage in COVID-19: Therapeutic Approaches Targetting the Renin-Angiotensin-Aldosterone System. Int. J. Mol. Sci. 2020, 21, 6471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Zhang, Y.H.; Dong, X.F.; Hao, Q.Q.; Zhou, X.M.; Yu, Q.Y.; Li, S.Y.; Chen, X.; Tengbeh, A.F.; Dong, B.; et al. ACE2 and Ang-(1–7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm. Res. 2015, 64, 253–260. [Google Scholar] [CrossRef] [PubMed]
- El-Arif, G.; Farhat, A.; Khazaal, S.; Annweiler, C.; Kovacic, H.; Wu, Y.; Cao, Z.; Fajloun, Z.; Khattar, Z.A.; Sabatier, J.M. The Renin-Angiotensin System: A Key Role in SARS-CoV-2- Induced COVID-19. Molecules 2021, 26, 6945. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, 1377–1384. [Google Scholar] [CrossRef]
- I’ñiguez, M.; Pérez-Matute, P.; Villoslada-Blanco, P.; Recio-Fernandez, E.; Ezquerro-Pérez, D.; Alba, J.; Ferreira-Laso, M.L.; Oteo, J.A. ACE Gene Variants Rise the Risk of Severe COVID-19 in Patients with Hypertension, Dyslipidemia or Diabetes: A Spanish Pilot Study. Front. Endocrinol. 2021, 12, 688071. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaoi, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System:Focus on Angiotensin-(1-7). Physiology 2017, 98, 505–553. [Google Scholar] [CrossRef]
- D’Ardes, D.; Boccatonda, A.; Rossi, I.; Guagnano, M.T.; Santilli, F.; Cipollone, F.; Bucci, M. COVID-19 and RAS: Unravelling an Unclear Relationship. Int. J. Mol. Sci. 2020, 21, 3. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet 2020, 8, 21. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Khan, M.S.; Abraham, W.T.; Bauersachs, J.; Bocchi, E.; Bozkurt, B.; Braunwald, E.; Chopra, V.K.; Cleland, J.G.; et al. Conducting clinical trials in heart failure during (and after) the COVID-19 pandemic: An Expert Consensus Position Paper from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 41, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Deshotels, M.R.; Xia, H.; Sriramula, S.; Lazartigues, E.; Filipeanu, C.M. Angiotensin II Mediates Angiotensin Converting Enzyme Type 2 Internalization and Degradation Through an Angiotensin II Type I Receptor–Dependent Mechanism. Hypertension 2014, 64, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef]
- Portales, A.E.; Mustafá, E.R.; McCarthy, C.I.; Cornejo, M.P.; Couto, P.M.; Gironacci, M.M.; Caramelo, J.J.; Perelló, M.; Raingo, J. ACE2 internalization induced by a SARS-CoV-2 recombinant protein is modulated by angiotensin II type 1 and bradykinin 2 receptors. Life Sci. 2021, 293, 120284. [Google Scholar] [CrossRef]
- Efficacy of Catopril in Covid-19 Patients with Severe Acute Respiratory Syndrome (SARS) CoV-2 Pneumonia (CAPTOCOVID). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04355429 (accessed on 9 December 2021).
- Ramipril for the Treatment of COVID-19 (RAMIC). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04366050 (accessed on 16 January 2022).
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Mirsaeidi, M.; Davies, C.; Telischi, F.F.; Chaudhari, N.; Mittal, R. Potential Mechanisms for COVID-19 Induced Anosmia and Dysgeusia. Front. Physiol. 2020, 11, 1039. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Castro, P.J.; Estivill-Torrus, G.; Cabezudo-Garcia, P.; Reyes-Bueno, J.A.; Petersen, C.; Aguilar-Castillo, M.J.; Suarez-Perez, J.; Jiménez-Hernández, M.D.; Moya-Molina, M.A.; Oliver-Martos, B.; et al. Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: A delayed pandemic? Neurologia 2020, 35, 245–251. [Google Scholar] [CrossRef]
- Khezri, M.R.; Ghasemnejad-Berenji, M. Neurological effects of elevated levels of angiotensin II in COVID-19 patients. Hum. Cell 2021, 34, 1941–1942. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Pogue, A.; Hill, J.M. SARS-CoV-2 Infectivity and Neurological Targets in the Brain. Cell. Mol. Neurobiol. 2020, 42, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yiu, A.; Zhou, X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef] [PubMed]
- Biancardi, V.C.; Stern, J.E. Compromised blood-brain barrier permeability: Novel mechanism by which circulating angiotensin II signals to sympathoexcitataory centres during hypertension. J. Physiol. 2015, 594, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhan, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef] [PubMed]
- Kastrup, A.; Groshcel, K.; Ringer, T.M.; Redecker, C.; Cordesmeyer, R.; Witte, O.W.; Terborg, C. Early Disruption of the Blood-Brain Barrier After Thrombolytic Therapy Predicts Hemorrhage in Patients with Acute Stroke. Stroke 2008, 39, 2385–2387. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.; Chappell, M.C.; Ferrario, C.; Tallant, E.A. Distinct roles for ANG II and Ang-(1–7) in the regulation of angiotensin-converting enzyme 2 in rat enterocytes. Am. J. Physiol. Physiol. 2006, 290, 420–426. [Google Scholar] [CrossRef]
- Grammatopoulos, T.N.; Jones, S.M.; Ahmadi, F.A.; Hoover, B.R.; Snell, L.D.; Skoch, J.; Jhaveri, V.V.; Poczobutt, A.M.; Weyhenmeyer, J.A.; Zawada, W.M. Angiotensin type 1 receptor antagononist losartan, reduces MPTP-induced degeneration of dopaminergic neurons in substantia nigra. Mol. Neurodegener. 2007, 2, 1. [Google Scholar] [CrossRef]
- Miller, A.J.; Arnold, A.C. The renin-angiotensin system in cardiovascular autonomic control: Recent developments and clinical implications. Clin. Auton. Res. 2019, 29, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Doobay, M.F.; Talman, L.S.; Obr, T.D.; Tian, X.; Davisson, R.L.; Lazartigues, E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Porzionato, A.; Emmi, A.; Barbon, S.; Boscolo-Berto, R.; Stecco, C.; Stocco, E.; Macchi, V.; De Caro, R. Sympathetic activation:a potential link between comorbidities and COVID-19. FEBS J. 2020, 287, 3681–3688. [Google Scholar] [CrossRef]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Alenina, N.; Bader, M. ACE2 in Brain Physiology and pathophysiology: Evidence from Transgenic Animal Models. Nerochem. Res. 2019, 44, 1323–1329. [Google Scholar] [CrossRef]
- Anand, H.; Ende, V.; Singh, G.; Qureshi, I.; Duon, T.; Mehler, M.F. Nervous System-Sytemic Crosstalk in SARS-CoV-2/COVID-19: A Unique Dyshomeostasis Syndrome. Front. Neurosci. 2021, 15, 727060. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Lian, J.S.; Hu, J.H.; Gao, J.; Zheng, L.; Zhang, Y.M.; Hao, S.R.; Jia, H.Y.; Cai, H.; Zhang, X.L.; et al. Epidemiological, clinical and virological characteristics of 74 cases of coronavirus-infected disease 2019 (COVID-19) with gastrointestinal symptoms. Gut 2020, 69, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Mehr, A.P.; Kreutz, R. Physiology of Local Renin-Angiotensin Systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Angus, P.W.; Burrell, L.M.; Herath, C.; Gibson, P.R.; Lubel, J.S. Review article: The pathophyiological roles of the renin-angiotensin system in the gastrointestinal tract. Aliment. Pharmacol. Ther. 2012, 35, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.P.; Ho, K.Y.; Ng, E.K.W.; Debnam, E.S.; Leung, P.S. Upregulation of ACE2-Ang-(1–7)-Mas axis in jejunal enterocytes of type I diabetic rats: Implications for glucose transport. Am. J. Physiol. Metab. 2012, 303, 669–681. [Google Scholar] [CrossRef]
- Pasanen, L.; Launonen, H.; Siltari, A.; Korpela, R.; Vapaatalo, H.; Salmenkari, H.; Forsgard, R.A. Age-related changes in the local intestinal Renin-Angiotensin System in normotensive and spontaneously hypertensive rats. J. Physiol. Pharmacol. 2019, 70, 199–208. [Google Scholar] [CrossRef]
- Shorning, B.; Jarde, T.; Carthy, A.; Ashworth, A.; WJ de Leng, W.; Offerhaus, G.J.A.; Resta, N.; Dale, T.; Clarke, A.R. Intestinal renin-angiotensin system is stimulated after detection of Lkb1. Gut 2011, 61, 202–213. [Google Scholar] [CrossRef]
- Ewert, S.; Spak, E.; Olbers, T.; Johnsson, E.; Edebo, A.; Fandriks, L. Angiotensin II induced contraction of rat and human small intestinal wall musculature in vitro. Acta Physiol. 2006, 188, 33–40. [Google Scholar] [CrossRef]
- Spak, E.; Casselbrant, A.; Olbers, T.; Lonroth, H.; Fandriks, L. Angiotensin II-induced contractions in human jejunal wall musculature in vitro. Acta Physiol. 2008, 193, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.C.; Zhang, H.X.; Zhang, Z.; Rinkiko, S.; Cui, Y.M.; Zhu, Y.Z. The Two-Way Switch Role of ACE2 in the Treatment of Novel Coronavirus Pneumonia and Underlying Comorbidities. Molecules 2021, 26, 142. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Korteweg, C. Pathology and Pathogenesis of Severe Acute Respiratory Syndrome. Am. J. Pathol. 2007, 170, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Ding, Y.; Zhang, Q.; Che, X.; He, Y.; Shen, H.; Wang, H.; Li, Z.; Zhao, L.; Geng, J.; et al. Expression of elevated levels of pre-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: Relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 2006, 210, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, Y.; Moraohashi, T.; Kawana, I.; Nagashima, Y.; Kihara, M.; Umemura, S. Amelioration of 2,4,6-trinitrobenzene sulphonic acid induced colitis in angiotensinogen gene knockout mice. Gut 2005, 54, 349–356. [Google Scholar] [CrossRef]
- Rossig, L.; Dimmeler, S.; Zeiher, A.M. Apoptosis in the vascular wall and atherosclerosis. Basic Res. Cardiol. 2001, 96, 11–22. [Google Scholar] [CrossRef]
- Penninger, J.M.; Grant, M.B.; Sung, J.J.Y. The Role of Angiotensin Converting Enzyme 2 in Modulating Gut Microbiota, Intestinal Inflammation, and Coronavirus Infection. Gastroenterology 2021, 160, 39–46. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology 2020, 159, 34701–34706. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Lu, X.; Jan Danser, A.H. Revisiting the Brain Renin Angiotensin System-Focus on Novel Therapies. Curr. Hypertens. Rep. 2019, 21, 28. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An update on the tissue Renin Angiotensin System and Its Rolein Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, D.; Sperhake, J.P.; Lutgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schröder, A.S.; et al. Autopsy findings and venous thromboembolism in patients with COVID-19: A prospective cohort study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef]
- Qi, F.; Qian, S.; Zhang, S.; Zhang, Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020, 526, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Hu, F.B.; Qi, L.; Curhan, G.C. Genetic polymorphisms of angiotensin-2 type 1 receptor and angiotensinogen and risk of renal dysfunction and coronary heart disease in type 2 diabetes mellitus. BMC Nephrol. 2009, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Rüster, C.; Wolf, G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J. Am. Soc. Nephrol. 2011, 22, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGF-dependent renal fibrosis. J. Am. Soc. Nephrol. 2011, 23, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Brands, M.W.; Banes-Berceli, A.K.; Inscho, E.W.; Al-Azawi, H.; Allen, A.J.; Labazi, H. Interleukin 6 knockout prevents angiotensin II hypertension: Role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 2010, 56, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Thai, K.; Scholey, J.W. Angiotensin II type 1 receptor gene polymorphism predicts response to losartan and angiotensin II. Vasc. Biol.-Hemodyn.-Hypertens. 1999, 56, 2173–2180. [Google Scholar] [CrossRef]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020, 81, 537–540. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Ahmad, S.; Groban, L. Mechanisms by which angiotensin-receptor blockers increase ACE2 levels. Nature 2020, 17, 378. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Gallagher, P.E.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Smith, R.D.; Chappell, M.C. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005, 68, 2189–2196. [Google Scholar] [CrossRef]
- Devaux, C.A.; Lagier, J.C.; Raoult, D. New Insights into the Physiopathology of COVID-19: SARS-CoV-2-Associated Gastrointestinal Illness. Front. Med. 2021, 8, 640073. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Pires de Souza, G.A.; Osman, I.O.; Le Bideau, M.; Baudoin, J.P.; Jaafar, R.; Devaux, C.; La Scola, B. Angiotensin II Receptor Blockers (ARBs Antihypertensive Agents) Increase Replication of SARS-CoV-2 in Vero E6 Cells. Front. Cell. Infect. Microbiol. 2021, 11, 639177. [Google Scholar] [CrossRef]
- Gommans, D.H.F.; Nas, J.; Pinto-Sietsma, S.J.; Koop, Y.; Konst, R.E.; Mensink, F.; Aarts, G.W.A.; Konijnenberg, L.S.F.; Cortenbach, K.; Verhaert, D.V.M. Rationale and design of the PRAETORIAN-COVID trial: A double-blind, placebo-controlled randomized clinical trial with valsartan for prevention of acute respiratory dIstress syndrome in hospitalized patients with SARS-CoV-2 Infection Disease. Am. Heart J. 2020, 226, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, A.; Kazemian, S.; Saleh, S.K.; Aminorroaya, A.; Shajari, Z.; Hadadi, A.; Talebpour, M.; Sadeghian, H.; Payandemehr, P.; Sotoodehnia, M. Effects of Angiotensin Receptor Blockers (ARBs) on In-Hospital Outcomes of Patients with Hypertension and Confirmed or Clinically Suspected COVID-19. Am. J. Hypertens. 2020, 33, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.D.; Macedo, A.V.S.; Silva, G.M.; Moll-Bernardes, R.J.; Dos Santos, T.M.; Mazza, L.; Feldman, A.; D’Andréa Saba Arruda, G.; De Albuquerque, D.C.; Camiletti, A.S. Effect of Discontinuing vs Continuing Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers on Days Alive and Out of the Hospital in Patients Admitted With COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Genet, B.; Vidal, J.S.; Cohen, A.; Duron, E.; Girerd, X.; Hanon, O. COVID-19 In-Hospital Mortality and Use of Renin-Angiotensin System Blockers in Geriatrics Patients. J. Am. Med. Dir. Assoc. 2020, 21, 1539–1545. [Google Scholar] [CrossRef]
- Cohen, J.; Hanff, T.C.; William, P.; Čižmek, L.S.N.; Rosado-Santander, N.R.; Medina, C.; Rodriguez-Mori, J.E.; Renna, N.; Chang, T.; Corrales-Medina, V.; et al. Continuation versus discontinuation of renin-angiotensin system inhibitors in patients admmitted to hospital with COVID-19: A prospective randomised, open-label trial. Lancet 2021, 9, 275–284. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Chen, J.; Zhang, H.; Deng, A. Assiociation of Renin-Angiotensin System Inhibitors with Severity or Risk of deaths in patients With Hypertension Hospitalized for coronavirus Disease 2019 (COVID-19) infection in Whuan, China. JAMA Cardiol. 2020, 5, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Piccinocchi, G.; Mandaliti, V.; Annunziata, S.; Cimmino, G.; Attena, E.; Moio, N.; Di Micco, P.; Severino, S.; Trotta, R.; et al. Cardiovascular comorbidities and pharmacological treatments of COVID-19 Patients Not Requiring Hospitalization. Int. J. Environ. Res. Public Health 2021, 18, 102. [Google Scholar] [CrossRef]
- Mancusi, C.; Grassi, G.; Borghi, C.; Carugo, S.; Fallo, F.; Ferri, C.; Giannattasio, C.; Grassi, D.; Letizia, C.; Minuz, P.; et al. Determinants of healing among patiemnts with COVID-19: The results of the SARS-RAS-study of the Italian Society of Hypertension. J. Hypertens. 2021, 39, 376–380. [Google Scholar] [CrossRef]
- Palazzuoli, A.; Mancone, M.; De Ferrari, G.M.; Forleo, G.; Secco, G.G.; Ruocco, G.M.; D’Ascenzo, F.; Monticone, S.; Paggi, A.; Vicenzi, M.; et al. Antecedent Administartion of Angiotensin-Converting Enzyme Inhibitors or Angiotensin II Receptor Antagonists and survival after Hospitalization for COVID-19 syndrome. J. Am. Heart Assoc. 2020, 9, 017364. [Google Scholar] [CrossRef]
- Watkins, J. Preventing a COVID-19 pandemic. Br. Med. J. 2020, 368. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H. Urinary Angiotensin-Converting Enzyme 2 in Hypertensive Patients May Be Increased by Olmesartan, an Angiotensin II Receptor Blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef]
- Klimas, J.; Olvedy, M.; Ochodnicka-Mackovicova, K.; Kruzliak, P.; Cacanyiova, S.; Kristek, F.; Krenek, P.; Ochodnicky, P. Perinatally administered losartan augments renal ACE2 expression but not cardiac or renal Mas receptor in spontaneously hypertensive rats. J. Cell. Mol. Med. 2015, 19, 1965–1975. [Google Scholar] [CrossRef]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of Angiotensin-Converting Enzyme 2 After Myocardial Infarction by Blockade of Angiotensin II Receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef]
- Bavishi, C.; Maddox, T.M.; Messerli, F.H. Coronavirus Disease 2019 (COVID-19) Infection and Renin Angiotensin System Blockers. JAMA Cardiol. 2020, 5, 745–747. [Google Scholar] [CrossRef]
- Patel, A.B.; Verma, A. COVID-19 and Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers. What Is the Evidence? JAMA 2020, 323, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with COVID-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Battistoni, A.; Volpe, M. Might renin–angiotensin system blockers play a role in the COVID-19 pandemic? Eur. Heart J. Cardiovasc. Pharmacother. 2020, 6, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Danser, A.H.J.; Epstein, M.; Batlle, D. Renin-Angiotensin System Blockers and the COVID-19 Pandemic. Hypertension 2020, 75, 1382–1385. [Google Scholar] [CrossRef]
- Wu, C.; Ye, D.; Mullick, A.E.; Li, Z.; Jan Danser, A.H.; Daugherty, A.; Lu, H.S. Effects of Renin-Angiotensin Inhibition on ACE2 (Angiotensin-Converting Enzyme 2) and TMPRSS2 (Transmembrane Protease Serine 2) Expression. Hypertension 2020, 76, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Muslin, A.J. MAPK signalling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin. Sci. 2008, 115, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Nitta, K.; Uchida, K.; Yumura, W.; Nihei, H. Possible involvement of mitogen-activated protein kinase in the angiotensin II-induced fibronectin synthesis in renal interstitial fibroblasts. Arch. Biochem. Biophys. 2003, 415, 63–68. [Google Scholar] [CrossRef]
- Zhong, J.C.; Ye, J.Y.; Jin, H.Y.; Yu, X.; Yud, H.M.; Zhu, D.L.; Gao, P.J.; Huang, D.Y.; Shuster, M.; Loibner, H. Telmisartan attenuates aortic hypertrophy in hypertensive rats by the modulation of ACE2 and profilin-1 expression. Regul. Pept. 2011, 166, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Baba, R.; Oki, K.; Itcho, K.; Kobuke, K.; Nagano, G.; Ohno, H.; Yoneda, M.; Hattori, N. Angiotensin-converting enzyme 2 expression is not induced by the renin–angiotensin system in the lung. ERJ Open Res. 2020, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Conversano, A.; Melillo, F.; Napolano, A.; Fominskiy, E.; Spessot, M.; Ciceri, F.; Agricola, E. Renin-Angiotensin-Aldosterone System Inhibitors and Outcome in Patients With SARS-CoV-2 Pneumonia. Hypertension 2020, 76, 10–12. [Google Scholar] [CrossRef]
- Lam, K.W.; Chow, K.W.; Vo, J.; Hou, W.; Li, H.; Richman, P.S.; Mallipattu, S.K.; Skopicki, H.A.; Singer, A.J.; Duong, T.Q. Continued In-Hospital Angiotensin-Converting Enzyme Inhibitor and Angiotensin II Receptor Blocker Use in Hypertensive COVID-19 Patients Is Associated with Positive Clinical Outcome. J. Infect. Dis. 2020, 222, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Cippà, P.E.; Cugnata, F.; Ferrari, P.; Brombin, C.; Ruinelli, L.; Bianchi, G.; Beria, N.; Schulz, L.; Bernasconi, E.; Merlani, P.; et al. A data-driven approach to identify risk profiles and protective drugs in COVID-19. Proc. Natl. Acad. Sci. USA 2021, 18, 2016877118. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, Y.; Hong, Y. Decreased Mortality of COVID-19 With Renin-Angiotensin-Aldosterone System Inhibitors Therapy in Patients with Hypertension: A Meta-Analysis. Hypertension 2020, 76, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Sookoian, S. Estimation of Renin-Angiotensin-Aldosterone-System (RAAS)-Inhibitor effect on COVID-19 outcome: A Meta-analysis. J. Infect. 2020, 81, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Baral, R.; Tsampasian, V.; Debski, M.; Moran, B.; Garg, P.; Clark, A.; Vassiliou, V.S. Association Between Renin-Angiotensin-Aldosterone System Inhibitors and Clinical Outcomes in Patients With COVID-19: A Systematic Review and Meta-analysis. JAMA 2021, 4, 213594. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, R.P.; Duarte, M.; Pelorosso, G.; Nicolosi, L.; Salgado, V.; VetullI, M.; Spitzer, E. Angiotensin Receptor Blockers for COVID-19: Pathophysiological and Pharmacological Considerations About Ongoing and Future Prospective Clinical Trials. Front. Pharmacol. 2021, 12, 603736. [Google Scholar] [CrossRef]
- Rothlin, R.P.; Vetulli, H.M.; Duarte, M.; Pelorosso, G. Telmisartan as tentative angiotensin receptor blocker therapeutic for COVID-19. Drug Dev. Res. 2020, 81, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.; Pelorosso, F.; Nicolosi, L.N.; Salgado, M.V.; Vetulli, H.; Aquieri, A.; Azzato, F.; Castro, M.; Coyle, J.; Davolos, I. Telmisartan for treatment of Covid-19 patients: An open multicenter randomized clinical trial. Lancet 2021, 37, 100962. [Google Scholar] [CrossRef]
- Chen, W.; Yang, H.; Jiang, X. Effects of romosozumab on low bone mineral density or osteoporosis in postmenopausal women: A systematic review. Ann. Joint 2020, 5, 18. [Google Scholar] [CrossRef]
- Oh, K.; Adnan, M.; Cho, D.H. Network pharmacology approach to decipher signaling pathways associated with target proteins of NSAIDs against COVID-19. Sci. Rep. 2021, 11, 9606. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.W.; Adnan, M.; Cho, D.H. Network Pharmacology Study on Morus alba L. Leaves: Pivotal Functions of Bioactives on RAS Signaling Pathway and Its Associated Target Proteins against Gout. Int. J. Mol. Sci. 2021, 22, 9372. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antihypertensive Drugs Used (% of Population) | Period of ARB Intake before COVID-19 Infection | Main Outcomes | References |
|---|---|---|---|
| ARBs (48%) | ≥7 days after hospital admission | ARB treatment did not worsen clinical outcomes during COVID-19 infection in hypertensive patients. | [119] |
| ARBs (50.6%) | Not mentioned | Previous administration of ARBs had no association with the number of days alive and out of the hospital in mild COVID-19. | [120] |
| ARBs (31.34%) | ≥1 week before the onset of infection | ARB administration, before the onset of infection, significantly lowered the mortality rate in COVID-19 patients. | [121] |
| ARBs (49.3%) | Not mentioned | ARB administration had no effect on the severity and mortality of COVID-19. | [122] |
| ARBs (40.5%) | For >1 y | ARB administration improved clinical outcomes of COVID-19 patients with hypertension. | [70] |
| ARBs (31.8%) | Not mentioned | ARBs were not associated with the severity or mortality of COVID-19 patients with hypertension. | [123] |
| ARB (30.8%) | ≥1 month before hospital admission | ARB administration reduced the risk of death during hospitalization in COVID-19 hypertensive patients. | [119] |
| ARBs (51%) | ≥3 months before study conduction | ARB administration lowered the risk of hospitalization and intubation or death with COVID-19; long-term use of ARBs might decrease the risk of COVID-19 in hypertensive patients. | [124] |
| ARBs (29.7%) | Unknown | Administration of ARBs did not affect the chance of recovery in COVID-19 patients with hypertension or heart failure. | [125] |
| ARBs (17%) | Not mentioned | Previous use of ARBs reduced the risk of mortality in patients hospitalized with COVID-19 infection. | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Arif, G.; Khazaal, S.; Farhat, A.; Harb, J.; Annweiler, C.; Wu, Y.; Cao, Z.; Kovacic, H.; Abi Khattar, Z.; Fajloun, Z.; et al. Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases. Molecules 2022, 27, 2048. https://doi.org/10.3390/molecules27072048
El-Arif G, Khazaal S, Farhat A, Harb J, Annweiler C, Wu Y, Cao Z, Kovacic H, Abi Khattar Z, Fajloun Z, et al. Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases. Molecules. 2022; 27(7):2048. https://doi.org/10.3390/molecules27072048
Chicago/Turabian StyleEl-Arif, George, Shaymaa Khazaal, Antonella Farhat, Julien Harb, Cédric Annweiler, Yingliang Wu, Zhijian Cao, Hervé Kovacic, Ziad Abi Khattar, Ziad Fajloun, and et al. 2022. "Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases" Molecules 27, no. 7: 2048. https://doi.org/10.3390/molecules27072048
APA StyleEl-Arif, G., Khazaal, S., Farhat, A., Harb, J., Annweiler, C., Wu, Y., Cao, Z., Kovacic, H., Abi Khattar, Z., Fajloun, Z., & Sabatier, J.-M. (2022). Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases. Molecules, 27(7), 2048. https://doi.org/10.3390/molecules27072048