
The Pathophysiology of Long COVID throughout the Renin-Angiotensin System

 ,
, 
 ,
,  , , and
, , and 
Abstract
1. Introduction
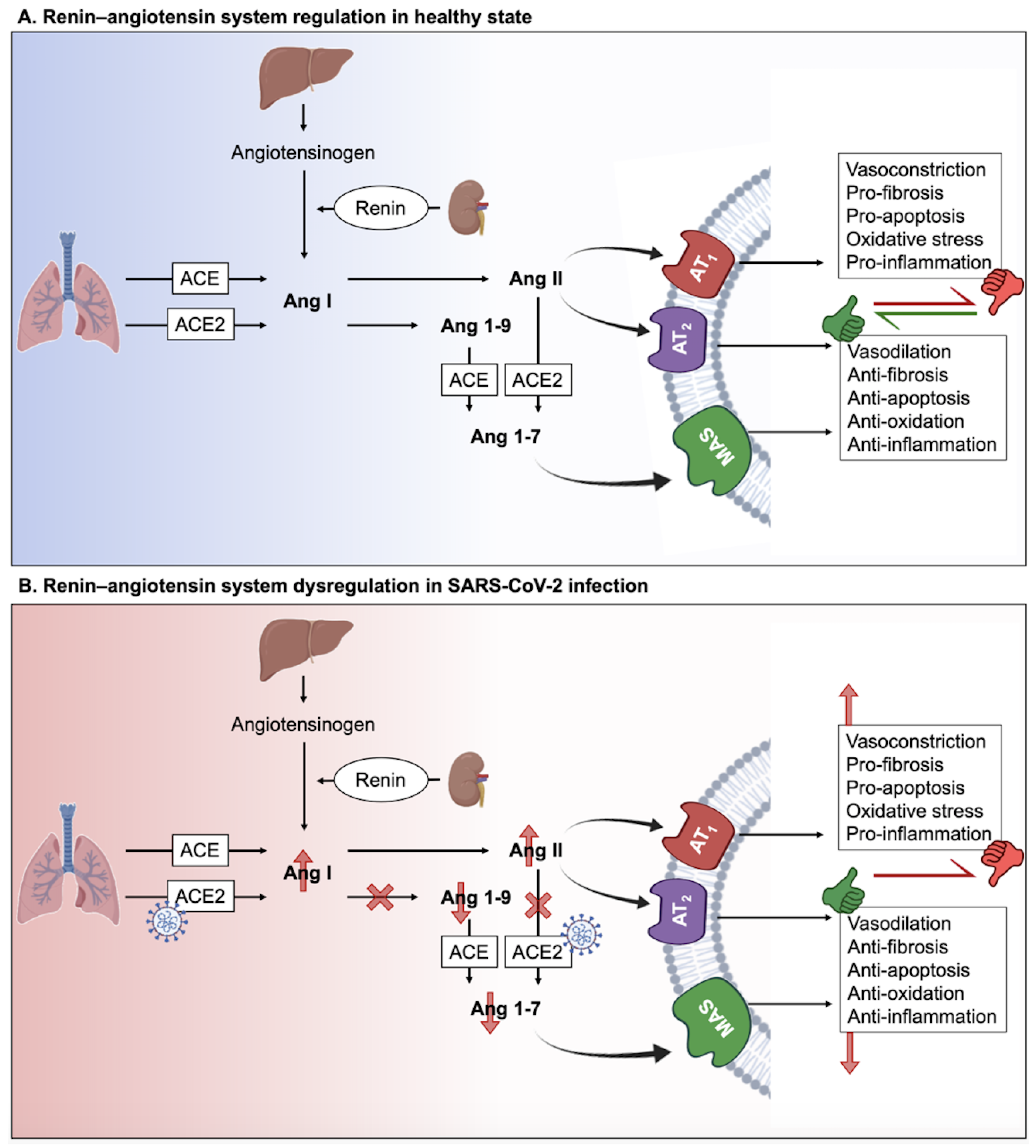
2. The Interplay between Long COVID and Renin-Angiotensin System (RAS)
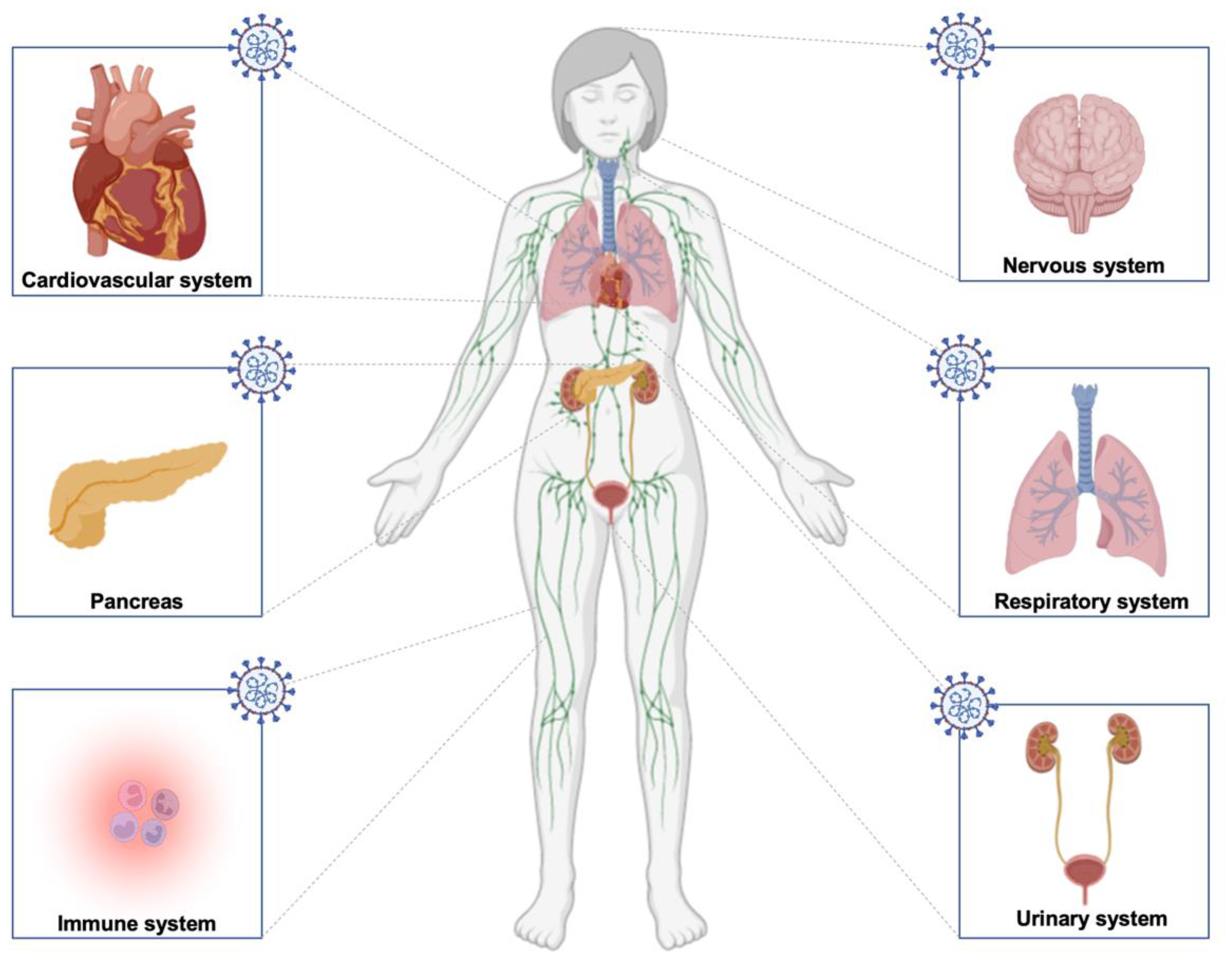
3. Long COVID in the Nervous System
3.1. The Central Nervous System
3.2. The Peripheral Nervous System
3.3. The Outcomes of Neuroinflammation
3.4. SARS-CoV-2 Entry into the CNS and PNS
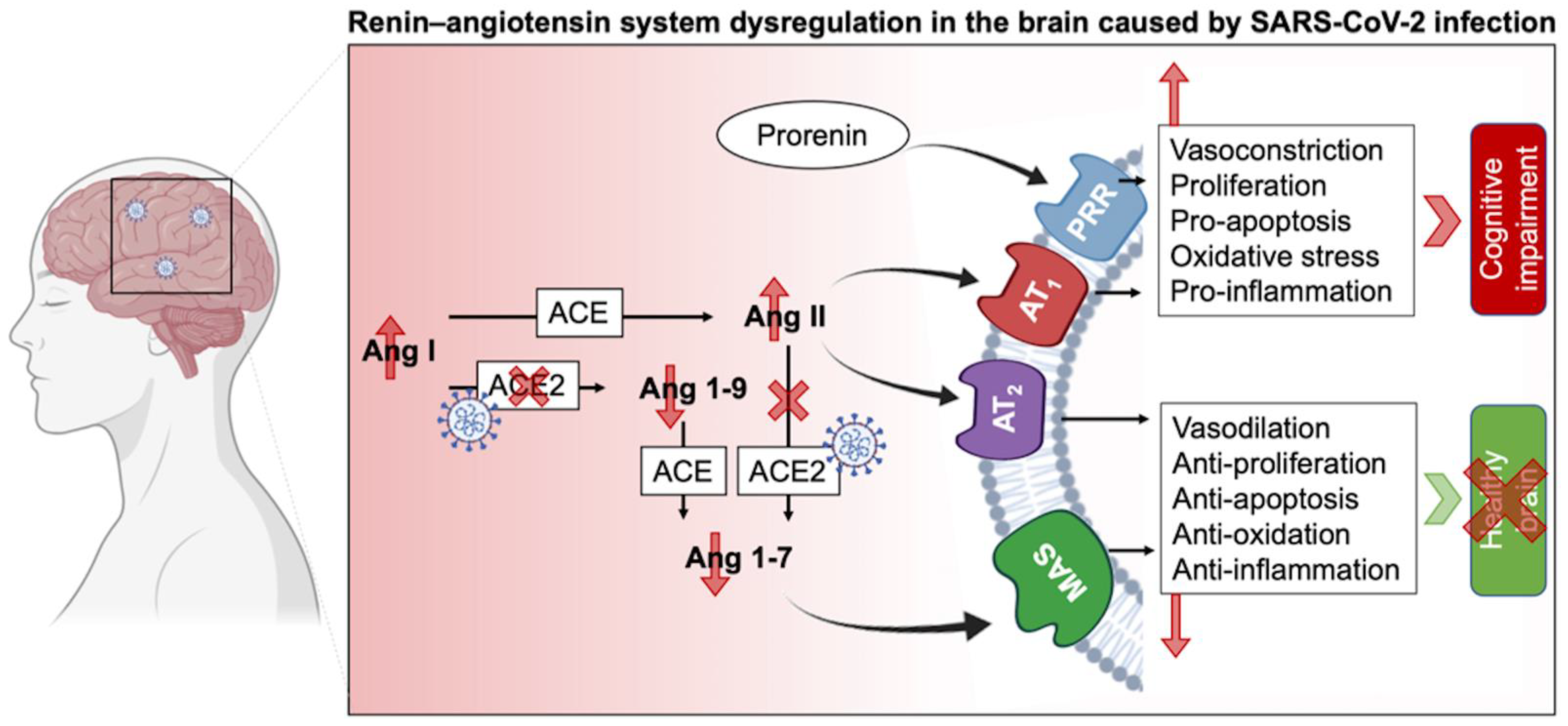
3.5. The Complexity of RAS in the CNS and Its Impairing in Long COVID
4. Evidence of RAS in the Cardiovascular System Highlight the Vulnerability of the System to Long COVID
5. RAS in Respiratory System, the Main Target of SARS-CoV-2, Therefore, Long COVID
6. Long COVID and RAS in Immune System
- (i).
- Intricate connections between stressor-induced cytokine storms and epigenetic variant-induced states of genomic fragility trigger further somatic mutations in stem cells or other mast cell progenitors [91].
- (ii).
- (iii).
- SARS-CoV-2-induced gene dysregulation, resulting in mast cell genetic regulatory loss [95].
- (iv).
- Autoantibodies reacting with immunoglobulin receptors on mast cells [96].
- (v).
- SARS-CoV-2-induced increase in Toll-like receptor activity leading to mast cell activation [97].
7. The Expression of RAS in the Urinary System Extends Long COVID Symptoms
8. The Outcomes of Long COVID on Metabolic Disorders, a Focus on Diabetes
9. Long COVID-19: Follow Up and Potential Treatment
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China. JAMA 2020, 323, 1239. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Arora, U.; Kumar, A.; Wig, N. The “post-COVID” syndrome: How deep is the damage? J. Med. Virol. 2021, 93, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, N. Long COVID: How to define it and how to manage it. BMJ 2020, 370, m3489. [Google Scholar] [CrossRef] [PubMed]
- Mehandru, S.; Merad, M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23, 194–202. [Google Scholar] [CrossRef]
- Greenhalgh, T.; Knight, M.; A’Court, C.; Buxton, M.; Husain, L. Management of post-acute COVID-19 in primary care. BMJ 2020, 370, m3026. [Google Scholar] [CrossRef] [PubMed]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 Long-term effects of COVID-19: A systematic review and meta-analysis. medRxiv Prepr. Serv. Health Sci. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Carfì, A.; Bernabei, R.; Landi, F. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603. [Google Scholar] [CrossRef]
- Arnold, D.T.; Hamilton, F.W.; Milne, A.; Morley, A.J.; Viner, J.; Attwood, M.; Noel, A.; Gunning, S.; Hatrick, J.; Hamilton, S.; et al. Patient outcomes after hospitalisation with COVID-19 and implications for follow-up: Results from a prospective UK cohort. Thorax 2021, 76, 399–401. [Google Scholar] [CrossRef]
- Raveendran, A.V.; Jayadevan, R.; Sashidharan, S. Long COVID: An overview. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 869–875. [Google Scholar] [CrossRef]
- El-Arif, G.; Farhat, A.; Khazaal, S.; Annweiler, C.; Kovacic, H.; Wu, Y.; Cao, Z.; Fajloun, Z.; Khattar, Z.A.; Sabatier, J.M. The Renin-Angiotensin System: A Key Role in SARS-CoV-2-Induced COVID-19. Molecules 2021, 26, 6945. [Google Scholar] [CrossRef] [PubMed]
- Rysz, S.; Al-Saadi, J.; Sjöström, A.; Farm, M.; Campoccia Jalde, F.; Plattén, M.; Eriksson, H.; Klein, M.; Vargas-Paris, R.; Nyrén, S.; et al. COVID-19 pathophysiology may be driven by an imbalance in the renin-angiotensin-aldosterone system. Nat. Commun. 2021, 12, 2417. [Google Scholar] [CrossRef] [PubMed]
- Perlot, T.; Penninger, J.M. ACE2—From the renin–angiotensin system to gut microbiota and malnutrition. Microbes Infect. 2013, 15, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Cao, Z.; Wu, Y.; Faucon, E.; Mouhat, S.; Kovacic, H.; Sabatier, J.-M. Counter-regulatory ‘Renin-Angiotensin’ System-based Candidate Drugs to Treat COVID-19 Diseases in SARS-CoV-2-infected Patients. Infect. Disord.-Drug Targets 2020, 20, 407–408. [Google Scholar] [CrossRef]
- Coto, E.; Avanzas, P.; Gómez, J. The Renin–Angiotensin–Aldosterone System and Coronavirus Disease 2019. Eur. Cardiol. Rev. 2021, 16, e07. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, 1–9. [Google Scholar] [CrossRef]
- Fajloun, Z.; Kovacic, H.; Annweiler, C.; Wu, Y.; Cao, Z.; Sabatier, J.-M. SARS-CoV-2-Induced Neurological Disorders in Symptomatic COVID-19 and Long COVID Patients: Key Role of Brain Renin-Angiotensin System. Infect. Disord.-Drug Targets 2022, 22. [Google Scholar] [CrossRef]
- Barré, J.; Sabatier, J.-M.; Annweiler, C. Montelukast Drug May Improve COVID-19 Prognosis: A Review of Evidence. Front. Pharmacol. 2020, 11, 1344. [Google Scholar] [CrossRef]
- Jakovac, H. COVID-19: Is the ACE2 just a foe? Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L1025–L1026. [Google Scholar] [CrossRef]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Fagyas, M.; Fejes, Z.; Sütő, R.; Nagy, Z.; Székely, B.; Pócsi, M.; Ivády, G.; Bíró, E.; Bekő, G.; Nagy, A.; et al. Circulating ACE2 activity predicts mortality and disease severity in hospitalized COVID-19 patients. Int. J. Infect. Dis. 2022, 115, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Dani, M.; Dirksen, A.; Taraborrelli, P.; Torocastro, M.; Panagopoulos, D.; Sutton, R.; Lim, P.B. Autonomic dysfunction in ‘long COVID’: Rationale, physiology and management strategies. Clin. Med. 2021, 21, e63–e67. [Google Scholar] [CrossRef]
- Annweiler, C.; Bourgeais, A.; Faucon, E.; Cao, Z.; Wu, Y.; Sabatier, J.-M. Neurological, Cognitive, and Behavioral Disorders during COVID-19: The Nitric Oxide Track. J. Am. Geriatr. Soc. 2020, 68, 1922–1923. [Google Scholar] [CrossRef] [PubMed]
- Spudich, S.; Nath, A. Nervous system consequences of COVID-19. Science 2022, 375, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Kandemirli, S.G.; Altundag, A.; Yildirim, D.; Tekcan Sanli, D.E.; Saatci, O. Olfactory Bulb MRI and Paranasal Sinus CT Findings in Persistent COVID-19 Anosmia. Acad. Radiol. 2021, 28, 28–35. [Google Scholar] [CrossRef]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef]
- Goldstein, D.S. The extended autonomic system, dyshomeostasis, and COVID-19. Clin. Auton. Res. 2020, 30, 299–315. [Google Scholar] [CrossRef]
- Li, H.; Kem, D.C.; Reim, S.; Khan, M.; Vanderlinde-Wood, M.; Zillner, C.; Collier, D.; Liles, C.; Hill, M.A.; Cunningham, M.W.; et al. Agonistic Autoantibodies as Vasodilators in Orthostatic Hypotension. Hypertension 2012, 59, 402–408. [Google Scholar] [CrossRef]
- Yu, X.; Stavrakis, S.; Hill, M.A.; Huang, S.; Reim, S.; Li, H.; Khan, M.; Hamlett, S.; Cunningham, M.W.; Kem, D.C. Autoantibody activation of beta-adrenergic and muscarinic receptors contributes to an “autoimmune” orthostatic hypotension. J. Am. Soc. Hypertens. 2012, 6, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ruzieh, M.; Batizy, L.; Dasa, O.; Oostra, C.; Grubb, B. The role of autoantibodies in the syndromes of orthostatic intolerance: A systematic review. Scand. Cardiovasc. J. 2017, 51, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, X.; Liles, C.; Khan, M.; Vanderlinde-Wood, M.; Galloway, A.; Zillner, C.; Benbrook, A.; Reim, S.; Collier, D.; et al. Autoimmune Basis for Postural Tachycardia Syndrome. J. Am. Heart Assoc. 2014, 3, e000755. [Google Scholar] [CrossRef]
- Gunning, W.T.; Kvale, H.; Kramer, P.M.; Karabin, B.L.; Grubb, B.P. Postural Orthostatic Tachycardia Syndrome Is Associated With Elevated G-Protein Coupled Receptor Autoantibodies. J. Am. Heart Assoc. 2019, 8, e013602. [Google Scholar] [CrossRef]
- Uncini, A.; Vallat, J.-M.; Jacobs, B.C. Guillain-Barré syndrome in SARS-CoV-2 infection: An instant systematic review of the first six months of pandemic. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, M.; Zakaria, F.; Ragab, Y.; Saad, A.; Bamaga, A.; Emad, Y.; Rasker, J.J. Guillain-Barré Syndrome Associated With Severe Acute Respiratory Syndrome Coronavirus 2 Detection and Coronavirus Disease 2019 in a Child. J. Pediatric Infect. Dis. Soc. 2020, 9, 510–513. [Google Scholar] [CrossRef]
- Wallukat, G.; Hohberger, B.; Wenzel, K.; Fürst, J.; Schulze-Rothe, S.; Wallukat, A.; Hönicke, A.-S.; Müller, J. Functional autoantibodies against G-protein coupled receptors in patients with persistent Long-COVID-19 symptoms. J. Transl. Autoimmun. 2021, 4, 100100. [Google Scholar] [CrossRef]
- Fedorowski, A. Postural orthostatic tachycardia syndrome: Clinical presentation, aetiology and management. J. Intern. Med. 2019, 285, 352–366. [Google Scholar] [CrossRef]
- Port, J.D.; Gilbert, E.M.; Larrabee, P.; Mealey, P.; Volkman, K.; Ginsburg, R.; Hershberger, R.E.; Murray, J.; Bristow, M.R. Neurotransmitter depletion compromises the ability of indirect-acting amines to provide inotropic support in the failing human heart. Circulation 1990, 81, 929–938. [Google Scholar] [CrossRef]
- Barizien, N.; Le Guen, M.; Russel, S.; Touche, P.; Huang, F.; Vallée, A. Clinical characterization of dysautonomia in long COVID-19 patients. Sci. Rep. 2021, 11, 14042. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Eshak, N.; Abdelnabi, M.; Ball, S.; Elgwairi, E.; Creed, K.; Test, V.; Nugent, K. Dysautonomia: An Overlooked Neurological Manifestation in a Critically ill COVID-19 Patient. Am. J. Med. Sci. 2020, 360, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Balcom, E.F.; Nath, A.; Power, C. Acute and chronic neurological disorders in COVID-19: Potential mechanisms of disease. Brain 2021, 144, 3576–3588. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.M. Brain Angiotensin II: New Developments, Unanswered Questions and Therapeutic Opportunities. Cell. Mol. Neurobiol. 2005, 25, 485–512. [Google Scholar] [CrossRef] [PubMed]
- El-Arif, G.; Khazaal, S.; Farhat, A.; Harb, J.; Annweiler, C.; Wu, Y.; Cao, Z.; Kovacic, H.; Abi Khattar, Z.; Fajloun, Z.; et al. Angiotensin II Type I Receptor (AT1R): The Gate towards COVID-19-Associated Diseases. Molecules 2022, 27, 2048. [Google Scholar] [CrossRef]
- Grobe, J.L.; Xu, D.; Sigmund, C.D. An Intracellular Renin-Angiotensin System in Neurons: Fact, Hypothesis, or Fantasy. Physiology 2008, 23, 187–193. [Google Scholar] [CrossRef]
- Annweiler, C.; Papon, N.; Sabatier, J.-M.; Barré, J. DAMPening Severe COVID-19 with Dexamethasone. Infect. Disord.-Drug Targets 2022, 22, 11–12. [Google Scholar] [CrossRef]
- Abadir, P.M. The Frail Renin-Angiotensin System. Clin. Geriatr. Med. 2011, 27, 53–65. [Google Scholar] [CrossRef]
- Kobori, H.; Nangaku, M.; Navar, L.G.; Nishiyama, A. The Intrarenal Renin-Angiotensin System: From Physiology to the Pathobiology of Hypertension and Kidney Disease. Pharmacol. Rev. 2007, 59, 251–287. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Lyoo, K.-S.; Kim, H.M.; Lee, B.; Che, Y.H.; Kim, S.-J.; Song, D.; Hwang, W.; Lee, S.; Park, J.-H.; Na, W.; et al. Direct neuronal infection of SARS-CoV-2 reveals cellular and molecular pathology of chemosensory impairment of COVID-19 patients. Emerg. Microbes Infect. 2022, 11, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Kumari, P.; Stone, S.; Natekar, J.P.; Arora, K.; Auroni, T.T.; Kumar, M. SARS-CoV-2 Infects Primary Neurons from Human ACE2 Expressing Mice and Upregulates Genes Involved in the Inflammatory and Necroptotic Pathways. Pathogens 2022, 11, 257. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.; Delarue, F.; Burcklé, C.; Bouzhir, L.; Giller, T.; Sraer, J.-D. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Investig. 2002, 109, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, P.; Gomez, J.; Grobe, J.L.; Sigmund, C.D. The Renin-Angiotensin System in the Central Nervous System and Its Role in Blood Pressure Regulation. Curr. Hypertens. Rep. 2020, 22, 7. [Google Scholar] [CrossRef]
- Jackson, L.; Eldahshan, W.; Fagan, S.; Ergul, A. Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef]
- Cao, Z.; Wu, Y.; Faucon, E.; Sabatier, J.-M. SARS-CoV-2 & COVID-19: Key-Roles of the ‘Renin-Angiotensin’ System / Vitamin D Impacting Drug and Vaccine Developments. Infect. Disord.-Drug Targets 2020, 20, 348–349. [Google Scholar] [CrossRef]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Guedj, E.; Campion, J.Y.; Dudouet, P.; Kaphan, E.; Bregeon, F.; Tissot-Dupont, H.; Guis, S.; Barthelemy, F.; Habert, P.; Ceccaldi, M.; et al. 18F-FDG brain PET hypometabolism in patients with long COVID. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2823–2833. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host–Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef]
- Marvel, C.L.; Morgan, O.P.; Kronemer, S.I. How the motor system integrates with working memory. Neurosci. Biobehav. Rev. 2019, 102, 184–194. [Google Scholar] [CrossRef]
- Bodranghien, F.; Bastian, A.; Casali, C.; Hallett, M.; Louis, E.D.; Manto, M.; Mariën, P.; Nowak, D.A.; Schmahmann, J.D.; Serrao, M.; et al. Consensus Paper: Revisiting the Symptoms and Signs of Cerebellar Syndrome. The Cerebellum 2016, 15, 369–391. [Google Scholar] [CrossRef] [PubMed]
- Borsook, D.; Sava, S.; Becerra, L. The Pain Imaging Revolution: Advancing Pain Into the 21st Century. Neuroscientist 2010, 16, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Guedj, E.; Cammilleri, S.; Niboyet, J.; Dupont, P.; Vidal, E.; Dropinski, J.-P.; Mundler, O. Clinical Correlate of Brain SPECT Perfusion Abnormalities in Fibromyalgia. J. Nucl. Med. 2008, 49, 1798–1803. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.B.; Onuki, Y.; Bruinsma, B.; van der Werf, Y.D.; De Zeeuw, C.I. The Sleeping Cerebellum. Trends Neurosci. 2017, 40, 309–323. [Google Scholar] [CrossRef]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain Glucose Hypometabolism and Oxidative Stress in Preclinical Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef]
- Farshidfar, F.; Koleini, N.; Ardehali, H. Cardiovascular complications of COVID-19. JCI Insight 2021, 6, 13. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Jakovac, H.; Ferenčić, A.; Stemberger, C.; Mohar Vitezić, B.; Cuculić, D. Detection of SARS-CoV-2 Antigens in the AV-Node of a Cardiac Conduction System—A Case Report. Trop. Med. Infect. Dis. 2022, 7, 43. [Google Scholar] [CrossRef]
- Kochav, S.M.; Coromilas, E.; Nalbandian, A.; Ranard, L.S.; Gupta, A.; Chung, M.K.; Gopinathannair, R.; Biviano, A.B.; Garan, H.; Wan, E.Y. Cardiac Arrhythmias in COVID-19 Infection. Circ. Arrhythmia Electrophysiol. 2020, 13, e310–e314. [Google Scholar] [CrossRef]
- Patel, N.H.; Rutland, J.; Tecson, K.M. Arrhythmias and Intraventricular Conduction Disturbances in Patients Hospitalized With Coronavirus Disease 2019. Am. J. Cardiol. 2022, 162, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef]
- Chung, M.K.; Zidar, D.A.; Bristow, M.R.; Cameron, S.J.; Chan, T.; Harding, C.V.; Kwon, D.H.; Singh, T.; Tilton, J.C.; Tsai, E.J.; et al. COVID-19 and Cardiovascular Disease. Circ. Res. 2021, 128, 1214–1236. [Google Scholar] [CrossRef]
- Zhang, L.; Richards, A.; Barrasa, M.I.; Hughes, S.H.; Young, R.A.; Jaenisch, R. Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc. Natl. Acad. Sci. USA 2021, 118, e2105968118. [Google Scholar] [CrossRef]
- Di Toro, A.; Bozzani, A.; Tavazzi, G.; Urtis, M.; Giuliani, L.; Pizzoccheri, R.; Aliberti, F.; Fergnani, V.; Arbustini, E. Long COVID: Long-term effects? Eur. Hear. J. Suppl. 2021, 23, E1–E5. [Google Scholar] [CrossRef]
- Cho, J.L.; Villacreses, R.; Nagpal, P.; Guo, J.; Pezzulo, A.A.; Thurman, A.L.; Hamzeh, N.Y.; Blount, R.J.; Fortis, S.; Hoffman, E.A.; et al. Quantitative Chest CT Assessment of Small Airways Disease in Post-Acute SARS-CoV-2 Infection. Radiology 2022, 2022, 212170. [Google Scholar] [CrossRef]
- Elicker, B.M. What Are the Long-term Pulmonary Sequelae of COVID-19 Infection? Radiology 2022, 2022, 220449. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Colombo, C.; Burgel, P.-R.; Gartner, S.; van Koningsbruggen-Rietschel, S.; Naehrlich, L.; Sermet-Gaudelus, I.; Southern, K.W. Impact of COVID-19 on people with cystic fibrosis. Lancet Respir. Med. 2020, 8, e35–e36. [Google Scholar] [CrossRef]
- Grist, J.T.; Chen, M.; Collier, G.J.; Raman, B.; Abueid, G.; McIntyre, A.; Matthews, V.; Fraser, E.; Ho, L.-P.; Wild, J.M.; et al. Hyperpolarized 129 Xe MRI Abnormalities in Dyspneic Patients 3 Months after COVID-19 Pneumonia: Preliminary Results. Radiology 2021, 301, E353–E360. [Google Scholar] [CrossRef] [PubMed]
- Galiatsatos, P. COVID-19 Lung Damage. Personal communication. 2022. Available online: https://www.hopkinsmedicine.org/health/conditions-and-diseases/coronavirus/what-coronavirus-does-to-the-lungs (accessed on 28 April 2022).
- Darley, D.R.; Dore, G.J.; Cysique, L.; Wilhelm, K.A.; Andresen, D.; Tonga, K.; Stone, E.; Byrne, A.; Plit, M.; Masters, J.; et al. Persistent symptoms up to four months after community and hospital-managed SARS-CoV-2 infection. Med. J. Aust. 2021, 214, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Darley, D.R.; Dore, G.J.; Byrne, A.L.; Plit, M.L.; Brew, B.J.; Kelleher, A.; Matthews, G.V. Limited recovery from post-acute sequelae of SARS-CoV-2 at 8 months in a prospective cohort. ERJ Open Res. 2021, 7, 00384–02021. [Google Scholar] [CrossRef]
- Patel, S.K.; Juno, J.A.; Lee, W.S.; Wragg, K.M.; Hogarth, P.M.; Kent, S.J.; Burrell, L.M. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: Implications for COVID-19 pathogenesis and consequences. Eur. Respir. J. 2021, 57, 2003730. [Google Scholar] [CrossRef]
- Kritas, S.K.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Conti, P. Mast cells contribute to coronavirus-induced inflammation: New anti-inflammatory strategy. J. Biol. Regul. Homeost. Agents 2020, 34, 9–14. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Conti, P. COVID-19 and Multisystem Inflammatory Syndrome, or is it Mast Cell Activation Syndrome? J. Biol. Regul. Homeost. Agents 2020, 34, 1633–1636. [Google Scholar] [CrossRef]
- Afrin, L.B.; Self, S.; Menk, J.; Lazarchick, J. Characterization of Mast Cell Activation Syndrome. Am. J. Med. Sci. 2017, 353, 207–215. [Google Scholar] [CrossRef]
- Afrin, L.B.; Ackerley, M.B.; Bluestein, L.S.; Brewer, J.H.; Brook, J.B.; Buchanan, A.D.; Cuni, J.R.; Davey, W.P.; Dempsey, T.T.; Dorff, S.R.; et al. Diagnosis of mast cell activation syndrome: A global “consensus-2". Diagnosis 2021, 8, 137–152. [Google Scholar] [CrossRef]
- Molderings, G.J.; Haenisch, B.; Bogdanow, M.; Fimmers, R.; Nöthen, M.M. Familial Occurrence of Systemic Mast Cell Activation Disease. PLoS ONE 2013, 8, e76241. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, L.B.; Brook, J.B.; Walters, A.S.; Goris, A.; Afrin, L.B.; Molderings, G.J. Mast cell activation symptoms are prevalent in Long-COVID. Int. J. Infect. Dis. 2021, 112, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, A.J.; Ding, J.; You, Y.; Dong, Z.; Chehade, H.; Alvero, A.; Mor, Y.; Draghici, S.; Mor, G. Identification of key signaling pathways induced by SARS-CoV-2 that underlie thrombosis and vascular injury in COVID-19 patients. J. Leukoc. Biol. 2021, 109, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Reaction of Human Monoclonal Antibodies to SARS-CoV-2 Proteins With Tissue Antigens: Implications for Autoimmune Diseases. Front. Immunol. 2021, 11, 617089. [Google Scholar] [CrossRef]
- Mukherjee, R.; Bhattacharya, A.; Bojkova, D.; Mehdipour, A.R.; Shin, D.; Khan, K.S.; Hei-Yin Cheung, H.; Wong, K.-B.; Ng, W.-L.; Cinatl, J.; et al. Famotidine inhibits toll-like receptor 3-mediated inflammatory signaling in SARS-CoV-2 infection. J. Biol. Chem. 2021, 297, 100925. [Google Scholar] [CrossRef]
- Dhar, N.; Dhar, S.; Timar, R.; Lucas, S.; Lamb, L.E.; Chancellor, M.B. De Novo Urinary Symptoms Associated With COVID-19: COVID-19-Associated Cystitis. J. Clin. Med. Res. 2020, 12, 681–682. [Google Scholar] [CrossRef]
- Lamb, L.E.; Dhar, N.; Timar, R.; Wills, M.; Dhar, S.; Chancellor, M.B. COVID-19 inflammation results in urine cytokine elevation and causes COVID-19 associated cystitis (CAC). Med. Hypotheses 2020, 145, 110375. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef]
- Mumm, J.-N.; Osterman, A.; Ruzicka, M.; Stihl, C.; Vilsmaier, T.; Munker, D.; Khatamzas, E.; Giessen-Jung, C.; Stief, C.; Staehler, M.; et al. Urinary Frequency as a Possibly Overlooked Symptom in COVID-19 Patients: Does SARS-CoV-2 Cause Viral Cystitis? Eur. Urol. 2020, 78, 624–628. [Google Scholar] [CrossRef]
- Lamb, L.E.; Timar, R.; Wills, M.; Dhar, S.; Lucas, S.M.; Komnenov, D.; Chancellor, M.B.; Dhar, N. Long COVID and COVID-19-associated cystitis (CAC). Int. Urol. Nephrol. 2022, 54, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Fan, J.; Hu, L.-F.; Zhang, Y.; Ooi, J.D.; Meng, T.; Jin, P.; Ding, X.; Peng, L.-K.; Song, L.; et al. Single-cell analysis of angiotensin-converting enzyme II expression in human kidneys and bladders reveals a potential route of 2019 novel coronavirus infection. Chin. Med. J. 2021, 134, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Frithiof, R.; Bergqvist, A.; Järhult, J.D.; Lipcsey, M.; Hultström, M. Presence of SARS-CoV-2 in urine is rare and not associated with acute kidney injury in critically ill COVID-19 patients. Crit. Care 2020, 24, 587. [Google Scholar] [CrossRef] [PubMed]
- Kashi, A.H.; De la Rosette, J.; Amini, E.; Abdi, H.; Fallah-Karkan, M.; Vaezjalali, M. Urinary Viral Shedding of COVID-19 and its Clinical Associations: A Systematic Review and Meta-analysis of Observational Studies. Urol. J. 2020, 17, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.L.; Ferrao, F.M.; Zheng, Y.; Li, X.C. New Frontiers in the Intrarenal Renin-Angiotensin System: A Critical Review of Classical and New Paradigms. Front. Endocrinol. 2013, 4, 166. [Google Scholar] [CrossRef]
- Lee, J.H.; Jang, S.J.; Rhie, S. Antinatriuretic phenomena seen in children with acute pyelonephritis may be related to the activation of intrarenal RAAS. Medicine 2018, 97, e12152. [Google Scholar] [CrossRef]
- Heyman, S.N.; Walther, T.; Abassi, Z. Angiotensin-(1-7)—A Potential Remedy for AKI: Insights Derived from the COVID-19 Pandemic. J. Clin. Med. 2021, 10, 1200. [Google Scholar] [CrossRef]
- Bowe, B.; Xie, Y.; Xu, E.; Al-Aly, Z. Kidney Outcomes in Long COVID. J. Am. Soc. Nephrol. 2021, 32, 2851–2862. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Unnikrishnan, R.; Misra, A. Diabetes and COVID19: A bidirectional relationship. Nutr. Diabetes 2021, 11, 21. [Google Scholar] [CrossRef]
- Misra, A.; Ghosh, A.; Gupta, R. Heterogeneity in presentation of hyperglycaemia during COVID-19 pandemic: A proposed classification. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Anjana, R.M.; Shanthi Rani, C.S.; Jeba Rani, S.; Gupta, R.; Jha, A.; Gupta, V.; Kuchay, M.S.; Luthra, A.; Durrani, S.; et al. Glycemic parameters in patients with new-onset diabetes during COVID-19 pandemic are more severe than in patients with new-onset diabetes before the pandemic: NOD COVID India Study. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Samuel, S.M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Diabetes and coronavirus (SARS-CoV-2): Molecular mechanism of Metformin intervention and the scientific basis of drug repurposing. PLOS Pathog. 2021, 17, e1009634. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.E.; Koyama, A.K.; Alvarez, P.; Chow, W.; Lundeen, E.A.; Perrine, C.G.; Pavkov, M.E.; Rolka, D.B.; Wiltz, J.L.; Bull-Otterson, L.; et al. Risk for Newly Diagnosed Diabetes >30 Days After SARS-CoV-2 Infection Among Persons Aged <18 Years—United States, March 1, 2020–June 28, 2021. MMWR. Morb. Mortal. Wkly. Rep. 2022, 71, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhang, Z.; Wang, Q.; Chen, Y.; Lu, D.; Wu, W. COVID-19 and Diabetes: A Comprehensive Review of Angiotensin Converting Enzyme 2, Mutual Effects and Pharmacotherapy. Front. Endocrinol. 2021, 12, 1541. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.D.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef]
- Hulme, K.D.; Gallo, L.A.; Short, K.R. Influenza Virus and Glycemic Variability in Diabetes: A Killer Combination? Front. Microbiol. 2017, 8, 861. [Google Scholar] [CrossRef]
- Filippi, C.M.; von Herrath, M.G. Viral Trigger for Type 1 Diabetes. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef]
- Fernandez, C.; Rysä, J.; Almgren, P.; Nilsson, J.; Engström, G.; Orho-Melander, M.; Ruskoaho, H.; Melander, O. Plasma levels of the proprotein convertase furin and incidence of diabetes and mortality. J. Intern. Med. 2018, 284, 377–387. [Google Scholar] [CrossRef]
- Zipeto, D.; Palmeira, J.D.F.; Argañaraz, G.A.; Argañaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 11, 2642. [Google Scholar] [CrossRef]
- Ji, H.-L.; Zhao, R.; Matalon, S.; Matthay, M.A. Elevated Plasmin(ogen) as a Common Risk Factor for COVID-19 Susceptibility. Physiol. Rev. 2020, 100, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Al-Aly, Z. Risks and burdens of incident diabetes in long COVID: A cohort study. Lancet Diabetes Endocrinol. 2022, 10, 311–321. [Google Scholar] [CrossRef]
- Bindom, S.M.; Lazartigues, E. The sweeter side of ACE2: Physiological evidence for a role in diabetes. Mol. Cell. Endocrinol. 2009, 302, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Faiq, M.A.; Pareek, V.; Raza, K.; Narayan, R.K.; Prasoon, P.; Kumar, P.; Kulandhasamy, M.; Kumari, C.; Kant, K.; et al. Relevance of SARS-CoV-2 related factors ACE2 and TMPRSS2 expressions in gastrointestinal tissue with pathogenesis of digestive symptoms, diabetes-associated mortality, and disease recurrence in COVID-19 patients. Med. Hypotheses 2020, 144, 110271. [Google Scholar] [CrossRef] [PubMed]
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Koufakis, T.; Metallidis, S.; Zebekakis, P.; Kotsa, K. Intestinal SGLT1 as a therapeutic target in COVID-19-related diabetes: A “two-edged sword” hypothesis. Br. J. Clin. Pharmacol. 2021, 87, 3643–3646. [Google Scholar] [CrossRef] [PubMed]
- Sathish, T.; Kapoor, N.; Cao, Y.; Tapp, R.J.; Zimmet, P. Proportion of newly diagnosed diabetes in COVID -19 patients: A systematic review and meta-analysis. Diabetes Obes. Metab. 2021, 23, 870–874. [Google Scholar] [CrossRef]
- Unsworth, R.; Wallace, S.; Oliver, N.S.; Yeung, S.; Kshirsagar, A.; Naidu, H.; Kwong, R.M.W.; Kumar, P.; Logan, K.M. New-Onset Type 1 Diabetes in Children During COVID-19: Multicenter Regional Findings in the U.K. Diabetes Care 2020, 43, e170–e171. [Google Scholar] [CrossRef]
- Vlad, A.; Serban, V.; Timar, R.; Sima, A.; Botea, V.; Albai, O.; Timar, B.; Vlad, M. Increased Incidence of Type 1 Diabetes during the COVID-19 Pandemic in Romanian Children. Medicina 2021, 57, 973. [Google Scholar] [CrossRef]
- Kamrath, C.; Mönkemöller, K.; Biester, T.; Rohrer, T.R.; Warncke, K.; Hammersen, J.; Holl, R.W. Ketoacidosis in Children and Adolescents With Newly Diagnosed Type 1 Diabetes During the COVID-19 Pandemic in Germany. JAMA 2020, 324, 801. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Schwartz, M.D. Admissions to Veterans Affairs Hospitals for Emergency Conditions During the COVID-19 Pandemic. JAMA 2020, 324, 96. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, M.; Barbi, E.; Apicella, A.; Marchetti, F.; Cardinale, F.; Trobia, G. Delayed access or provision of care in Italy resulting from fear of COVID-19. Lancet Child Adolesc. Health 2020, 4, e10–e11. [Google Scholar] [CrossRef]
- Duca, L.M.; Reboussin, B.A.; Pihoker, C.; Imperatore, G.; Saydah, S.; Mayer-Davis, E.; Rewers, A.; Dabelea, D. Diabetic ketoacidosis at diagnosis of type 1 diabetes and glycemic control over time: The SEARCH for diabetes in youth study. Pediatr. Diabetes 2019, 20, 172–179. [Google Scholar] [CrossRef]
- Sosale, A.; Sosale, B.; Kesavadev, J.; Chawla, M.; Reddy, S.; Saboo, B.; Misra, A. Steroid use during COVID-19 infection and hyperglycemia—What a physician should know. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 102167. [Google Scholar] [CrossRef]
- Jakovac, H.; Ferenčić, A.; Stemberger, C.; Mohar Vitezić, B.; Cuculić, D. Detection of SARS-CoV-2 antigens in thyroid gland showing histopathological features of subacute thyroiditis. Eur. Thyroid J. 2022, 11, e220005. [Google Scholar] [CrossRef]
- Feghali, K.; Atallah, J.; Norman, C. Manifestations of thyroid disease post COVID-19 illness: Report of Hashimoto thyroiditis, Graves’ disease, and subacute thyroiditis. J. Clin. Transl. Endocrinol. Case Rep. 2021, 22, 100094. [Google Scholar] [CrossRef]
- Townsend, L.; Dyer, A.H.; McCluskey, P.; O’Brien, K.; Dowds, J.; Laird, E.; Bannan, C.; Bourke, N.M.; Ní Cheallaigh, C.; Byrne, D.G.; et al. Investigating the Relationship between Vitamin D and Persistent Symptoms Following SARS-CoV-2 Infection. Nutrients 2021, 13, 2430. [Google Scholar] [CrossRef]
- Myall, K.J.; Mukherjee, B.; Castanheira, A.M.; Lam, J.L.; Benedetti, G.; Mak, S.M.; Preston, R.; Thillai, M.; Dewar, A.; Molyneaux, P.L.; et al. Persistent Post–COVID-19 Interstitial Lung Disease. An Observational Study of Corticosteroid Treatment. Ann. Am. Thorac. Soc. 2021, 18, 799–806. [Google Scholar] [CrossRef]
- Moores, L.K.; Tritschler, T.; Brosnahan, S.; Carrier, M.; Collen, J.F.; Doerschug, K.; Holley, A.B.; Jimenez, D.; Le Gal, G.; Rali, P.; et al. Prevention, Diagnosis, and Treatment of VTE in Patients With Coronavirus Disease 2019. Chest 2020, 158, 1143–1163. [Google Scholar] [CrossRef]
- Postolache, T.T.; Benros, M.E.; Brenner, L.A. Targetable Biological Mechanisms Implicated in Emergent Psychiatric Conditions Associated With SARS-CoV-2 Infection. JAMA Psychiatry 2021, 78, 353. [Google Scholar] [CrossRef] [PubMed]
- Kirby, R.S.; Kirby, J.A. Correlation of COVID-19 Mortality with Clinical Parameters in an Urban and Suburban Nursing Home Population. medRxiv 2020. [Google Scholar] [CrossRef]
- Jones, A.G.; Hattersley, A.T. The clinical utility of C-peptide measurement in the care of patients with diabetes. Diabet. Med. 2013, 30, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Baral, R.; White, M.; Vassiliou, V.S. Effect of Renin-Angiotensin-Aldosterone System Inhibitors in Patients with COVID-19: A Systematic Review and Meta-analysis of 28,872 Patients. Curr. Atheroscler. Rep. 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Samavati, L.; Uhal, B.D. ACE2, Much More Than Just a Receptor for SARS-CoV-2. Front. Cell. Infect. Microbiol. 2020, 10, 317. [Google Scholar] [CrossRef]
- Chung, O.; Csikós, T.; Unger, T. Angiotensin II receptor pharmacology and AT1-receptor blockers. J. Hum. Hypertens. 1999, 13, S11–S20. [Google Scholar] [CrossRef][Green Version]
- Ferrario, C.M.; Ahmad, S.; Groban, L. Mechanisms by which angiotensin-receptor blockers increase ACE2 levels. Nat. Rev. Cardiol. 2020, 17, 378. [Google Scholar] [CrossRef]
- Devaux, C.A.; Lagier, J.-C.; Raoult, D. New Insights Into the Physiopathology of COVID-19: SARS-CoV-2-Associated Gastrointestinal Illness. Front. Med. 2021, 8, 640073. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Jessup, J.; Gallagher, P.E.; Averill, D.B.; Brosnihan, K.B.; Ann Tallant, E.; Smith, R.D.; Chappell, M.C. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005, 68, 2189–2196. [Google Scholar] [CrossRef]
- D’Ardes, D.; Boccatonda, A.; Rossi, I.; Guagnano, M.T.; Santilli, F.; Cipollone, F.; Bucci, M. COVID-19 and RAS: Unravelling an Unclear Relationship. Int. J. Mol. Sci. 2020, 21, 3003. [Google Scholar] [CrossRef]
- Wu, Y. Compensation of ACE2 Function for Possible Clinical Management of 2019-nCoV-Induced Acute Lung Injury. Virol. Sin. 2020, 35, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Hanotte, B.; Grandin de l’Eprevier, C.; Sabatier, J.-M.; Lafaie, L.; Célarier, T. Vitamin D and survival in COVID-19 patients: A quasi-experimental study. J. Steroid Biochem. Mol. Biol. 2020, 204, 105771. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Cao, Z.; Sabatier, J.-M. Point of view: Should COVID-19 patients be supplemented with vitamin D? Maturitas 2020, 140, 24–26. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Symptoms | Systems |
|---|---|
| Fatigue | Nervous system |
| Attention deficit | |
| Sleep disturbances | |
| Cognitive impairment | |
| Post-traumatic stress disorder | |
| Muscle pain | |
| Concentration problems | |
| Headache | |
| Pins and needles sensation | |
| Anxiety | |
| Depression | |
| Delusions Paranoia | |
| Anosmia | |
| Ageusia | |
| Pericarditis | Cardiovascular system |
| Myocarditis | |
| Heart failure | |
| Thromboembolic illness | |
| Ischemic and non-ischemic heart disease | |
| Arrhythmias and palpitations | |
| Cerebrovascular disorders | |
| Eye disorders Skin lesions | |
| Cardiac abnormalities | |
| Cardiac arrest | |
| Sore throat | Respiratory system |
| Dyspnea | |
| Persistent cough | |
| Interstitial pneumonia | |
| Chest pain | |
| Activation of innate immune cells | Immune system |
| Inflammation | |
| Mast cell activation syndrome | |
| Macrophage activation syndrome | |
| Overactive bladder symptoms | Urinary system |
| Cystitis | |
| Renal damage |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khazaal, S.; Harb, J.; Rima, M.; Annweiler, C.; Wu, Y.; Cao, Z.; Abi Khattar, Z.; Legros, C.; Kovacic, H.; Fajloun, Z.; et al. The Pathophysiology of Long COVID throughout the Renin-Angiotensin System. Molecules 2022, 27, 2903. https://doi.org/10.3390/molecules27092903
Khazaal S, Harb J, Rima M, Annweiler C, Wu Y, Cao Z, Abi Khattar Z, Legros C, Kovacic H, Fajloun Z, et al. The Pathophysiology of Long COVID throughout the Renin-Angiotensin System. Molecules. 2022; 27(9):2903. https://doi.org/10.3390/molecules27092903
Chicago/Turabian StyleKhazaal, Shaymaa, Julien Harb, Mohamad Rima, Cédric Annweiler, Yingliang Wu, Zhijian Cao, Ziad Abi Khattar, Christian Legros, Hervé Kovacic, Ziad Fajloun, and et al. 2022. "The Pathophysiology of Long COVID throughout the Renin-Angiotensin System" Molecules 27, no. 9: 2903. https://doi.org/10.3390/molecules27092903
APA StyleKhazaal, S., Harb, J., Rima, M., Annweiler, C., Wu, Y., Cao, Z., Abi Khattar, Z., Legros, C., Kovacic, H., Fajloun, Z., & Sabatier, J.-M. (2022). The Pathophysiology of Long COVID throughout the Renin-Angiotensin System. Molecules, 27(9), 2903. https://doi.org/10.3390/molecules27092903