
Novel 1,2,3-Triazole-Coumarin Hybrid Glycosides and Their Tetrazolyl Analogues: Design, Anticancer Evaluation and Molecular Docking Targeting EGFR, VEGFR-2 and CDK-2

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
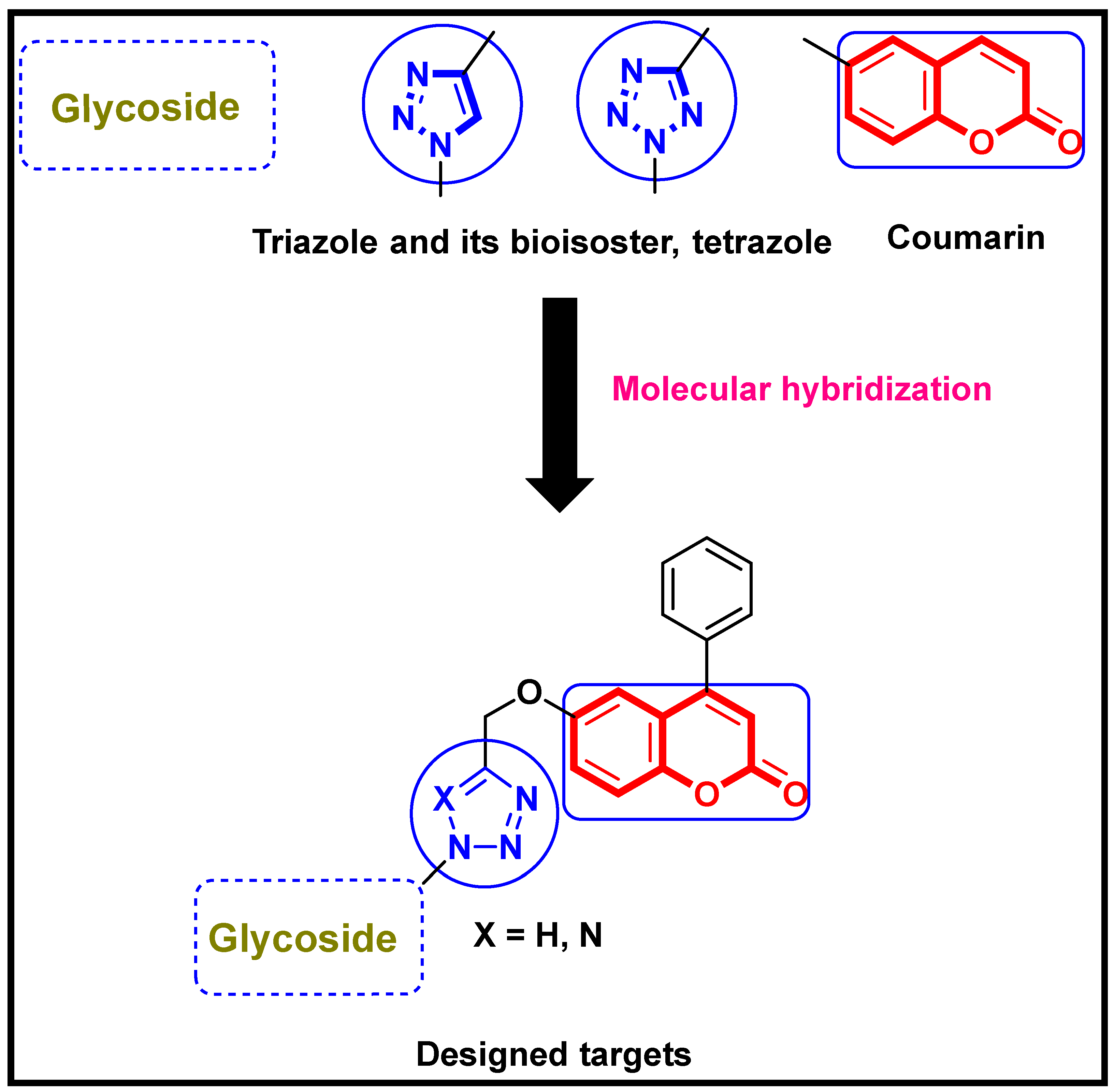
2. Results and Discussion
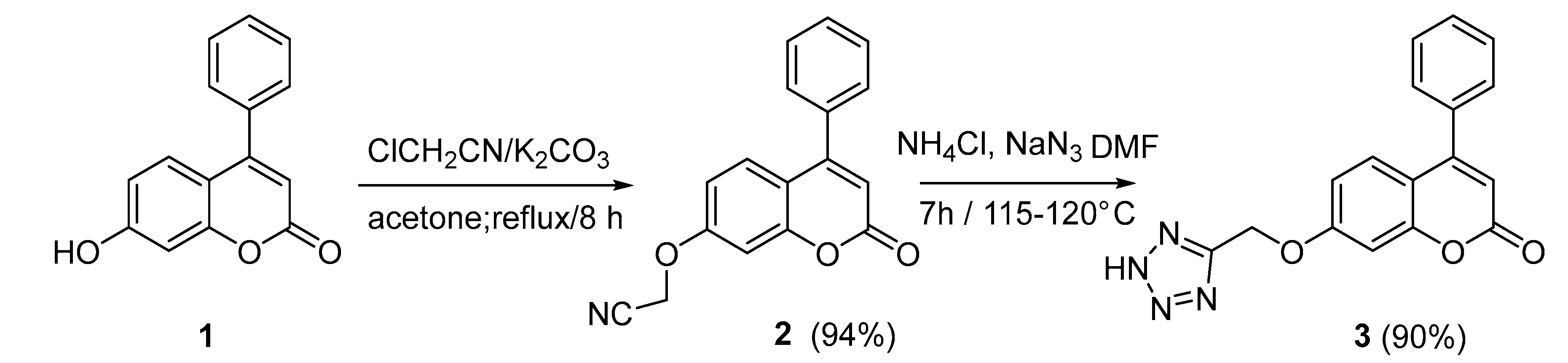
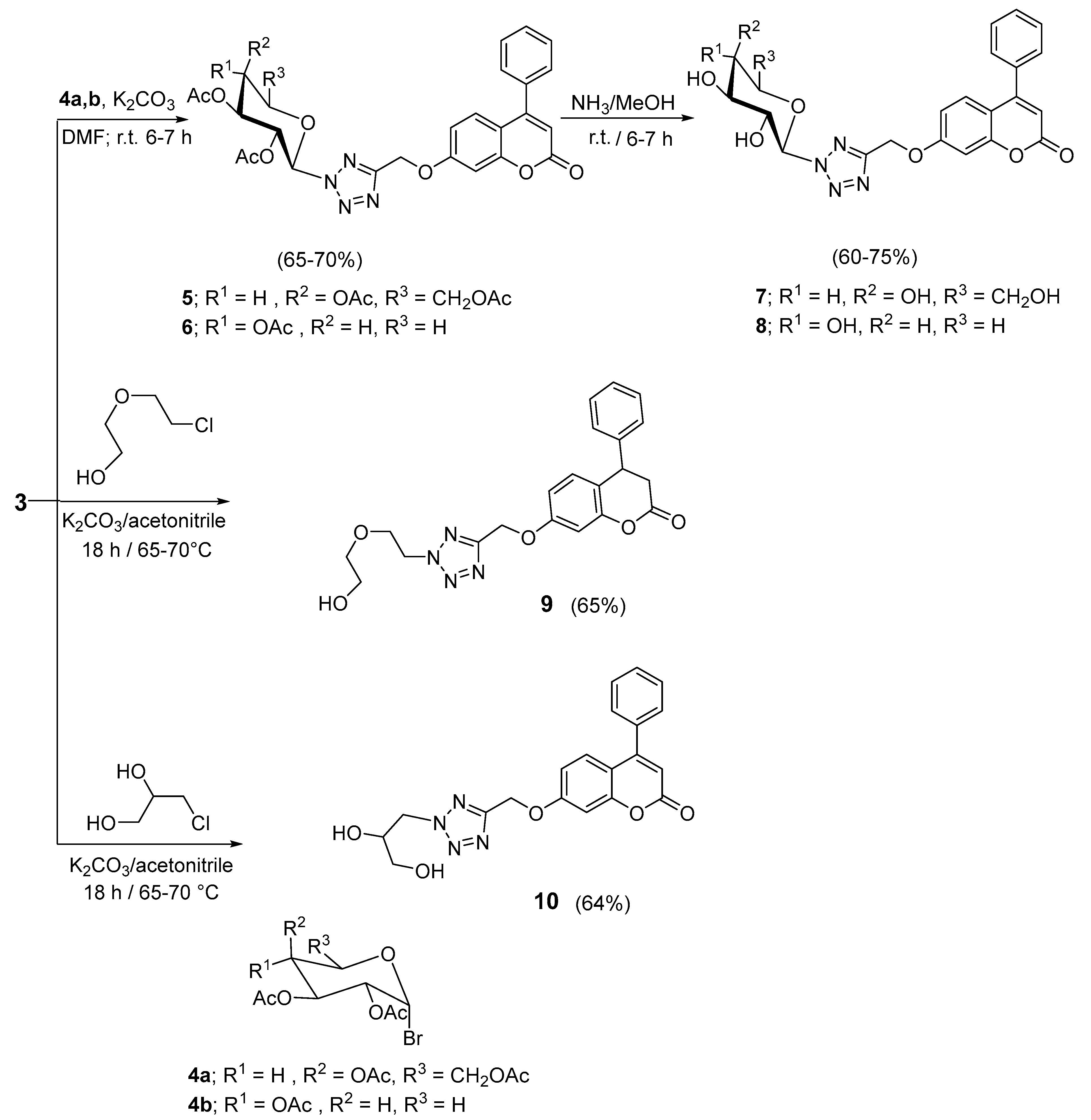
2.1. Chemistry
2.2. Biological Evaluation
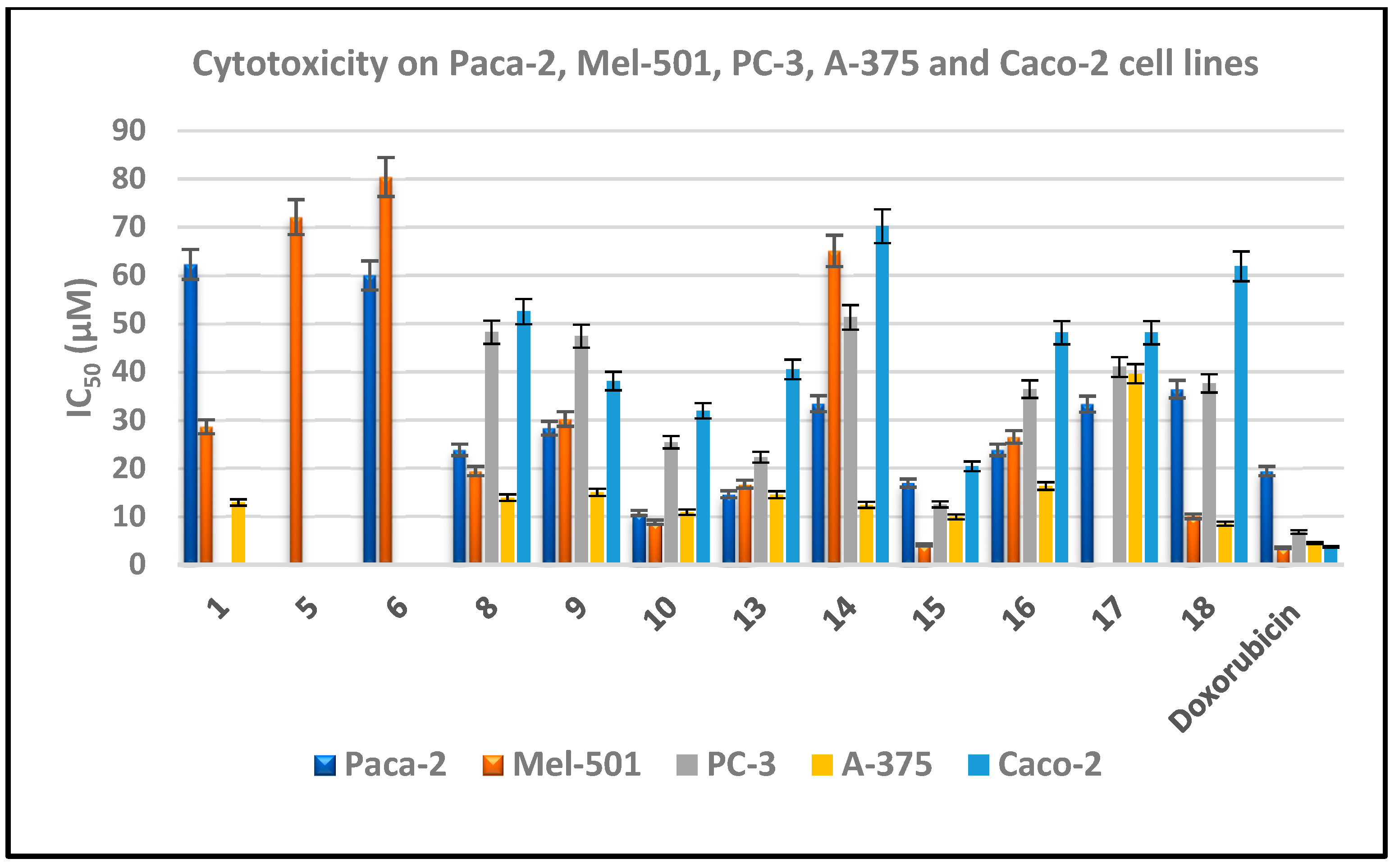
2.2.1. In Vitro Cytotoxicity
Structure Activity Relationship Study
2.2.2. In Vitro Enzyme Inhibitory Assessment against EGFR, VEGFR-2 and CDK-2
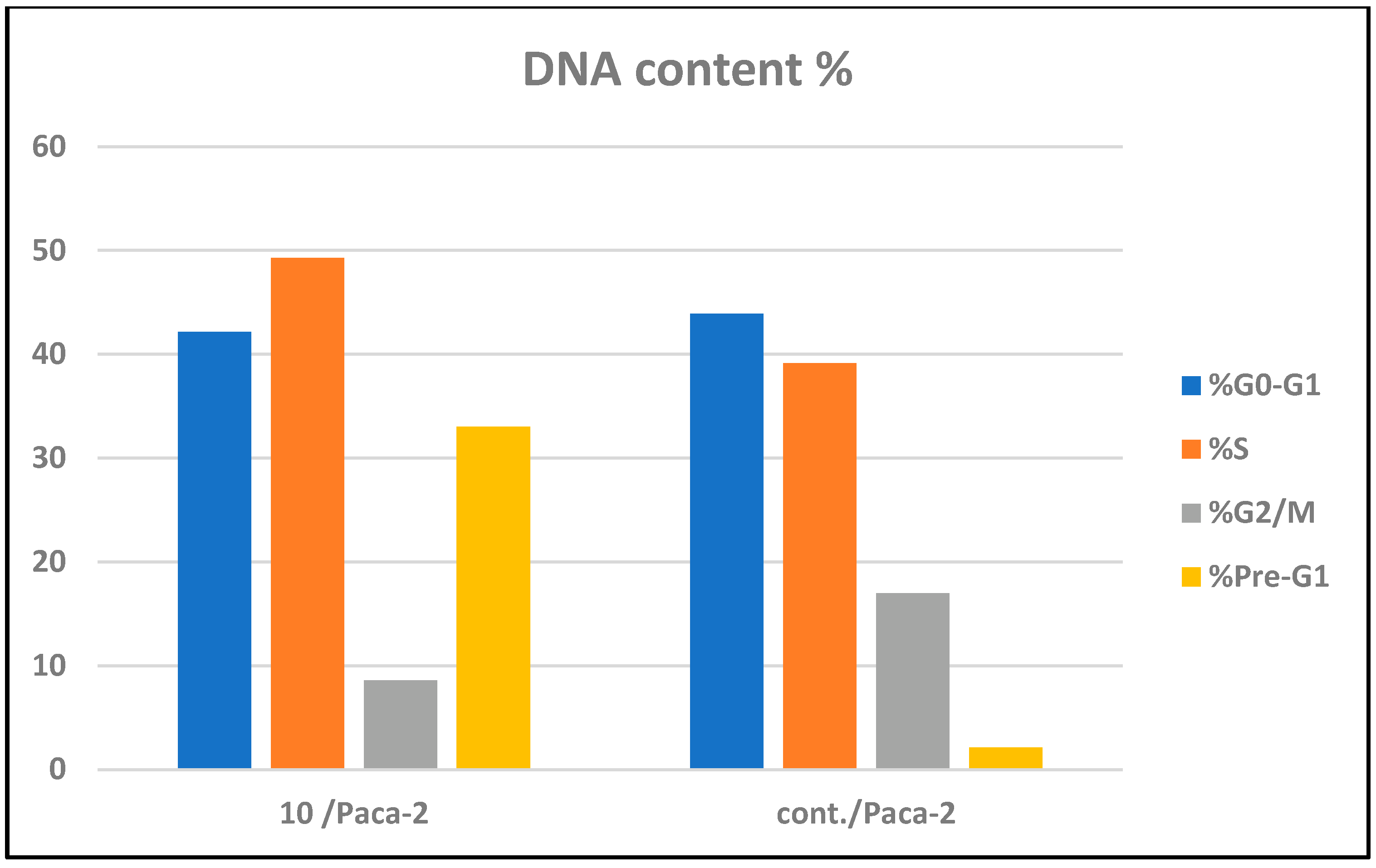
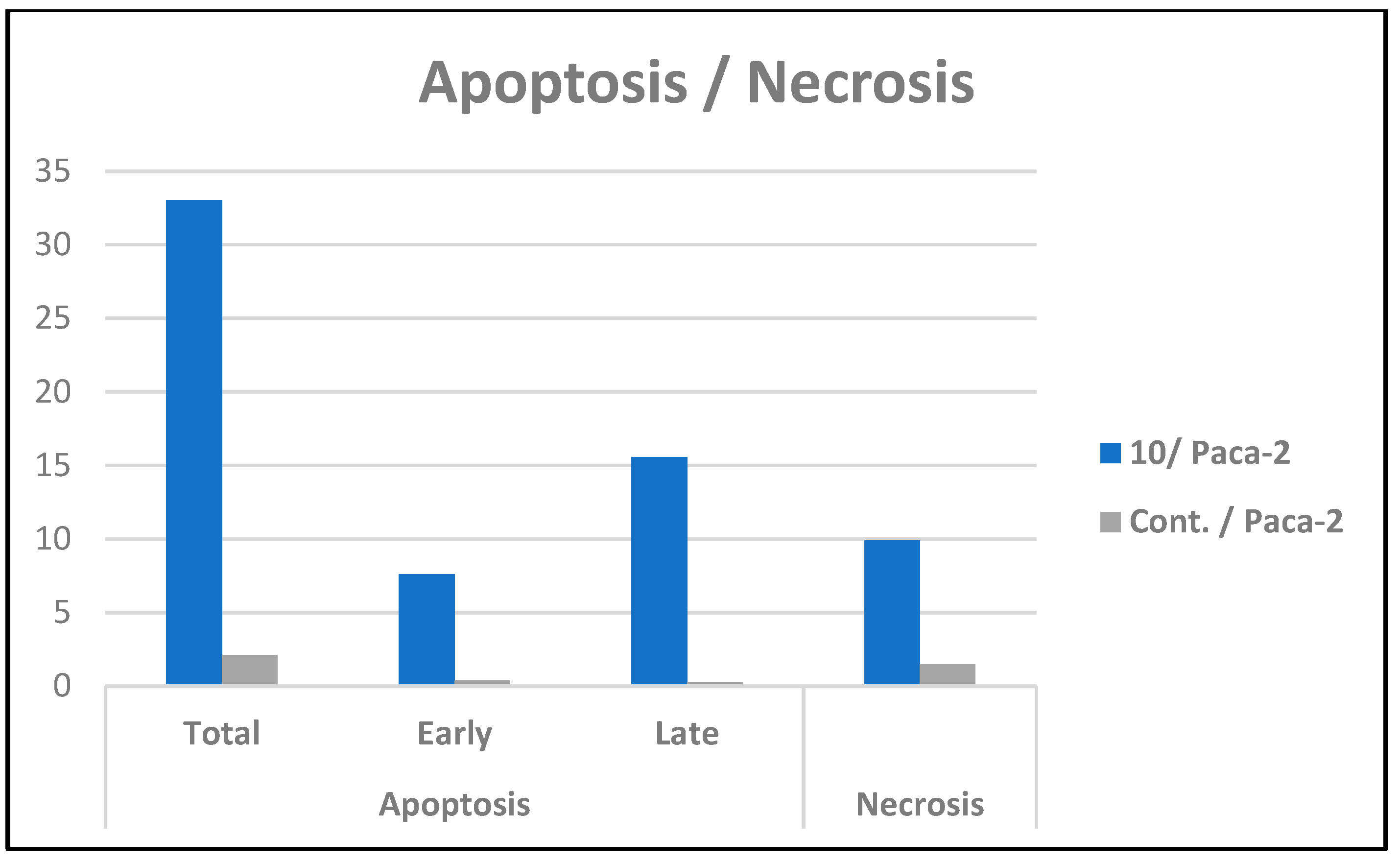
2.2.3. Cell Cycle Arrest and Apoptosis of Compound 10
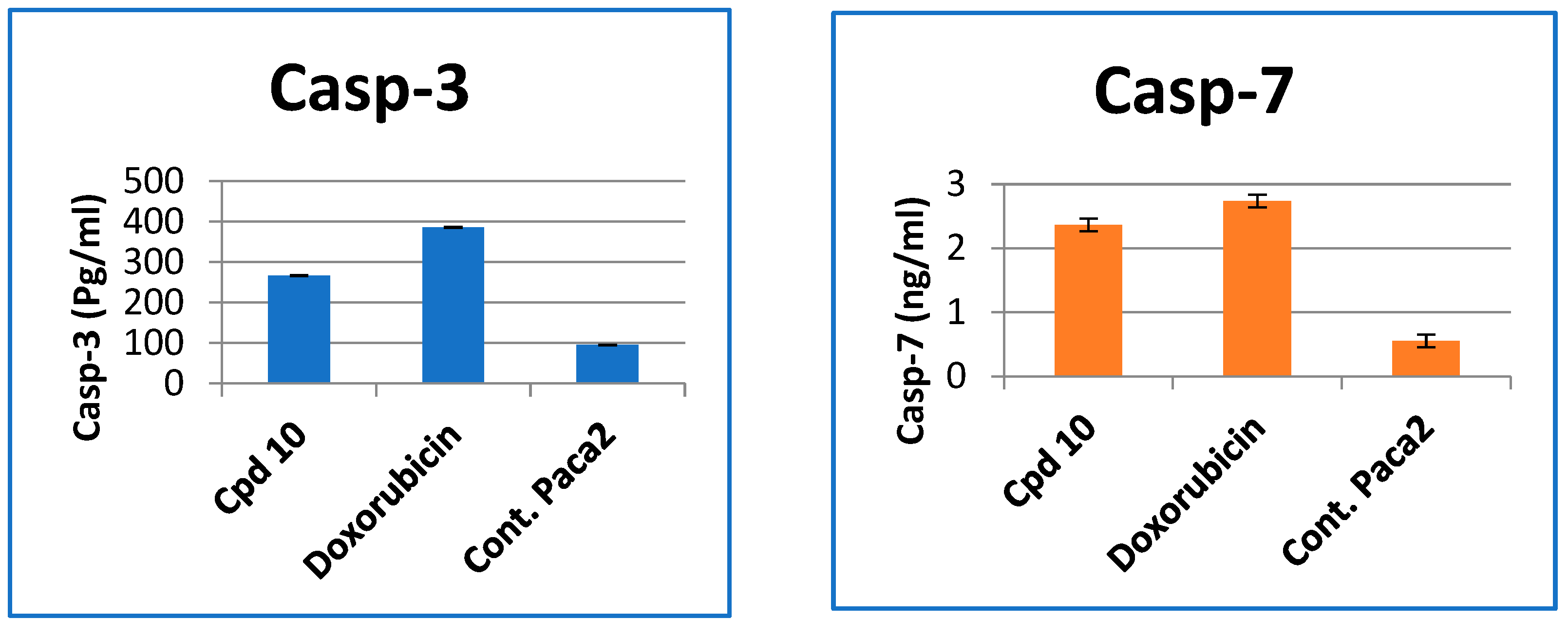
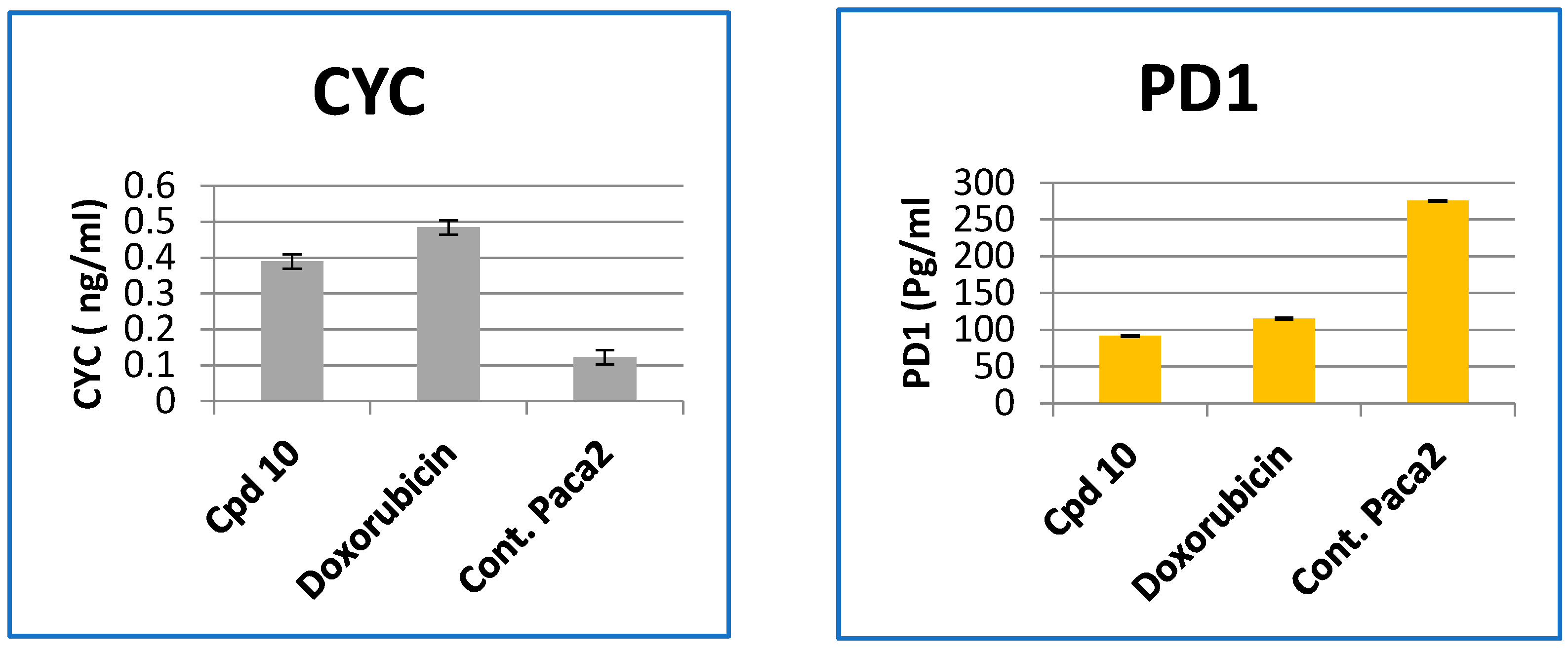
2.2.4. Effect of Compound 10 on the Levels of Caspase-3, 7, Cytochrome-c and PD1 Protein in Paca-2
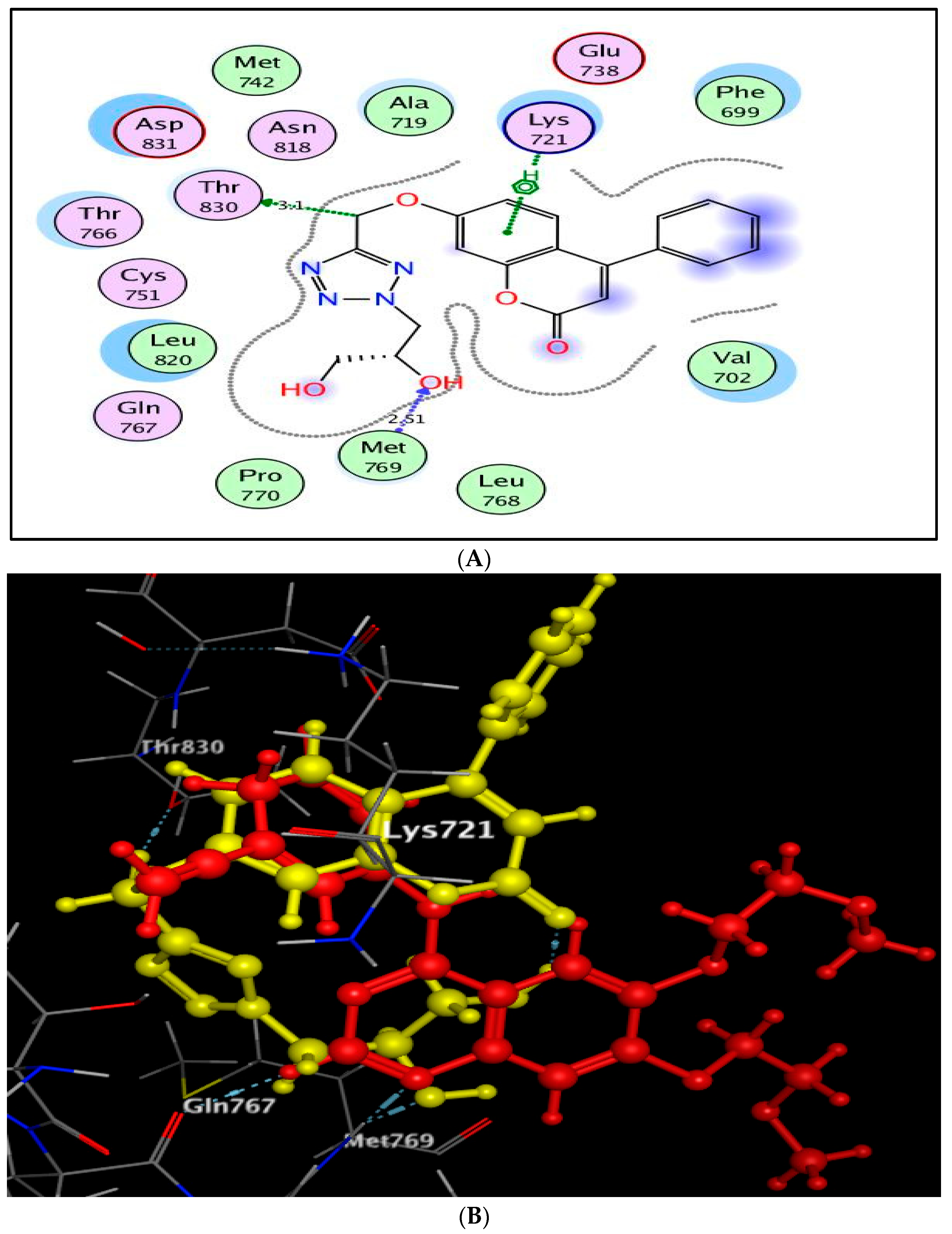
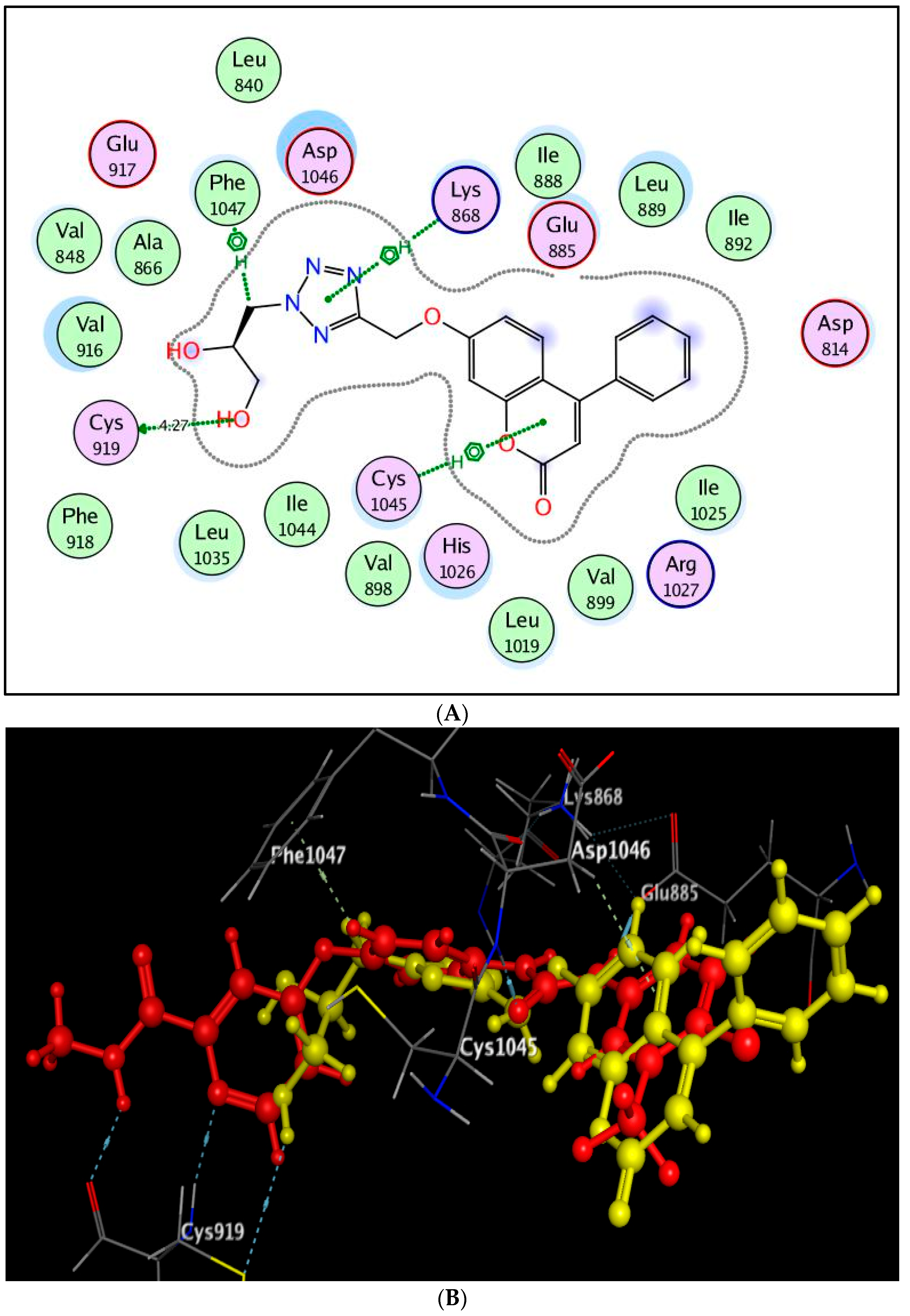
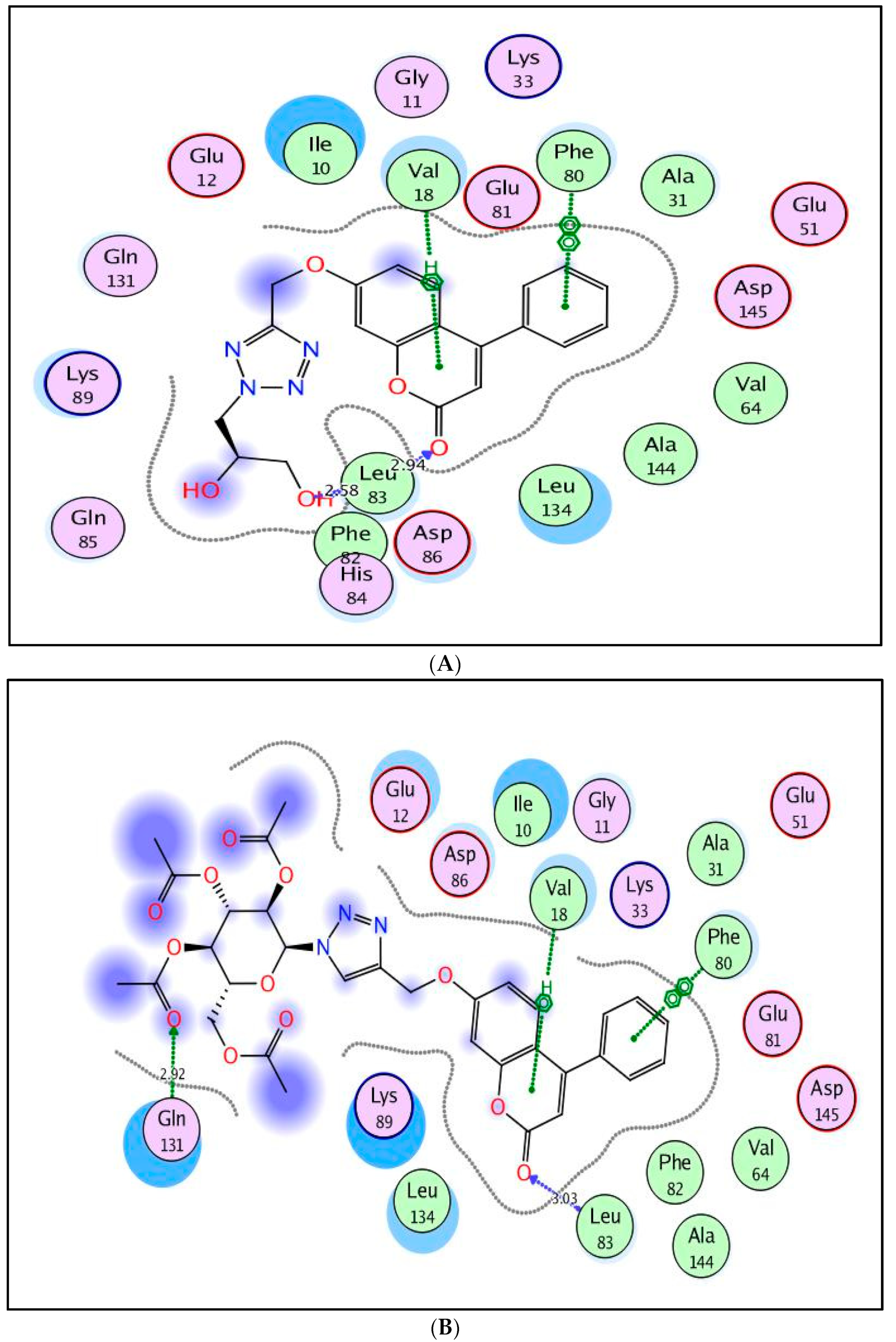
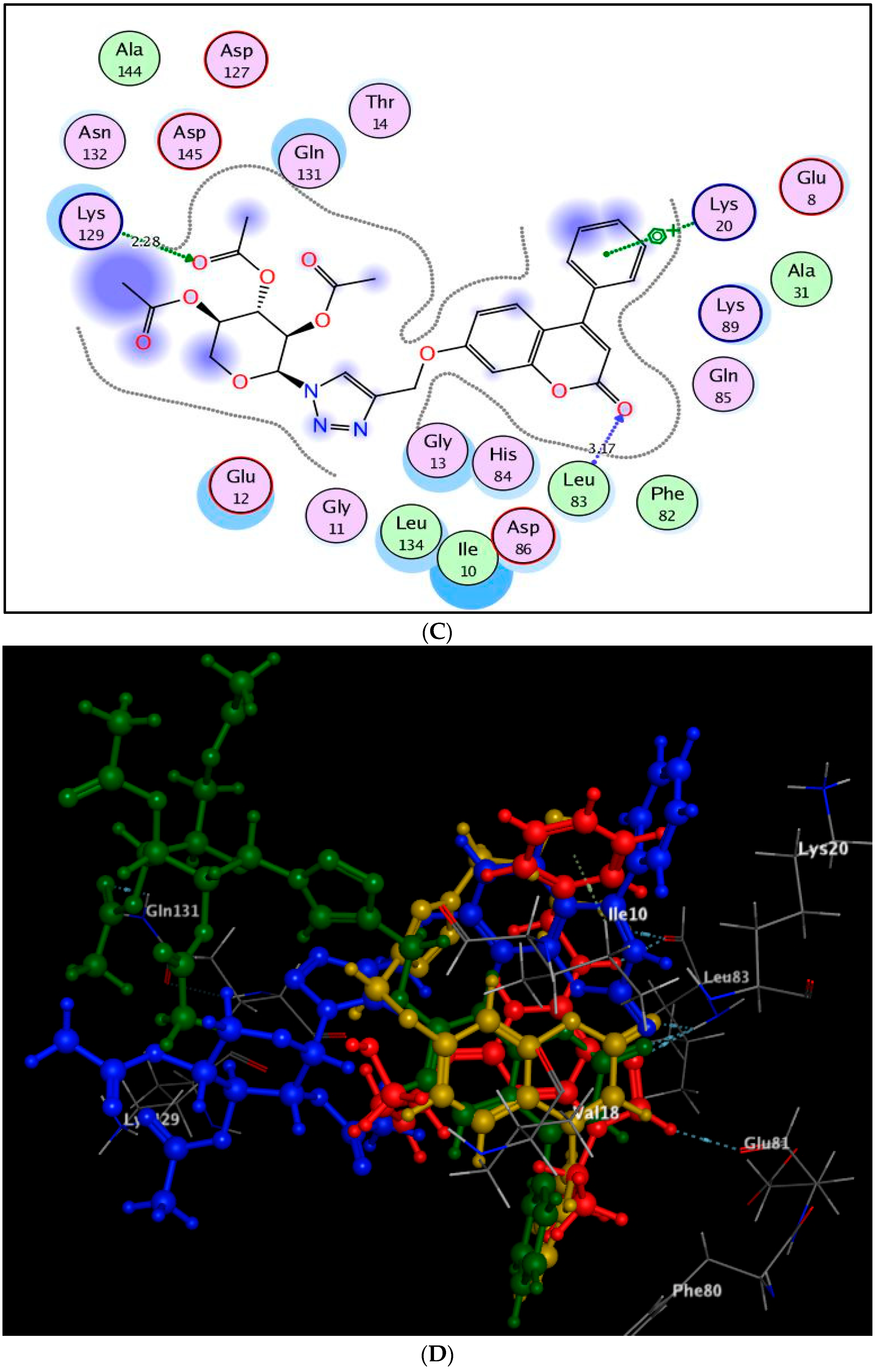
2.3. Molecular Docking Study
3. Experimental
3.1. Chemistry
3.2. Biological Evaluation
3.2.1. In Vitro Cytotoxicity
3.2.2. In Vitro Kinase Inhibitory Assessment against EGFR, VEGFR-2 and CDK-2
3.2.3. Cell Cycle Analysis and Apoptosis Detection of Compound 10
3.2.4. Effect of Compound 10 on the Levels of Caspase-3, 7, Cytochrome-c and PD1 Protein in Paca-2
3.3. Molecular Docking Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ferlay, J.; Laversanne, M.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Pinñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Tomorrow; International Agency for Research on Cancer: Lyon, France, 2020; Available online: https://gco.iarc.fr/tomorrow (accessed on 25 February 2020).
- Gerber, D.E. Targeted therapies: A new generation of cancer treatments. Am. Fam. Physician 2008, 77, 311–319. [Google Scholar]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef] [PubMed]
- El-Meguid, E.A.A.; Moustafa, G.O.; Awad, H.M.; Zaki, E.R.; Nossier, E.S. Novel benzothiazole hybrids targeting EGFR: Design, synthesis, biological evaluation and molecular docking studies. J. Mol. Struct. 2021, 1240, 130595. [Google Scholar] [CrossRef]
- Khattab, R.R.; Hassan, A.A.; Osman, D.A.A.; Abdel-Megeid, F.M.; Awad, H.M.; Nossier, E.S.; El-Sayed, W.A. Synthesis, anticancer activity and molecular docking of new triazolo [4,5-d] pyrimidines based thienopyrimidine system and their derived N-glycosides and thioglycosides. Nucleosides Nucleotides Nucleic Acids 2021, 40, 1090–1113. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar] [PubMed]
- Khattab, R.R.; Alshamari, A.K.; Hassan, A.A.; Elganzory, H.H.; El-Sayed, W.A.; Awad, H.M.; Nossier, E.S.; Hassan, N.A. Click chemistry based synthesis, cytotoxic activity and molecular docking of novel triazole-thienopyrimidine hybrid glycosides targeting EGFR. J. Enzym. Inhib. Med. Chem. 2021, 36, 504–516. [Google Scholar] [CrossRef]
- Othman, I.M.; Alamshany, Z.M.; Tashkandi, N.Y.; Gad-Elkareem, M.A.; Anwar, M.M.; Nossier, E.S. New pyrimidine and pyrazole-based compounds as potential EGFR inhibitors: Synthesis, anticancer, antimicrobial evaluation and computational studies. Bioorg. Chem. 2021, 114, 105078. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef]
- Otrock, Z.K.; Hatoum, H.A.; Musallam, K.M.; Awada, A.H.; Shamseddine, A.I. Is VEGF a predictive biomarker to an-ti-angiogenic therapy? Crit. Rev. Oncol. Hematol. 2011, 79, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Tugues, S.; Koch, S.; Gualandi, L.; Li, X.; Claesson-Welsh, L. Vascular endothelial growth factors and receptors: Anti-angiogenic therapy in the treatment of cancer. Mol. Asp. Med. 2011, 32, 88–111. [Google Scholar] [CrossRef]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Chohan, T.A.; Qian, H.; Pan, Y.; Chen, J.Z. Cyclin-dependent kinase-2 as a target for cancer therapy: Progress in the devel-opment of CDK2 inhibitors as anti-cancer agents. Curr. Med. Chem. 2015, 22, 237–263. [Google Scholar] [CrossRef] [PubMed]
- Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From Small molecules to peptide inhibitors. Cancers 2015, 7, 179–237. [Google Scholar] [CrossRef]
- Pathoor, R.; Bahulayan, D. MCR-click synthesis, molecular docking and cytotoxicity evaluation of a new series of indole–triazole–coumarin hybrid peptidomimetics. New J. Chem. 2018, 42, 6810–6816. [Google Scholar] [CrossRef]
- Gediya, L.K.; Njar, V.C. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2009, 4, 1099–1111. [Google Scholar] [CrossRef]
- Ghoneim, A.A.; El-Farargy, A.F.; Bakr, R.B. Design, synthesis, molecular docking of Novel substituted pyrimidinone derivatives as anticancer agents. Polycycl. Aromat. Compd. 2020, 1–17. [Google Scholar] [CrossRef]
- Hebishy, A.M.S.; Abdelfattah, M.S.; Elmorsy, A.; Elwahy, A.H.M. ZnO nanoparticles catalyzed synthesis ofbis- and poly(imidazoles) as potential anticancer agents. Synth. Commun. 2020, 50, 980–996. [Google Scholar] [CrossRef]
- Sheikhi-Mohammareh, S.; Shiri, A.; Maleki, E.H.; Matin, M.M.; Beyzaei, H.; Baranipour, P.; Oroojalian, F.; Memariani, T. Synthesis of various derivatives of [1,3] selenazolo [4,5-d] pyrimidine and exploitation of these heterocyclic systems as antibacterial, antifungal, and anticancer agents. ChemistrySelect 2020, 5, 10060–10066. [Google Scholar] [CrossRef]
- Scattolin, T.; Bortolamiol, E.; Rizzolio, F.; Demitri, N.; Visentin, F. Allyl palladium complexes bearing carbohydrate-based N-heterocyclic carbenes: Anticancer agents for selective and potent in vitro cytotoxicity. Appl. Organomet. Chem. 2020, 34, 5876. [Google Scholar] [CrossRef]
- Barraja, P.; Caracausi, L.; Diana, P.; Spano, V.; Montalbano, A.; Carbone, A.; Parrino, B.; Cirrincione, G. Synthesis and anti-proliferative activity of the ring system [1,2]Oxazolo[4,5-g] indole. Chem. Med. Chem. 2012, 7, 1901–1904. [Google Scholar] [CrossRef]
- El-Meguid, E.A.A.; El-Deen, E.M.M.; Moustafa, G.O.; Awad, H.M.; Nossier, E.S. Synthesis, anticancer evaluation and molecular docking of new benzothiazole scaffolds targeting FGFR-1. Bioorg. Chem. 2021, 119, 105504. [Google Scholar] [CrossRef] [PubMed]
- Spanò, V.; Rocca, R.; Barreca, M.; Giallombardo, D.; Montalbano, A.; Carbone, A.; Raimondi, M.V.; Gaudio, E.; Bortolozzi, R.; Bai, R.; et al. Pyrrolo [2′,3′:3,4] cyclohepta [1,2-d] [1,2] oxazoles, a new class of antimitotic agents active against multiple malignant cell types. J. Med. Chem. 2020, 63, 12023–12042. [Google Scholar] [CrossRef]
- Khan, M.S.; Agrawal, R.; Ubaidullah, M.; Hassan, I.; Tarannum, N. Design, synthesis and validation of anti-microbial coumarin derivatives: An efficient green approach. Heliyon 2019, 5, e02615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshibl, H.M.; Al-Abdullah, E.S.; Haiba, M.E.; Alkahtani, H.M.; Awad, G.E.; Mahmoud, A.H.; Ibrahim, B.M.; Bari, A.; Villinger, A. Synthesis and evaluation of new coumarin derivatives as antioxidant, antimicrobial, and anti-inflammatory agents. Molecules 2020, 25, 3251. [Google Scholar] [CrossRef]
- Bhattarai, N.; Kumbhar, A.A.; Pokharel, Y.R.; Yadav, P.N. Anticancer potential of coumarin and its derivatives. Mini-Rev. Med. Chem. 2021, 21, 2996–3029. [Google Scholar] [CrossRef]
- Vu, N.T.; Nguyen, D.T. Synthesis and antiproliferative activity of hybrid thiosemicarbazone derivatives bearing coumarin and D-galactose moieties with EGFR inhibitory activity and molecular docking study. Med. Chem. Res. 2021, 30, 1868–1885. [Google Scholar]
- Ahmed, E.Y.; Elserwy, W.S.; El-Mansy, M.F.; Serry, A.M.; Salem, A.M.; Abdou, A.M.; Abdelrahman, B.A.; Elsayed, K.H.; Elaziz, M.R.A. Angiokinase inhibition of VEGFR-2, PDGFR and FGFR and cell growth inhibition in lung cancer: Design, synthesis, biological evaluation and molecular docking of novel azaheterocyclic coumarin derivatives. Bioorg. Med. Chem. Lett. 2021, 48, 128258. [Google Scholar] [CrossRef]
- Mohamed, T.K.; Batran, R.Z.; Elseginy, S.A.; Ali, M.M.; Mahmoud, A.E. Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis. Bioorg. Chem. 2019, 85, 253–273. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Syam, Y.M.; El Kerdawy, A.M.; Abdelghany, T.M. New thiazol-hydrazono-coumarin hybrids targeting human cervical cancer cells: Synthesis, CDK2 inhibition, QSAR and molecular docking studies. Bioorg. Chem. 2019, 86, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Kassem, A.F.; Abbas, E.M.H.; El-Kady, D.S.; Awad, H.; El-Sayed, W.A. Design, synthesis and anticancer activity of new thiazole-tetrazole or triazole hybrid glycosides targeting CDK-2 via structure-based virtual screening. Mini-Rev. Med. Chem. 2019, 19, 933–948. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; Khalaf, H.; Mohamed, S.F.; Hussien, H.A.; Kutkat, O.; Amr, A.E. Synthesis and antiviral activity of 1,2,3-triazole glycosides based substituted pyridine via click cycloaddition. Russ. J. Gen. Chem. 2017, 87, 2444–2453. [Google Scholar] [CrossRef]
- Li, J.; Zheng, M.; Tang, W.; He, P.-L.; Zhu, W.; Li, T.; Zuo, J.-P.; Liu, H.; Jiang, H. Syntheses of triazole-modified zanamivir analogues via click chemistry and anti-AIV activities. Bioorg. Med. Chem. Lett. 2006, 16, 5009–5013. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Bourne, Y.; Kolb, H.C.; Radić, Z.; Sharpless, K.B.; Taylor, P.; Marchot, P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. USA 2004, 101, 1449–1454. [Google Scholar] [CrossRef] [Green Version]
- Popova, E.; Protas, A.; Trifonov, R. Tetrazole Derivatives as Promising Anticancer Agents. Anti-Cancer Agents Med. Chem. 2018, 17, 1856–1868. [Google Scholar] [CrossRef]
- Mohite, P.B.; Bhaskar, V.H. Potential pharmacological activities of tetrazoles in the new millennium. Int. J. PharmTech. Res. 2011, 3, 1557–1566. [Google Scholar]
- Szulczyk, D.; Dobrowolski, M.; Roszkowski, P.; Bielenica, A.; Stefańska, J.; Kolinski, M.; Kmiecik, S.; Jóźwiak, M.; Wrzosek, M.; Olejarz, W.; et al. Design and synthesis of novel 1H-tetrazol-5-amine based potent antimicrobial agents: DNA topoisomerase IV and gyrase affinity evaluation supported by molecular docking studies. Eur. J. Med. Chem. 2018, 156, 631–640. [Google Scholar] [CrossRef]
- Mendez, Y.; De Armas, G.; Pérez, I.; Rojas, T.; Valdes-Tresanco, M.E.; Izquierdo, M.; Del Rivero, M.A.; Álvarez-Ginarte, Y.M.; Valiente, P.A.; Soto, C.; et al. Discovery of potent and selective inhibitors of the Escherichia coli M1-aminopeptidase via multicomponent solid-phase synthesis of tetrazole-peptidomimetics. Eur. J. Med. Chem. 2019, 163, 481–499. [Google Scholar] [CrossRef]
- Kaushik, N.; Kumar, N.; Kumar, A.; Singh, U.K. Tetrazole, synthesis and biological activity. Immunol. Endocr. Metab. Agents Med. Chem. 2018, 18, 3–21. [Google Scholar] [CrossRef]
- Gonzalez-Lara, M.F.; Sifuentes-Osornio, J.; Ostrosky-Zeichner, L. Drugs in clinical development for fungal infections. Drugs 2017, 77, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chang, L.; Xu, Z.; Yan, X.F.; Ding, C.; Zhao, F.; Wu, X.; Feng, L.S. Recent advances of tetrazole derivatives as po-tential anti-tubercular and antimalarial agents. Eur. J. Med. Chem. 2019, 163, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, P.N.; Karad, S.C.; Raval, D.K. A review on diverse heterocyclic compounds as the privileged scaffolds in anti-malarial drug discovery. Eur. J. Med. Chem. 2018, 158, 917e936. [Google Scholar]
- Karabanovich, G.; Nemecek, J.; Valaskova, L.; Carazo, A.; Konecna, K.; Klara, J.; Stolarikova, J.; Hrabalek, A.; Roh, J.; Klimesova, V. S-Substituted 3,5- dinitrophenyl 1,3,4-oxadiazole-2-thiols and tetrazole-5-thios as highly efficient antitubercular agents. Eur. J. Med. Chem. 2017, 126, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Němeček, J.; Sychra, P.; Macháček, M.; Benková, M.; Karabanovich, G.; Konečná, K.; Kavková, V.; Stolaříková, J.; Hrabálek, A.; Vávrová, K.; et al. Structure-activity relationship studies on 3,5-dinitrophenyl tetrazoles as antitubercular agents. Eur. J. Med. Chem. 2017, 130, 419–432. [Google Scholar] [CrossRef]
- Kushwaha, P.; Fatima, S.; Upadhyay, A.; Gupta, S.; Bhagwati, S.; Baghel, T.; Siddiqi, M.I.; Nazir, A.; Sashidhara, K.V. Synthesis, biological evaluation and molecular dynamic simulations of novel Benzofuran-tetrazole derivatives as potential agents against Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2019, 29, 66–72. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. Tetrazole hybrids with potential anticancer activity. Eur. J. Med. Chem. 2019, 178, 341–351. [Google Scholar] [CrossRef]
- Fahad, M.A.; Hussein, H.E.; Mohamed, N.B.; Hanem, M.A.; El-Sayed, W.A. Synthesis and cytotoxic activity of new 1,3,4-thiadiazole thioglycosides and 1,2,3-triazolyl-1,3,4-thiadiazole n-glycosides. Molecules 2019, 24, 3738–3752. [Google Scholar]
- Flefel, E.M.; Tantawy, W.A.; El-Sayed, W.A.; Sayed, H.H.; Fathy, N.M. Synthesis and anticancer activity of new substituted pyrazoles and their derived 1,2,4-triazoles and sugar derivatives. J. Heterocycl. Chem. 2013, 50, 344–350. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; Hemat, S.K.; Dalia, A.A.O.; Hebat-Allah, S.A.; Mahmoud, M.A. Synthesis and anticancer activity of new pyrazolyl and oxadiazolyl glycosides based on theinopyrimidine nucleus and their acyclic analogs. Acta Pol. Pharm. 2017, 74, 1739–1751. [Google Scholar]
- Abbas, H.-A.S.; El Sayed, W.A.; Fathy, N. Synthesis and antitumor activity of new dihydropyridine thioglycosides and their corresponding dehydrogenated forms. Eur. J. Med. Chem. 2010, 45, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.A.A.; Nassar, I.F.; Shaban, A.K.; El-Kady, D.S.; Awad, H.; El Sayed, W.A. Synthesis, docking studies into CDK-2 and anticancer activity of new derivatives based pyrimidine scaffold and their derived glycosides. Mini-Rev. Med. Chem. 2019, 19, 1093–1110. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; Fathi, N.M.; Gad, W.A.; El-Ashry, E.S.H. Synthesis and antiviral evaluation of some 5-n-arylaminomethyl-2-glycosylsulphanyl-1,3,4-oxadiazoles and their analogs against hepatitis a and herpes simplex viruses. J. Carbohydr. Chem. 2008, 27, 357–372. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; El-Essawy, F.A.; Ali, O.M.; Nasr, B.S.; Abdalla, M.M.; Abdel-Rahman, A.A.-H. Synthesis and antiviral evaluation of new 2,5-disubstituted 1,3,4-oxadiazole derivatives and their acyclic nucleoside analogues. Mon. Chem.-Chem. Mon. 2010, 141, 1021–1028. [Google Scholar] [CrossRef]
- El-Sayed, W.A.; Ramiz, M.M.M.; Abdel-Rahman, A.A.-H. Anti-Hepatitis B virus activity of new N4-β-D-Glycoside pyrazolo[3,4-d] pyrimidine derivatives. Z. Nat. C 2009, 64, 323–328. [Google Scholar] [CrossRef]
- Hassan, A.A.; Khattab, R.R.; Wasfy, A.A.F.; Abuzeid, K.M.; Hassan, N.A. A convenient one-pot synthesis of thieno[2′,3′:4,5]pyrimido[1,2-b]-[1,2,4,5]tetrazines. J. Heterocycl. Chem. 2018, 55, 907–912. [Google Scholar] [CrossRef]
- Khattab, R.R.; Hassan, A.A.; Kutkat, O.; Abuzeid, K.M. Synthesis and antiviral activity of novel thieno [2,3-d] pyrimidine hydrazones and their c-nucleosides. Russ. J. Gen. Chem. 2019, 89, 1707–1717. [Google Scholar] [CrossRef]
- López-Rojas, P.; Janeczko, M.; Kubiński, K.; Amesty, Á.; Masłyk, M.; Estévez-Braun, A. Synthesis and antimicrobial activity of 4-substituted 1,2,3-triazole-coumarin derivatives. Molecules 2018, 23, 199. [Google Scholar] [CrossRef] [Green Version]
- Mounier, M.M.; Shehata, S.H.; Soliman, T.N. Anticancer activity of nanoencapsulated ginger in whey proteins against human tumor cell lines. Egypt. Pharm. J. 2020, 19, 87. [Google Scholar] [CrossRef]
- Nossier, E.S.; El-Hallouty, S.M.; Zaki, E.R. Synthesis, anticancer evaluation and molecular modeling of some substituted thiazolidinonyl and thiazolyl pyrazole derivatives. Int. J. Pharm. Sci. 2015, 7, 353–359. [Google Scholar]
- Nossier, E.; El-Karim, S.S.A.; Khalifa, N.M.; El-Sayed, A.S.; Hassan, E.S.I.; El-Hallouty, S.M. Kinase inhibitory activities and molecular docking of a novel series of anticancer pyrazole derivatives. Molecules 2018, 23, 3074. [Google Scholar] [CrossRef] [Green Version]
- Dawood, D.H.; Nossier, E.S.; Ali, M.M.; Mahmoud, A.E. Synthesis and molecular docking study of new pyrazole derivatives as potent anti-breast cancer agents targeting VEGFR-2 kinase. Bioorg. Chem. 2020, 101, 103916. [Google Scholar] [CrossRef]
- El-Sayed, A.A.; Nossier, E.S.; Almehizia, A.A.; Amr, A.E.-G.E. Design, synthesis, anticancer evaluation and molecular docking study of novel 2,4-dichlorophenoxymethyl-based derivatives linked to nitrogenous heterocyclic ring systems as potential CDK-2 inhibitors. J. Mol. Struct. 2021, 1247, 131285. [Google Scholar] [CrossRef]
- Hassan, A.S.; Moustafa, G.O.; Awad, H.M.; Nossier, E.S.; Mady, M.F. Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole–indole hybrids. ACS Omega 2021, 6, 12361–12374. [Google Scholar] [CrossRef]
- Moustafa, G.O.; Shalaby, A.; Naglah, A.M.; Mounier, M.M.; El-Sayed, H.; Anwar, M.M.; Nossier, E.S. Synthesis, characterization, in vitro anticancer potentiality, and antimicrobial activities of novel peptide–glycyrrhetinic-acid-based derivatives. Molecules 2021, 26, 4573. [Google Scholar] [CrossRef]
- Hashem, H.E.; Amr, A.E.-G.E.; Nossier, E.S.; Elsayed, E.A.; Azmy, E.M. Synthesis, antimicrobial activity and molecular docking of novel thiourea derivatives tagged with thiadiazole, imidazole and triazine moieties as potential dna gyrase and topoisomerase iv inhibitors. Molecules 2020, 25, 2766. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Mohamed, H.S.; Abdelhameed, M.F.; El-Galil, E.; Amr, A.; Almehizia, A.A.; Nossier, E.S. Design, synthesis and molecular docking of new pyrazole-thiazolidinones as potent anti-inflammatory and analgesic agents with TNF-α inhibitory activity. Bioorg. Chem. 2021, 111, 104827. [Google Scholar] [CrossRef]
- El-Deen, E.M.M.; El-Meguid, E.A.A.; Hasabelnaby, S.; Karam, E.A.; Nossier, E.S.; El-Deen, E.M.; El-Meguid, E.A. Synthesis, docking studies, and in vitro evaluation of some novel thienopyridines and fused thienopyridine-quinolines as antibacterial agents and dna gyrase inhibitors. Molecules 2019, 24, 3650. [Google Scholar] [CrossRef] [Green Version]
- Hawata, M.A.; El-Sayed, W.A.; Nossier, E.S.; Abdel-Rahman, A.A.H. Synthesis and Cytotoxic Activity of New Pyrimido [1,2-c] quinazolines [1,2,4] triazolo [4,3-c] quinazolines and (quinazolin-4-yl)-1H-pyrazoles Hybrids. Biointerface Res. Appl. Chem. 2022, 12, 5217–5233. [Google Scholar]
- El-serwy, W.S.; Mohamed, H.S.; El-serwy, W.S.; Mohamed, N.A.; Kassem, E.M.; Mahmoud, K.; Nossier, E.S. Thiopyrimidine-5-carbonitrile Derivatives as VEGFR-2 Inhibitors: Synthesis, Anticancer Evaluation, Molecular Docking, ADME Predictions and QSAR Studies. ChemistrySelect 2020, 5, 15243–15253. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | % Inhibition | ||||
|---|---|---|---|---|---|
| Paca-2 | Mel-501 | PC-3 | A-375 | Caco-2 | |
| 1 | 79 | 96.2 | 55.7 | 95 | 40.5 |
| 2 | 44.1 | 35.6 | 10.5 | 6.5 | - |
| 3 | 55.6 | 48.4 | 12.2 | 55.3 | - |
| 5 | 56.9 | 68.4 | 15 | 23.1 | - |
| 7 | 21.3 | 22.3 | 0 | 13.8 | - |
| 6 | 72.1 | 71 | 9.1 | 45.8 | - |
| 8 | 96.5 | 98.1 | 98.1 | 95 | 97 |
| 9 | 94.7 | 97.7 | 95.9 | 84.1 | 99.5 |
| 10 | 99.9 | 95.8 | 99.2 | 98.2 | 99.6 |
| 11 | 58.1 | 44 | 16.7 | 9.1 | 0 |
| 13 | 97.6 | 98.5 | 95.6 | 96.9 | 99.6 |
| 14 | 94.9 | 77.1 | 80.2 | 96.8 | 89.8 |
| 15 | 95 | 97.8 | 98.2 | 94.7 | 99.3 |
| 16 | 96.9 | 99.1 | 95.8 | 97.5 | 99 |
| 17 | 96.3 | 98.6 | 97.3 | 98.5 | 95.6 |
| 18 | 96.9 | 98.5 | 91.4 | 97.9 | 99.4 |
| Compound No. | IC50 (Mean ± SEM) (μM) | ||||
|---|---|---|---|---|---|
| Paca-2 | Mel-501 | PC-3 | A-375 | Caco-2 | |
| 1 | 62.3 ± 1.3 | 28.6 ± 0.4 | --- | 12.9 ± 0.6 | --- |
| 5 | --- | 72.1 ± 1.2 | --- | --- | --- |
| 6 | 60 ± 1.4 | 80.4 ± 2.1 | --- | --- | --- |
| 8 | 23.8 ± 0.9 | 19.4 ± 0.4 | 48.2 ± 0.7 | 13.9 ± 0.2 | 52.5 ± 1.3 |
| 9 | 28.3 ± 0.7 | 30.2 ± 0.5 | 47.4 ± 1.4 | 15 ± 0.04 | 38.1 ± 0.4 |
| 10 | 10.7 ± 0.4 | 8.8 ± 0.2 | 25.4 ± 0.5 | 10.9 ± 0.1 | 31.9 ± 0.7 |
| 13 | 14.6 ± 1.1 | 16.7 ± 0.3 | 22.3 ± 0.6 | 14.5 ± 0.2 | 40.5 ± 0.9 |
| 14 | 33.4 ± 0.6 | 65.1 ± 1.2 | 51.3 ± 1.6 | 12.4 ± 0.3 | 70.2 ± 1.4 |
| 15 | 16.9 ± 0.9 | 4.1 ± 0.3 | 12.5 ± 0.9 | 9.9 ± 1 | 20.4 ± 0.6 |
| 16 | 23.8 ± 1.1 | 26.5 ± 0.7 | 36.4 ± 1.1 | 16.3 ± 0.6 | 48.1 ± 0.4 |
| 17 | 33.3 ± 0.5 | 35.8 ± 0.7 | 41 ± 2.1 | 39.6 ± 0.5 | 48.1 ± 0.8 |
| 18 | 36.4 ± 1.2 | 10 ± 0.1 | 37.6 ± 0.4 | 8.5 ± 0.3 | 61.9 ± 1.3 |
| Doxorubicin | 19.4 ± 0.5 | 3.5 ± 0.3 | 6.8 ± 0.2 | 4.5 ± 0.07 | 3.7 ± 0.2 |
| Compound No. | IC50 (Mean ± SEM) (µM) | ||
|---|---|---|---|
| EGFR | VEGFR-2 | CDK-2/Cyclin A2 | |
| Erlotinib | 0.13 ± 0.020 | - | - |
| Sorafenib | - | 1.050 ± 0.20 | - |
| Roscovitine | - | - | 0.37 ± 0.22 |
| 10 | 0.09 ± 0.21 | 0.86 ± 0.25 | 0.19 ± 0.05 |
| 13 | 66 ± 0.01 | 45 ± 0.10 | 0.50± 0.11 |
| 15 | 53 ± 0.15 | 40 ± 0.05 | 0.48± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sayed, W.A.; Alminderej, F.M.; Mounier, M.M.; Nossier, E.S.; Saleh, S.M.; Kassem, A.F. Novel 1,2,3-Triazole-Coumarin Hybrid Glycosides and Their Tetrazolyl Analogues: Design, Anticancer Evaluation and Molecular Docking Targeting EGFR, VEGFR-2 and CDK-2. Molecules 2022, 27, 2047. https://doi.org/10.3390/molecules27072047
El-Sayed WA, Alminderej FM, Mounier MM, Nossier ES, Saleh SM, Kassem AF. Novel 1,2,3-Triazole-Coumarin Hybrid Glycosides and Their Tetrazolyl Analogues: Design, Anticancer Evaluation and Molecular Docking Targeting EGFR, VEGFR-2 and CDK-2. Molecules. 2022; 27(7):2047. https://doi.org/10.3390/molecules27072047
Chicago/Turabian StyleEl-Sayed, Wael A., Fahad M. Alminderej, Marwa M. Mounier, Eman S. Nossier, Sayed M. Saleh, and Asmaa F. Kassem. 2022. "Novel 1,2,3-Triazole-Coumarin Hybrid Glycosides and Their Tetrazolyl Analogues: Design, Anticancer Evaluation and Molecular Docking Targeting EGFR, VEGFR-2 and CDK-2" Molecules 27, no. 7: 2047. https://doi.org/10.3390/molecules27072047
APA StyleEl-Sayed, W. A., Alminderej, F. M., Mounier, M. M., Nossier, E. S., Saleh, S. M., & Kassem, A. F. (2022). Novel 1,2,3-Triazole-Coumarin Hybrid Glycosides and Their Tetrazolyl Analogues: Design, Anticancer Evaluation and Molecular Docking Targeting EGFR, VEGFR-2 and CDK-2. Molecules, 27(7), 2047. https://doi.org/10.3390/molecules27072047