A Recent Ten-Year Perspective: Bile Acid Metabolism and Signaling

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Bile Acids in the Intestine
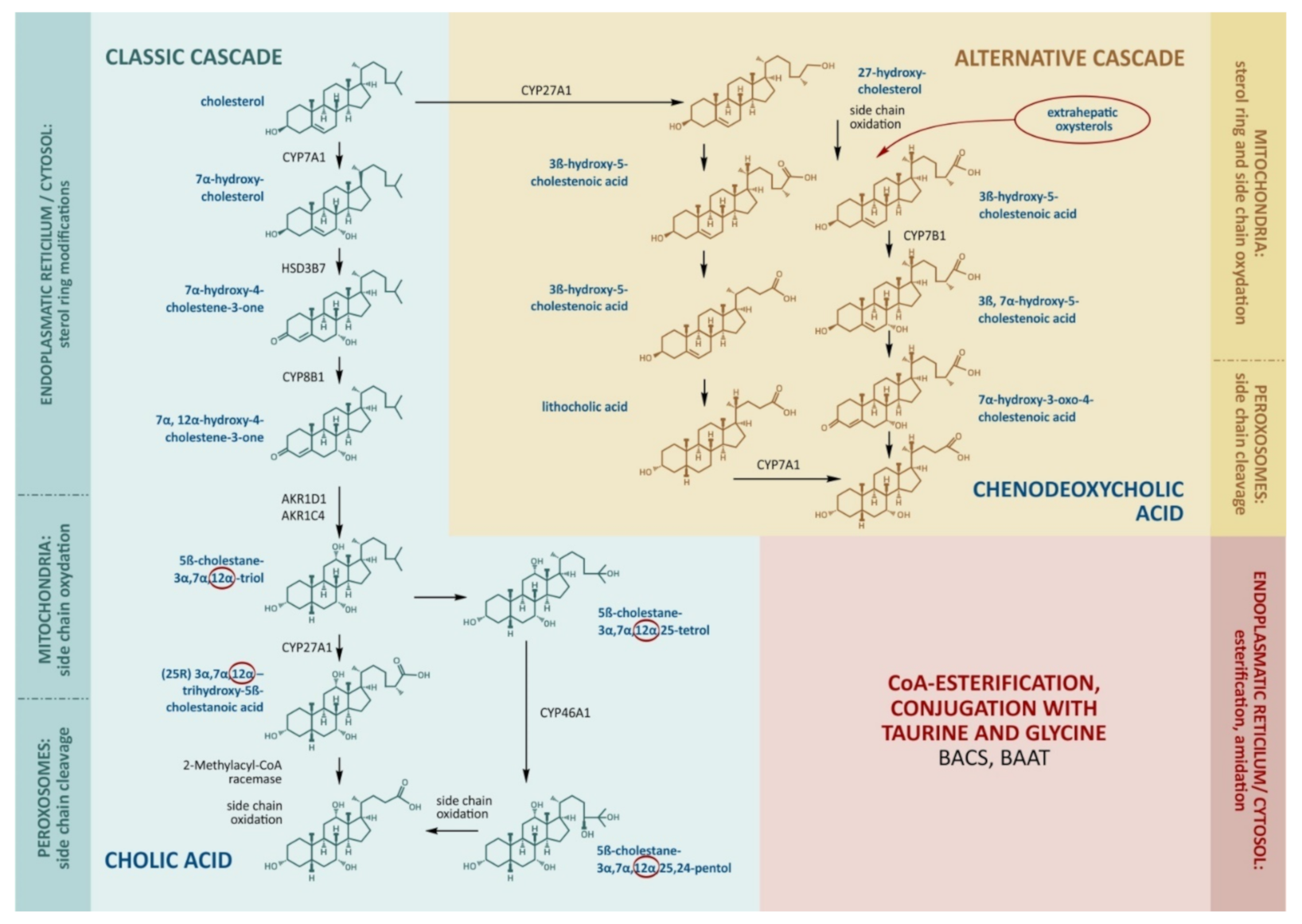
2.1. Bile Acid Synthesis in the Liver
2.2. Micelle Formation
2.3. Absorption
2.4. Microbial Transformations
2.5. Bile Acids in the Gallbladder and Extrahepatic Bile Ducts
3. Enterohepatic Circulation and Bile Acid Cell Transport
4. The Regulation of Bile Acid Synthesis
4.1. The Mechanism of the Regulation of the Classical Cascade Involving the Farnesoid X Receptor
4.2. Biliary Tract in the Regulation of Bile Acid Synthesis
4.3. Other Factors Affecting the Regulation of Bile Acids
5. Extensive Physiological Role of Bile Acids
5.1. Bile Acid Receptors
5.2. Bile Acids in Cholesterol Homeostasis
5.3. Bile Acids in the Brain
6. The Action of Bile Acids on Cell Membranes
7. Mutations of Genes Controlling the Cellular Metabolism and Transport of Bile Acids
7.1. Mutation in Genes Encoding Enzymes of the Classical and Alternative Cascades of Bile Acid Synthesis
7.2. Mutation of Genes Encoding Enzymes for the Esterification and Amidation of Bile Acids
7.3. Bile Acid Transporter Defects
8. Effects of Bile Acid Sequestrants
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arrese, M.; Accatino, L. From blood to bile: Recent advances in hepatobiliary transport. Ann. Hepatol. 2002, 1, 64–71. [Google Scholar] [CrossRef]
- Boyer, J.L. Bile Formation and Secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef]
- Arrese, M.; Trauner, M. Molecular aspects of bile formation and cholestasis. Trends Mol. Med. 2003, 9, 558–564. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J. Bile Acids as Metabolic Regulators and Nutrient Sensors. Annu. Rev. Nutr. 2019, 39, 175–200. [Google Scholar] [CrossRef]
- Stofan, M.; Guo, G.L. Bile Acids and FXR: Novel Targets for Liver Diseases. Front. Med. 2020, 7, 544. [Google Scholar] [CrossRef] [PubMed]
- Tarbeeva, S.; Lyamtseva, E.; Lisitsa, A.; Kozlova, A.; Ponomarenko, E.; Ilgisonis, E. ScanBious: Survey for Obesity Genes Using PubMed Abstracts and DisGeNET. J. Pers. Med. 2021, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Ilgisonis, E.; Lisitsa, A.; Kudryavtseva, V.; Ponomarenko, E. Creation of Individual Scientific Concept-Centered Semantic Maps Based on Automated Text-Mining Analysis of PubMed. Adv. Bioinform. 2018, 2018, 4625394. [Google Scholar] [CrossRef]
- Namazova-Baranova, L.S.; Polyakova, S.I. Violation of Primary Bile Acids. Russ. Pediatr. J. 2015, 6, 35–40. [Google Scholar]
- Šarenac, T.M.; Mikov, M. Bile Acid Synthesis: From Nature to the Chemical Modification and Synthesis and Their Applications as Drugs and Nutrients. Front. Pharmacol. 2018, 9, 939. [Google Scholar] [CrossRef]
- Duane, W.C.; Javitt, N.B. 27-Hydroxycholesterol: Production rates in normal human subjects. J. Lipid Res. 1999, 40, 1194–1199. [Google Scholar] [CrossRef]
- Chiang, J.Y. Hepatocyte nuclear factor 4α regulation of bile acid and drug metabolism. Expert Opin. Drug Metab. Toxicol. 2009, 5, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Duane, W.C.; Pooler, P.A.; Hamilton, J.N. Bile acid synthesis in man. In vivo activity of the 25-hydroxylation pathway. J. Clin. Investig. 1988, 82, 82–85. [Google Scholar] [CrossRef] [PubMed]
- del Castillo-Olivares, A.; Gil, G. Differential Effects of Sterol Regulatory Binding Proteins 1 and 2 on Sterol 12α-Hydroxylase. J. Biol. Chem. 2002, 277, 6750–6757. [Google Scholar] [CrossRef]
- Adachi, J.; Kudo, R.; Asano, M.; Ueno, Y.; Hunter, R.; Rajendram, R.; Martin, C.; Preedy, V.R. Skeletal muscle and liver oxysterols during fasting and alcohol exposure. Metabolism 2006, 55, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Biasi, F.; Leonarduzzi, G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013, 1, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- Russell, D.W. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef]
- Kevresan, S.; Kuhajda, K.; Kandrac, J.; Fawcett, J.P.; Mikov, M. Biosynthesis of bile acids in mammalian liver. Eur. J. Drug Metab. Pharmacokinet. 2006, 31, 145–156. [Google Scholar] [CrossRef]
- Setchell, K.D.R.; Schwarz, M.; O'Connell, N.C.; Lund, E.G.; Davis, D.L.; Lathe, R.; Thompson, H.R.; Tyson, R.W.; Sokol, R.J.; Russell, D.W. Identification of a new inborn error in bile acid synthesis: Mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J. Clin. Investig. 1998, 102, 1690–1703. [Google Scholar] [CrossRef]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef]
- Wilson, F.A.; Sallee, V.L.; Dietschy, J.M. Unstirred Water Layers in Intestine: Rate Determinant of Fatty Acid Absorption from Micellar Solutions. Science 1971, 174, 1031–1033. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.J.; Marin, J.J.; Antelo, A.; Vazquez-Tato, J. Bile acids: Chemistry, physiology, and pathophysiology. World J. Gastroenterol. 2009, 15, 804–816. [Google Scholar] [CrossRef]
- Carey, M.C. Measurement of the physical-chemical properties of bile salt solutions. In Proceedings of the International Symposium, Cortina d’Ampezzo, Italy, 17–20 March 1982; pp. 19–56. [Google Scholar] [CrossRef]
- Boron, W.; Boulpaep, E. Medical Physiology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2016; p. 1312. [Google Scholar]
- Goncalves, A.; Gontero, B.; Nowicki, M.; Margier, M.; Masset, G.; Amiot, M.J.; Reboul, E. Micellar lipid composition affects micelle interaction with class B scavenger receptor extracellular loops. J. Lipid Res. 2015, 56, 1123–1133. [Google Scholar] [CrossRef]
- Dietschy, J.M. Mechanisms for the intestinal absorption of bile acids. J. Lipid Res. 1968, 9, 297–309. [Google Scholar] [CrossRef]
- Aldini, R.; Montagnani, M.; Roda, A.; Hrelia, S.; Biagi, P.; Roda, E. Intestinal absorption of bile acids in the rabbit: Different transport rates in jejunum and ileum. Gastroenterology 1996, 110, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.N.; Krenz, H.K.; Modlin, I.M.; Ballantyne, G.H. Bile salt inhibition of motility in the isolated perfused rabbit terminal ileum. Gut 1993, 34, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Bourgin, M.; Kriaa, A.; Mkaouar, H.; Mariaule, V.; Jablaoui, A.; Maguin, E.; Rhimi, M. Bile Salt Hydrolases: At the Crossroads of Microbiota and Human Health. Microorganisms 2021, 9, 1122. [Google Scholar] [CrossRef]
- Naumann, S.; Haller, D.; Eisner, P.; Schweiggert-Weisz, U. Mechanisms of Interactions between Bile Acids and Plant Compounds—A Review. Int. J. Mol. Sci. 2020, 21, 6495. [Google Scholar] [CrossRef] [PubMed]
- Gérard, P. Metabolism of Cholesterol and Bile Acids by the Gut Microbiota. Pathogens 2013, 3, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Chatterjee, I.; Lu, R.; Zhang, Y.; Zhang, J.; Dai, Y.; Xia, Y.; Sun, J. Vitamin D receptor promotes healthy microbial metabolites and microbiome. Sci. Rep. 2020, 10, 7340. [Google Scholar] [CrossRef] [PubMed]
- Ahlberg, J.; Angelin, B.; Björkhem, I.; Einarsson, K. Individual Bile Acids in Portal Venous and Systemic Blood Serum of Fasting Man. Gastroenterology 1977, 73, 1377–1382. [Google Scholar] [CrossRef]
- Meier-Abt, F.; Mokrab, Y.; Mizuguchi, K. Organic Anion Transporting Polypeptides of the OATP/SLCO Superfamily: Identification of New Members in Nonmammalian Species, Comparative Modeling and a Potential Transport Mode. J. Membr. Biol. 2006, 208, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Kriegermeier, A.; Green, R. Pediatric Cholestatic Liver Disease: Review of Bile Acid Metabolism and Discussion of Current and Emerging Therapies. Front. Med. 2020, 7, 149. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Sips, F.L.P.; Eggink, H.M.; Hilbers, P.A.J.; Soeters, M.R.; Groen, A.K.; Van Riel, N.A.W. In Silico Analysis Identifies Intestinal Transit as a Key Determinant of Systemic Bile Acid Metabolism. Front. Physiol. 2018, 9, 631. [Google Scholar] [CrossRef] [PubMed]
- Jani, M.; Beéry, E.; Heslop, T.; Tóth, B.; Jagota, B.; Kis, E.; Park, B.K.; Krajcsi, P.; Weaver, R.J. Kinetic characterization of bile salt transport by human NTCP (SLC10A1). Toxicol. Vitr. 2018, 46, 189–193. [Google Scholar] [CrossRef]
- Sticova, E.; Jirsa, M.; Pawlowska, J. New Insights in Genetic Cholestasis: From Molecular Mechanisms to Clinical Implications. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2313675. [Google Scholar] [CrossRef]
- Stieger, B. The Role of the Sodium-Taurocholate Cotransporting Polypeptide (NTCP) and of the Bile Salt Export Pump (BSEP) in Physiology and Pathophysiology of Bile Formation. In Drug Transporters; Fromm, M., Kim, R., Eds.; Springer: Berlin, Germany, 2010; pp. 205–259. [Google Scholar]
- Mita, S.; Suzuki, H.; Akita, H.; Hayashi, H.; Onuki, R.; Hofmann, A.F.; Sugiyama, Y.; Nozaki, S. Vectorial transport of unconjugated and conjugated bile salts by monolayers of LLC-PK1 cells doubly transfected with human NTCP and BSEP or with rat Ntcp and Bsep. Am. J. Physiol. Liver Physiol. 2006, 290, G550–G556. [Google Scholar] [CrossRef]
- Russel, F.G.M.; Koenderink, J.B.; Masereeuw, R. Multidrug resistance protein 4 (MRP4/ABCC4): A versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol. Sci. 2008, 29, 200–207. [Google Scholar] [CrossRef]
- Wen, J.; Luo, J.; Huang, W.-H.; Tang, J.; Zhou, H.; Zhang, W. The Pharmacological and Physiological Role of Multidrug-Resistant Protein 4. J. Pharmacol. Exp. Ther. 2015, 354, 358–375. [Google Scholar] [CrossRef] [PubMed]
- Denk, G.U.; Soroka, C.J.; Takeyama, Y.; Chen, W.-S.; Schuetz, J.D.; Boyer, J.L. Multidrug resistance-associated protein 4 is up-regulated in liver but down-regulated in kidney in obstructive cholestasis in the rat. J. Hepatol. 2004, 40, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pineda, S.I.; Baylón-Pacheco, L.; Espíritu-Gordillo, P.; Tsutsumi, V.; Rosales-Encina, J.L. Effect of bile acids on the expression of MRP3 and MRP4: An In vitro study in HepG2 cell line. Ann. Hepatol. 2021, 24, 100325. [Google Scholar] [CrossRef]
- Wang, W.; Kim, M.T.; Sedykh, A.; Zhu, H. Developing Enhanced Blood–Brain Barrier Permeability Models: Integrating External Bio-Assay Data in QSAR Modeling. Pharm. Res. 2015, 32, 3055–3065. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Corlianò, M.; Singaraja, R.R. Bile Acids: A Communication Channel in the Gut-Brain Axis. NeuroMol. Med. 2021, 23, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chiang, J. Regulation of human sterol 27-hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4α (HNF4α). Gene 2003, 313, 71–82. [Google Scholar] [CrossRef]
- Zhang, M.; Chiang, J. Transcriptional Regulation of the Human Sterol 12α-Hydroxylase Gene (CYP8B1). J. Biol. Chem. 2001, 276, 41690–41699. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef]
- Lundåsen, T.; Gälman, C.; Angelin, B.; Rudling, M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 2006, 260, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Walters, J.R.; Tasleem, A.M.; Omer, O.S.; Brydon, W.G.; Dew, T.; le Roux, C.W. A New Mechanism for Bile Acid Diarrhea: Defective Feedback Inhibition of Bile Acid Biosynthesis. Clin. Gastroenterol. Hepatol. 2009, 7, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Kim, Y.-C.; Byun, S.; Kim, N.-H.; Seok, S.; Suino-Powell, K.; Xu, H.E.; Kemper, B.; Kemper, J.K. FXR Primes the Liver for Intestinal FGF15 Signaling by Transient Induction of β-Klotho. Mol. Endocrinol. 2016, 30, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Pandak, W.M.; Heuman, D.M.; Hylemon, P.B.; Chiang, J.Y.; Vlahcevic, Z.R. Failure of intravenous infusion of taurocholate to down-regulate cholesterol 7α-hydroxylase in rats with biliary fistulas. Gastroenterology 1995, 108, 533–544. [Google Scholar] [CrossRef]
- Wong, B.S.; Camilleri, M.; Carlson, P.J.; Guicciardi, M.E.; Burton, D.; McKinzie, S.; Rao, A.S.; Zinsmeister, A.R.; Gores, G.J. A Klothoβ Variant Mediates Protein Stability and Associates with Colon Transit in Irritable Bowel Syndrome with Diarrhea. Gastroenterology 2011, 140, 1934–1942. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Schaap, F.G.; van der Gaag, N.A.; Gouma, D.J.; Jansen, P.L.M. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology 2008, 49, 1228–1235. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y.L. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Nadolny, C.; Dong, X. Liver receptor homolog-1 (LRH-1): A potential therapeutic target for cancer. Cancer Biol. Ther. 2015, 16, 997–1004. [Google Scholar] [CrossRef]
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.; Karpen, S.J. The Orphan Nuclear Receptor, shp, Mediates Bile Acid-Induced Inhibition of the Rat Bile Acid Transporter, ntcp. Gastroenterology 2001, 121, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo-Olivares, A.; Gil, G. Suppression of sterol 12α-hydroxylase transcription by the short heterodimer partner: Insights into the repression mechanism. Nucleic Acids Res. 2001, 29, 4035–4042. [Google Scholar] [CrossRef] [PubMed]
- Pircher, P.C.; Kitto, J.L.; Petrowski, M.L.; Tangirala, R.K.; Bischoff, E.D.; Schulman, I.G.; Westin, S.K. Farnesoid X Receptor Regulates Bile Acid-Amino Acid Conjugation. J. Biol. Chem. 2003, 278, 27703–27711. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.-L.; Zhao, A.; Yu, J.; Huang, L.; de Pedro, N.; Peláez, F.; Wright, S.D.; Cui, J. The Farnesoid X Receptor Controls Gene Expression in a Ligand- and Promoter-selective Fashion. J. Biol. Chem. 2004, 279, 8856–8861. [Google Scholar] [CrossRef]
- Ren, S.; Hylemon, P.B.; Marques, D.; Gurley, E.; Bodhan, P.; Hall, E.; Redford, K.; Gil, G.; Pandak, W.M. Overexpression of cholesterol transporter StAR increasesin vivo rates of bile acid synthesis in the rat and mouse. Hepatology 2004, 40, 910–917. [Google Scholar] [CrossRef]
- Stocco, D.M.; Wang, X.; Jo, Y.; Manna, P.R. Multiple Signaling Pathways Regulating Steroidogenesis and Steroidogenic Acute Regulatory Protein Expression: More Complicated than We Thought. Mol. Endocrinol. 2005, 19, 2647–2659. [Google Scholar] [CrossRef]
- Qiu, Y.; Sui, X.; Zhan, Y.; Xu, C.; Li, X.; Ning, Y.; Zhi, X.; Yin, L. Steroidogenic acute regulatory protein (StAR) overexpression attenuates HFD-induced hepatic steatosis and insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 978–990. [Google Scholar] [CrossRef]
- Kanai, S.; Kitani, K.; Sato, Y. The nature of choleresis induced by deoxycholate and its conjugates in the rabbit. Jpn. J. Physiol. 1989, 39, 907–918. [Google Scholar] [CrossRef]
- Alpini, G.; Ueno, Y.; Glaser, S.S.; Marzioni, M.; Phinizy, J.L.; Francis, H.; Lesage, G. Bile acid feeding increased proliferative activity and apical bile acid transporter expression in both small and large rat cholangiocytes. Hepatology 2001, 34, 868–876. [Google Scholar] [CrossRef]
- Alpini, G.; Baiocchi, L.; Glaser, S.; Ueno, Y.; Marzioni, M.; Francis, H.; Phinizy, J.L.; Angelico, M.; Lesage, G. Ursodeoxycholate and tauroursodeoxycholate inhibit cholangiocyte growth and secretion of BDL rats through activation of PKC alpha. Hepatology 2002, 35, 1041–1052. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Tietz, P.; Wu, T.; Kip, S.; Dawson, P.A.; LaRusso, N.F. Alternative splicing of the rat sodium/bile acid transporter changes its cellular localization and transport properties. Proc. Natl. Acad. Sci. USA 2000, 97, 11092–11097. [Google Scholar] [CrossRef]
- De Vree, J.M.L.; Jacquemin, E.; Sturm, E.; Cresteil, D.; Bosma, P.J.; Aten, J.; Deleuze, J.-F.; Desrochers, M.; Burdelski, M.; Bernard, O.; et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc. Natl. Acad. Sci. USA 1998, 95, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Klaassen, C.D. Molecular Regulation of Bile Acid Homeostasis. Drug Metab. Dispos. 2021. [Google Scholar] [CrossRef]
- Oehler, N.; Volz, T.; Bhadra, O.D.; Kah, J.; Allweiss, L.; Giersch, K.; Bierwolf, J.; Riecken, K.; Pollok, J.M.; Lohse, A.W.; et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology 2014, 60, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kishimoto, Y.; Konishi, T.; Fujita, Y.; Ito, M.; Shimokado, K.; Maruyama, N.; Ishigami, A. Ascorbic acid deficiency affects genes for oxidation–reduction and lipid metabolism in livers from SMP30/GNL knockout mice. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Jahan, A.; Chiang, J.Y.L. Cytokine regulation of human sterol 12α-hydroxylase (CYP8B1) gene. Am. J. Physiol. Liver Physiol. 2005, 288, G685–G695. [Google Scholar] [CrossRef]
- Xiao, Y.; Yan, W.; Zhou, K.; Cao, Y.; Cai, W. Glucocorticoid treatment alters systemic bile acid homeostasis by regulating the biosynthesis and transport of bile salts. Dig. Liver Dis. 2016, 48, 771–779. [Google Scholar] [CrossRef]
- Bile Acid Profile, Serum. Available online: https://www.mayocliniclabs.com/test-catalog/overview/62538#Clinical-and-Interpretive (accessed on 15 February 2022).
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Fiorucci, S.; Zampella, A.; Cirino, G.; Bucci, M.; Distrutti, E. Decoding the vasoregulatory activities of bile acid-activated receptors in systemic and portal circulation: Role of gaseous mediators. Am. J. Physiol. Circ. Physiol. 2017, 312, H21–H32. [Google Scholar] [CrossRef]
- Singh, N.; Yadav, M.; Singh, A.K.; Kumar, H.; Dwivedi, S.K.D.; Mishra, J.S.; Gurjar, A.; Manhas, A.; Chandra, S.; Yadav, P.N.; et al. Synthetic FXR Agonist GW4064 Is a Modulator of Multiple G Protein-Coupled Receptors. Mol. Endocrinol. 2014, 28, 659–673. [Google Scholar] [CrossRef]
- Wilson, A.; Almousa, A.; Teft, W.A.; Kim, R.B. Attenuation of bile acid-mediated FXR and PXR activation in patients with Crohn’s disease. Sci. Rep. 2020, 10, 1866. [Google Scholar] [CrossRef]
- Patel, M.; Oza, N.; Anand, I.; Deshpande, S.; Patel, C. Liver X Receptor: A novel therapeutic target. Indian J. Pharm. Sci. 2008, 70, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.G. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile Acid Regulation of Gene Expression: Roles of Nuclear Hormone Receptors. Endocr. Rev. 2002, 23, 443–463. [Google Scholar] [CrossRef]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D Receptor as an Intestinal Bile Acid Sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Kakizaki, S.; Yamazaki, Y.; Takizawa, D.; Negishi, M. New Insights on the Xenobiotic-Sensing Nuclear Receptors in Liver Diseases-CAR and PXR. Curr. Drug Metab. 2008, 9, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Kachaylo, E.M.; Pustylnyak, V.O.; Lyakhovich, V.V.; Gulyaeva, L.F. Constitutive androstane receptor (CAR) is a xenosensor and target for therapy. Biochemistry 2011, 76, 1087–1097. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Y.; Guo, C.; Wang, J.; Boral, D.; Nie, D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012, 83, 1112–1126. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-κB target gene expression. Am. J. Physiol. Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef]
- Ibrahim, E.; Diakonov, I.; Arunthavarajah, D.; Swift, T.; Goodwin, M.; McIlvride, S.; Nikolova, V.; Williamson, C.; Gorelik, J. Bile acids and their respective conjugates elicit different responses in neonatal cardiomyocytes: Role of Gi protein, muscarinic receptors and TGR5. Sci. Rep. 2018, 8, 7110. [Google Scholar] [CrossRef] [PubMed]
- Baiocchi, L.; Zhou, T.; Liangpunsakul, S.; Lenci, I.; Santopaolo, F.; Meng, F.; Kennedy, L.; Glaser, S.; Francis, H.; Alpini, G. Dual Role of Bile Acids on the Biliary Epithelium: Friend or Foe? Int. J. Mol. Sci. 2019, 20, 1869. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Yuza, K.; Hirose, Y.; Nakajima, M.; Ramanathan, R.; Hait, N.C.; Hylemon, P.B.; Zhou, H.; Takabe, K.; Wakai, T. The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J. Lipid Res. 2016, 57, 1636–1643. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Qiang, X.; Luo, L.; Hylemon, P.B.; Jiang, Z.; Zhang, L.; Zhou, H. Taurocholate Induces Cyclooxygenase-2 Expression via the Sphingosine 1-phosphate Receptor 2 in a Human Cholangiocarcinoma Cell Line. J. Biol. Chem. 2015, 290, 30988–31002. [Google Scholar] [CrossRef]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P.N. Bile acid: A potential inducer of colon cancer stem cells. Stem Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef]
- Ali, O.; Tolaymat, M.; Hu, S.; Xie, G.; Raufman, J.-P. Overcoming Obstacles to Targeting Muscarinic Receptor Signaling in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 716. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.-P.; Chen, Y.; Cheng, K.; Compadre, C.; Compadre, L.; Zimniak, P. Selective interaction of bile acids with muscarinic receptors: A case of molecular mimicry. Eur. J. Pharmacol. 2002, 457, 77–84. [Google Scholar] [CrossRef]
- Kadir, S.H.S.A.; Miragoli, M.; Abu-Hayyeh, S.; Moshkov, A.; Xie, Q.; Keitel, V.; Nikolaev, V.O.; Williamson, C.; Gorelik, J. Bile Acid-Induced Arrhythmia Is Mediated by Muscarinic M2 Receptors in Neonatal Rat Cardiomyocytes. PLoS ONE 2010, 5, e9689. [Google Scholar] [CrossRef]
- Kundu, S.; Bansal, S.; Muthukumarasamy, K.M.; Sachidanandan, C.; Motiani, R.K.; Bajaj, A. Deciphering the role of hydrophobic and hydrophilic bile acids in angiogenesis using in vitro and in vivo model systems. MedChemComm 2017, 8, 2248–2257. [Google Scholar] [CrossRef]
- Glaser, S.S.; Gaudio, E.; Alpini, G. Vascular factors, angiogenesis and biliary tract disease. Curr. Opin. Gastroenterol. 2010, 26, 246–250. [Google Scholar] [CrossRef]
- Li, T.; Francl, J.M.; Boehme, S.; Chiang, J.Y.L. Regulation of cholesterol and bile acid homeostasis by the cholesterol 7α-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology 2013, 58, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.; Kimmel, R.; Stroup, D. Regulation of cholesterol 7α-hydroxylase gene (CYP7A1) transcription by the liver orphan receptor (LXRα). Gene 2001, 262, 257–265. [Google Scholar] [CrossRef]
- Björkhem, I.; Andersson, U.; Ellis, E.; Alvelius, G.; Ellegård, L.; Diczfalusy, U.; Sjövall, J.; Einarsson, C. From Brain to Bile. J. Biol. Chem. 2001, 276, 37004–37010. [Google Scholar] [CrossRef]
- Li-Hawkins, J.; Lund, E.G.; Bronson, A.D.; Russell, D.W. Expression Cloning of an Oxysterol 7α-Hydroxylase Selective for 24-Hydroxycholesterol. J. Biol. Chem. 2000, 275, 16543–16549. [Google Scholar] [CrossRef]
- Fuchs, M. III. Regulation of bile acid synthesis: Past progress and future challenges. Am. J. Physiol. Liver Physiol. 2003, 284, G551–G557. [Google Scholar] [CrossRef] [PubMed]
- Björkhem, I. Do oxysterols control cholesterol homeostasis? J. Clin. Investig. 2002, 110, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; Goto, T.; Uchida, M.; Nishimura, K.; Ando, M.; Kobayashi, N.; Goto, J. Presence of protein-bound unconjugated bile acids in the cytoplasmic fraction of rat brain. J. Lipid Res. 2004, 45, 295–300. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, T.; Zhao, A.; Wang, X.; Xie, G.; Huang, F.; Liu, J.; Zhao, Q.; Wang, S.; Wang, C.; et al. The Brain Metabolome of Male Rats across the Lifespan. Sci. Rep. 2016, 6, 24125. [Google Scholar] [CrossRef]
- Higashi, T.; Watanabe, S.; Tomaru, K.; Yamazaki, W.; Yoshizawa, K.; Ogawa, S.; Nagao, H.; Minato, K.; Maekawa, M.; Mano, N. Unconjugated bile acids in rat brain: Analytical method based on LC/ESI-MS/MS with chemical derivatization and estimation of their origin by comparison to serum levels. Steroids 2017, 125, 107–113. [Google Scholar] [CrossRef]
- Pan, X.; Elliott, C.T.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Hölscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites 2017, 7, 28. [Google Scholar] [CrossRef]
- Meaney, S.; Heverin, M.; Panzenböck, U.; Ekström, L.; Axelsson, M.; Andersson, U.; Diczfalusy, U.; Pikuleva, I.; Wahren, J.; Sattler, W.; et al. Novel route for elimination of brain oxysterols across the blood-brain barrier: Conversion into 7α-hydroxy-3-oxo-4-cholestenoic acid. J. Lipid Res. 2007, 48, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef]
- Hofmann, A.; Mysels, K. Bile acid solubility and precipitation in vitro and in vivo: The role of conjugation, pH, and Ca2+ ions. J. Lipid Res. 1992, 33, 617–626. [Google Scholar] [CrossRef]
- Hanafi, N.I.; Mohamed, A.S.; Kadir, S.H.S.A.; Othman, M.H.D. Overview of Bile Acids Signaling and Perspective on the Signal of Ursodeoxycholic Acid, the Most Hydrophilic Bile Acid, in the Heart. Biomolecules 2018, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Billington, D.; Evans, C.E.; Godfrey, P.P.; Coleman, R. Effects of bile salts on the plasma membranes of isolated rat hepatocytes. Biochem. J. 1980, 188, 321–327. [Google Scholar] [CrossRef]
- Coleman, R.; Rahman, K. Lipid flow in bile formation. Biochim. Biophys. Acta Lipids Lipid Metab. 1992, 1125, 113–133. [Google Scholar] [CrossRef]
- Elferink, R.P.J.O.; Tytgat, G.N.J.; Groen, A.K. The role of mdr2 P-glycoprotein in hepatobiliary lipid transport. FASEB J. 1997, 11, 19–28. [Google Scholar] [CrossRef]
- Werlin, S.; Scotet, V.; Uguen, K.; Audrezet, M.-P.; Cohen, M.; Yaakov, Y.; Safadi, R.; Ilan, Y.; Konikoff, F.; Galun, E.; et al. Primary sclerosing cholangitis is associated with abnormalities in CFTR. J. Cyst. Fibros. 2018, 17, 666–671. [Google Scholar] [CrossRef]
- Zhou, Y.; Doyen, R.; Lichtenberger, L.M. The role of membrane cholesterol in determining bile acid cytotoxicity and cytoprotection of ursodeoxycholic acid. Biochim. Biophys. Acta Biomembr. 2009, 1788, 507–513. [Google Scholar] [CrossRef][Green Version]
- Ionova, I.V.; Livshits, V.A.; Marsh, D. Phase Diagram of Ternary Cholesterol/Palmitoylsphingomyelin/Palmitoyloleoyl-Phosphatidylcholine Mixtures: Spin-Label EPR Study of Lipid-Raft Formation. Biophys. J. 2012, 102, 1856–1865. [Google Scholar] [CrossRef]
- Powell, A.A.; LaRue, J.M.; Batta, A.K.; Martinez, J.D. Bile acid hydrophobicity is correlated with induction of apoptosis and/or growth arrest in HCT116 cells. Biochem. J. 2001, 356, 481–486. [Google Scholar] [CrossRef]
- Coreta-Gomes, F.M.; Martins, P.A.T.; Velazquez-Campoy, A.; Vaz, W.L.C.; Geraldes, C.F.G.; Moreno, M.J. Interaction of Bile Salts with Model Membranes Mimicking the Gastrointestinal Epithelium: A Study by Isothermal Titration Calorimetry. Langmuir 2015, 31, 9097–9104. [Google Scholar] [CrossRef] [PubMed]
- Heuman, D.M.; Bajaj, R. Ursodeoxycholate conjugates protect against disruption of cholesterol-rich membranes by bile salts. Gastroenterology 1994, 106, 1333–1341. [Google Scholar] [CrossRef]
- Im, E.; Akare, S.; Powell, A.; Martinez, J.D. Ursodeoxycholic Acid Can Suppress Deoxycholic Acid-Induced Apoptosis by Stimulating Akt/PKB-Dependent Survival Signaling. Nutr. Cancer 2005, 51, 110–116. [Google Scholar] [CrossRef]
- Rodrigues, C.; Fan, G.; Wong, P.Y.; Kren, B.T.; Steer, C.J. Ursodeoxycholic Acid May Inhibit Deoxycholic Acid-Induced Apoptosis by Modulating Mitochondrial Transmembrane Potential and Reactive Oxygen Species Production. Mol. Med. 1998, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sharma, K.; Gupta, A.; Yadav, A.; Kumar, A. Gallbladder cancer epidemiology, pathogenesis and molecular genetics: Recent update. World J. Gastroenterol. 2017, 23, 3978–3998. [Google Scholar] [CrossRef]
- Paumgartner, G.; Sauerbruch, T. Gallstones: Pathogenesis. Lancet 1991, 338, 1117–1121. [Google Scholar] [CrossRef]
- Chawla, A.; Saez, E.; Evans, R. Don’t Know Much Bile-ology. Cell 2000, 103, 1–4. [Google Scholar] [CrossRef]
- Liu, H.; Pathak, P.; Boehme, S.; Chiang, J.Y.L. Cholesterol 7α-hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J. Lipid Res. 2016, 57, 1831–1844. [Google Scholar] [CrossRef]
- Li, T.; Chanda, D.; Zhang, Y.; Choi, H.-S.; Chiang, J.Y. Glucose stimulates cholesterol 7α-hydroxylase gene transcription in human hepatocytes. J. Lipid Res. 2010, 51, 832–842. [Google Scholar] [CrossRef]
- Sharma, V.; Hiller, M. Loss of Enzymes in the Bile Acid Synthesis Pathway Explains Differences in Bile Composition among Mammals. Genome Biol. Evol. 2018, 10, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Atallah, I.; Millán, D.S.; Benoît, W.; Campos-Xavier, B.; Superti-Furga, A.; Tran, C. Spinal cerebrotendinous xanthomatosis: A case report and literature review. Mol. Genet. Metab. Rep. 2021, 26, 100719. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.; Heubi, J.E.; Shah, S.; Lavine, J.E.; Suskind, D.; Al–Edreesi, M.; Potter, C.; Russell, D.W.; O'Connell, N.C.; Wolfe, B.; et al. Genetic Defects in Bile Acid Conjugation Cause Fat-Soluble Vitamin Deficiency. Gastroenterology 2013, 144, 945–955.e6. [Google Scholar] [CrossRef] [PubMed]
- Carlton, V.E.H.; Harris, B.Z.; Puffenberger, E.; Batta, A.K.; Knisely, A.S.; Robinson, D.L.; Strauss, K.A.; Shneider, B.L.; Lim, W.A.; Salen, G.; et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat. Genet. 2003, 34, 91–96. [Google Scholar] [CrossRef]
- Beauséjour, Y.; Alvarez, F.; Beaulieu, M.; Bilodeau, M. Description of Two NewABCB11Mutations Responsible for Type 2 Benign Recurrent Intrahepatic Cholestasis in a French-Canadian Family. Can. J. Gastroenterol. 2011, 25, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Takada, T.; Suzuki, H.; Akita, H.; Sugiyama, Y. Two common PFIC2 mutations are associated with the impaired membrane trafficking of BSEP/ABCB11. Hepatology 2005, 41, 916–924. [Google Scholar] [CrossRef]
- Keitel, V.; Burdelski, M.; Warskulat, U.; Kühlkamp, T.; Keppler, D.; Häussinger, D.; Kubitz, R. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology 2005, 41, 1160–1172. [Google Scholar] [CrossRef]
- Anzivino, C.; Odoardi, M.R.; Meschiari, E.; Baldelli, E.; Facchinetti, F.; Neri, I.; Ruggiero, G.; Zampino, R.; Bertolotti, M.; Loria, P.; et al. ABCB4 and ABCB11 mutations in intrahepatic cholestasis of pregnancy in an Italian population. Dig. Liver Dis. 2013, 45, 226–232. [Google Scholar] [CrossRef]
- Lam, P.; Pearson, C.L.; Soroka, C.J.; Xu, S.; Mennone, A.; Boyer, J.L. Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am. J. Physiol. Cell Physiol. 2007, 293, C1709–C1716. [Google Scholar] [CrossRef]
- Gan, L.; Pan, S.; Cui, J.; Bai, J.; Jiang, P.; He, Y. Functional analysis of the correlation between ABCB11 gene mutation and primary intrahepatic stone. Mol. Med. Rep. 2018, 19, 195–204. [Google Scholar] [CrossRef]
- Vaz, F.M.; Paulusma, C.; Huidekoper, H.; De Ru, M.; Lim, C.; Koster, J.; Ho-Mok, K.; Bootsma, A.H.; Groen, A.K.; Schaap, F.; et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: Conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2014, 61, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Smet, M.; Heath, S.; Curnow, J.; Lin, M.; George, J.; Alahakoon, T.I. Pregnancy complicated by refractory severe hypercholanaemia from sodium taurocholate co-transporting polypeptide deficiency. J. Obstet. Gynaecol. Res. 2020, 47, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef]
- Dawson, P.A.; Haywood, J.; Craddock, A.L.; Wilson, M.; Tietjen, M.; Kluckman, K.; Maeda, N.; Parks, J.S. Targeted Deletion of the Ileal Bile Acid Transporter Eliminates Enterohepatic Cycling of Bile Acids in Mice. J. Biol. Chem. 2003, 278, 33920–33927. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Inagaki, T.; Gerard, R.D.; Dawson, P.A.; Kliewer, S.A.; Mangelsdorf, D.; Moschetta, A. FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J. Lipid Res. 2007, 48, 2693–2700. [Google Scholar] [CrossRef] [PubMed]
- Vivian, D.; Cheng, K.; Khurana, S.; Xu, S.; Kriel, E.H.; Dawson, P.A.; Raufman, J.-P.; Polli, J.E. In Vivo Performance of a Novel Fluorinated Magnetic Resonance Imaging Agent for Functional Analysis of Bile Acid Transport. Mol. Pharm. 2014, 11, 1575–1582. [Google Scholar] [CrossRef] [PubMed]
- Oelkers, P.; Kirby, L.C.; Heubi, J.E.; Dawson, P.A. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J. Clin. Investig. 1997, 99, 1880–1887. [Google Scholar] [CrossRef]
- Wu, Y.; Aquino, C.J.; Cowan, D.J.; Anderson, D.L.; Ambroso, J.L.; Bishop, M.J.; Boros, E.E.; Chen, L.; Cunningham, A.; Dobbins, R.L.; et al. Discovery of a Highly Potent, Nonabsorbable Apical Sodium-Dependent Bile Acid Transporter Inhibitor (GSK2330672) for Treatment of Type 2 Diabetes. J. Med. Chem. 2013, 56, 5094–5114. [Google Scholar] [CrossRef]
- Tremont, S.J.; Lee, L.F.; Huang, H.-C.; Keller, B.T.; Banerjee, S.C.; Both, S.R.; Carpenter, A.J.; Wang, C.-C.; Garland, D.J.; Huang, W.; et al. Discovery of Potent, Nonsystemic Apical Sodium-Codependent Bile Acid Transporter Inhibitors (Part 1). J. Med. Chem. 2005, 48, 5837–5852. [Google Scholar] [CrossRef]
- Root, C.; Smith, C.D.; Sundseth, S.S.; Pink, H.M.; Wilson, J.G.; Lewis, M.C. Ileal bile acid transporter inhibition, CYP7A1 induction, and antilipemic action of 264W94. J. Lipid Res. 2002, 43, 1320–1330. [Google Scholar] [CrossRef]
- Out, C.; Groen, A.K.; Brufau, G. Bile acid sequestrants. Curr. Opin. Lipidol. 2012, 23, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.G. Cholestyramine. Can. Med. Assoc. J. 1971, 104, 305–309. [Google Scholar] [PubMed]
- Heel, R.C.; Brogden, R.N.; Pakes, G.E.; Speight, T.M.; Avery, G.S. Colestipol. Drugs 1980, 19, 161–180. [Google Scholar] [CrossRef]
- The Lipid Research Clinics Coronary Primary Prevention Trial Results: II. The Relationship of Reduction in Incidence of Coronary Heart Disease to Cholesterol Lowering. JAMA 1984, 251, 365–374. [CrossRef]
- Ferrebee, C.B.; Dawson, P.A. Metabolic effects of intestinal absorption and enterohepatic cycling of bile acids. Acta Pharm. Sin. B 2015, 5, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Zieve, F.J.; Kalin, M.F.; Schwartz, S.L.; Jones, M.R.; Bailey, W.L. Results of the glucose-lowering effect of WelChol study (GLOWS): A randomized, double-blind, placebo-controlled pilot study evaluating the effect of colesevelam hydrochloride on glycemic control in subjects with type 2 diabetes. Clin. Ther. 2007, 29, 74–83. [Google Scholar] [CrossRef]
- Chen, L.; McNulty, J.; Anderson, D.; Liu, Y.; Nystrom, C.; Bullard, S.; Collins, J.; Handlon, A.L.; Klein, R.; Grimes, A.; et al. Cholestyramine Reverses Hyperglycemia and Enhances Glucose-Stimulated Glucagon-Like Peptide 1 Release in Zucker Diabetic Fatty Rats. J. Pharmacol. Exp. Ther. 2010, 334, 164–170. [Google Scholar] [CrossRef]
- Sugimoto-Kawabata, K.; Shimada, H.; Sakai, K.; Suzuki, K.; Kelder, T.; Pieterman, E.J.; Cohen, L.H.; Havekes, L.M.; Princen, H.; Hoek, A.M.V.D. Colestilan decreases weight gain by enhanced NEFA incorporation in biliary lipids and fecal lipid excretion. J. Lipid Res. 2013, 54, 1255–1264. [Google Scholar] [CrossRef]
- Harach, T.; Pols, T.W.H.; Nomura, M.; Maida, A.; Watanabe, M.; Auwerx, J.; Schoonjans, K. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci. Rep. 2012, 2, 430. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Potts, A.; He, T.; Duarte, J.A.G.; Taussig, R.; Mangelsdorf, D.J.; Kliewer, S.A.; Burgess, S.C. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am. J. Physiol. Liver Physiol. 2013, 304, G371–G380. [Google Scholar] [CrossRef]
- West, K.L.; Zern, T.L.; Butteiger, D.N.; Keller, B.T.; Fernandez, M.L. SC-435, an ileal apical sodium co-dependent bile acid transporter (ASBT) inhibitor lowers plasma cholesterol and reduces atherosclerosis in guinea pigs. Atherosclerosis 2003, 171, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.G.; Rapp, S.R.; Beaudry, J.A.; Napawan, N.; Butteiger, D.N.; Hall, K.A.; Null, C.L.; Luo, Y.; Keller, B.T. Inhibition of ileal bile acid transport and reduced atherosclerosis in apoE mice by SC-435. J. Lipid Res. 2003, 44, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Rao, A.; Haywood, J.; Kock, N.D.; Dawson, P.A. Mouse organic solute transporter alpha deficiency alters FGF15 expression and bile acid metabolism. J. Hepatol. 2012, 57, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.G.; Hammond, C.L.; Jornayvaz, F.; Samuel, V.T.; Shulman, G.; Soroka, C.J.; Boyer, J.L.; Hinkle, P.M.; Ballatori, N. Ostα−/−mice exhibit altered expression of intestinal lipid absorption genes, resistance to age-related weight gain, and modestly improved insulin sensitivity. Am. J. Physiol. Liver Physiol. 2014, 306, G425–G438. [Google Scholar] [CrossRef] [PubMed]
- Lundåsen, T.; Andersson, E.-M.; Snaith, M.; Lindmark, H.; Lundberg, J.; Östlund-Lindqvist, A.-M.; Angelin, B.; Rudling, M. Inhibition of Intestinal Bile Acid Transporter Slc10a2 Improves Triglyceride Metabolism and Normalizes Elevated Plasma Glucose Levels in Mice. PLoS ONE 2012, 7, e37787. [Google Scholar] [CrossRef]


{kind=link}
| Receptor | Functions | Localization | Ref. |
|---|---|---|---|
| Nuclear receptors | |||
| Farnesoid X receptor | The main regulator of the enterohepatic circulation of BAs. Induces the production of FGF19, CYP3A4, and PXR in the ileum. Suppresses the transcription of the CYP7A1 and CYP8B1 genes and the synthesis of BAs. Suppresses the transcription of the NTCP gene and the uptake of BA by hepatocytes. Increases the activity of BACS, BAAT, BSEP, and MRP2 and simulates the export of BAs and bilirubin to bile. Suppresses ABST and OATP and the absorption of BAs by cholangiocytes by the ileal epithelium. Vasodilating action in the systemic and splanchnic circulation. | Epithelium of the ileum, hepatocytes, cholangiocytes, endothelium of sinusoids, renal epithelium, adrenal cortex, and cells of innate and adaptive immunity. | [82,83,84,85] |
| Nuclear receptor subfamily 1 group H member 3 | Regulates the remodeling of the phospholipids of the endoplasmic reticulum, and affects the processing of SREBF1 and the inclusion of triglycerides in VLDL. Suppresses the stress of the endoplasmic reticulum and acute phase reactions. Reduces the absorption of cholesterol in the intestines. Increases the activity of CYP7A1 and the synthesis of Bas; promotes the transport of cholesterol from peripheral tissues to the liver and its transformation into BA. Activates sterol response element-binding protein-1c, regulating lipogenesis. | Hepatocytes, enterocytes, renal epithelium, adipose tissue, skeletal muscles, and cells of innate and adaptive immunity. | [86,87,88] |
| Vitamin D receptor | Modulation of the intestinal microbiota composition and indirect influence on the conversion of secondary BAs. Potential impact on the risk of developing colorectal cancer. | The ileum, endocrine glands, skin, cells of innate and adaptive immunity. | [33,89] |
| Nuclear receptors—xenobiotic sensors | |||
| Constitutive activated receptor, nuclear receptor subfamily 1, group I, member 3 | Many effects are mediated by the HNF4α transcription factor. Suppression of CYP7A expression and BA synthesis with an increase in the content of LCA in the blood; activation of phase II enzymes for the detoxification of xenobiotics (sulfotransferases, glucoronosultransferases, glutathione S-transferases), including the activation of LCA sulfation and bilirubin conjugation. Activation of transporters (MRP, MDR, and OATP). Suppression of gluconeogenesis, development of steatosis, and decrease in thyroxine activity. | Hepatocytes and renal tubular epithelium. | [90,91,92] |
| Pregnane X receptor, nuclear receptor subfamily 1, group I, member 3 | The effects are similar to those of constitutive androstane receptor activation (mediated by the transcription factor HNF4α); CYP3A43 activation; suppression of the inflammatory cascade caused by the influence of NFκB and the maintenance of the intestinal epithelial barrier; suppression of CYP7A1. | Hepatocytes and intestinal epithelium. | [85,93,94] |
| Membrane receptors | |||
| G protein–coupled bile acid receptor 1, Takeda G-protein receptor 5 | Systemic effects of Bas; regulation of intestinal motility and metabolism; relaxation of the gallbladder during the interdigestive period (together with FGF19); vasodilating action in the systemic and splanchnic circulation; regulation of the proliferation of non-ciliated cholangiocytes, a possible role in the development of cholangiocellular cancer. | Epithelium of the ileum, cholangiocytes, smooth muscle cells, endothelium (in particular, the endothelium of sinusoids), adipose tissue, and cells of innate and adaptive immunity. | [83,95,96] |
| Sphingosine-1-phosphate receptor 2 | Increased activity of enzymes of lipid metabolism (SREBP1c, FAS, LDLR, FXRα, and PPARγ) and glucose (ERK1/2 and AKT signaling pathways and glycogen synthesis); regulates the differentiation of endothelial cells; promotes the growth and metastasis of cholangiocarcinoma. | Hepatocytes, intestinal epithelium, endothelium, vascular smooth muscle cells, myocardium, and fibroblasts. | [97,98] |
| Muscarinic receptors M2, M3 | Stimulation of intestinal motility, negative chronotropic action. Probably promote the growth of colon cancer. | Intestinal smooth muscle cells, exocrine glands, and myocardiu. | [99,100,101,102] |
| Vascular endothelial growth factor | Prevention of bile duct injury, possibly fibrosis. New vessel formation. | Cell lines of stomach and colon cancer. | [103,104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shulpekova, Y.; Shirokova, E.; Zharkova, M.; Tkachenko, P.; Tikhonov, I.; Stepanov, A.; Sinitsyna, A.; Izotov, A.; Butkova, T.; Shulpekova, N.; et al. A Recent Ten-Year Perspective: Bile Acid Metabolism and Signaling. Molecules 2022, 27, 1983. https://doi.org/10.3390/molecules27061983
Shulpekova Y, Shirokova E, Zharkova M, Tkachenko P, Tikhonov I, Stepanov A, Sinitsyna A, Izotov A, Butkova T, Shulpekova N, et al. A Recent Ten-Year Perspective: Bile Acid Metabolism and Signaling. Molecules. 2022; 27(6):1983. https://doi.org/10.3390/molecules27061983
Chicago/Turabian StyleShulpekova, Yulia, Elena Shirokova, Maria Zharkova, Pyotr Tkachenko, Igor Tikhonov, Alexander Stepanov, Alexandra Sinitsyna, Alexander Izotov, Tatyana Butkova, Nadezhda Shulpekova, and et al. 2022. "A Recent Ten-Year Perspective: Bile Acid Metabolism and Signaling" Molecules 27, no. 6: 1983. https://doi.org/10.3390/molecules27061983
APA StyleShulpekova, Y., Shirokova, E., Zharkova, M., Tkachenko, P., Tikhonov, I., Stepanov, A., Sinitsyna, A., Izotov, A., Butkova, T., Shulpekova, N., Nechaev, V., Damulin, I., Okhlobystin, A., & Ivashkin, V. (2022). A Recent Ten-Year Perspective: Bile Acid Metabolism and Signaling. Molecules, 27(6), 1983. https://doi.org/10.3390/molecules27061983