
Combined Analytical Study on Chemical Transformations and Detoxification of Model Phenolic Pollutants during Various Advanced Oxidation Treatment Processes


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and AOP Treatment
2.2. Instrumental Analysis
2.3. Inhibition on Daphnia magna
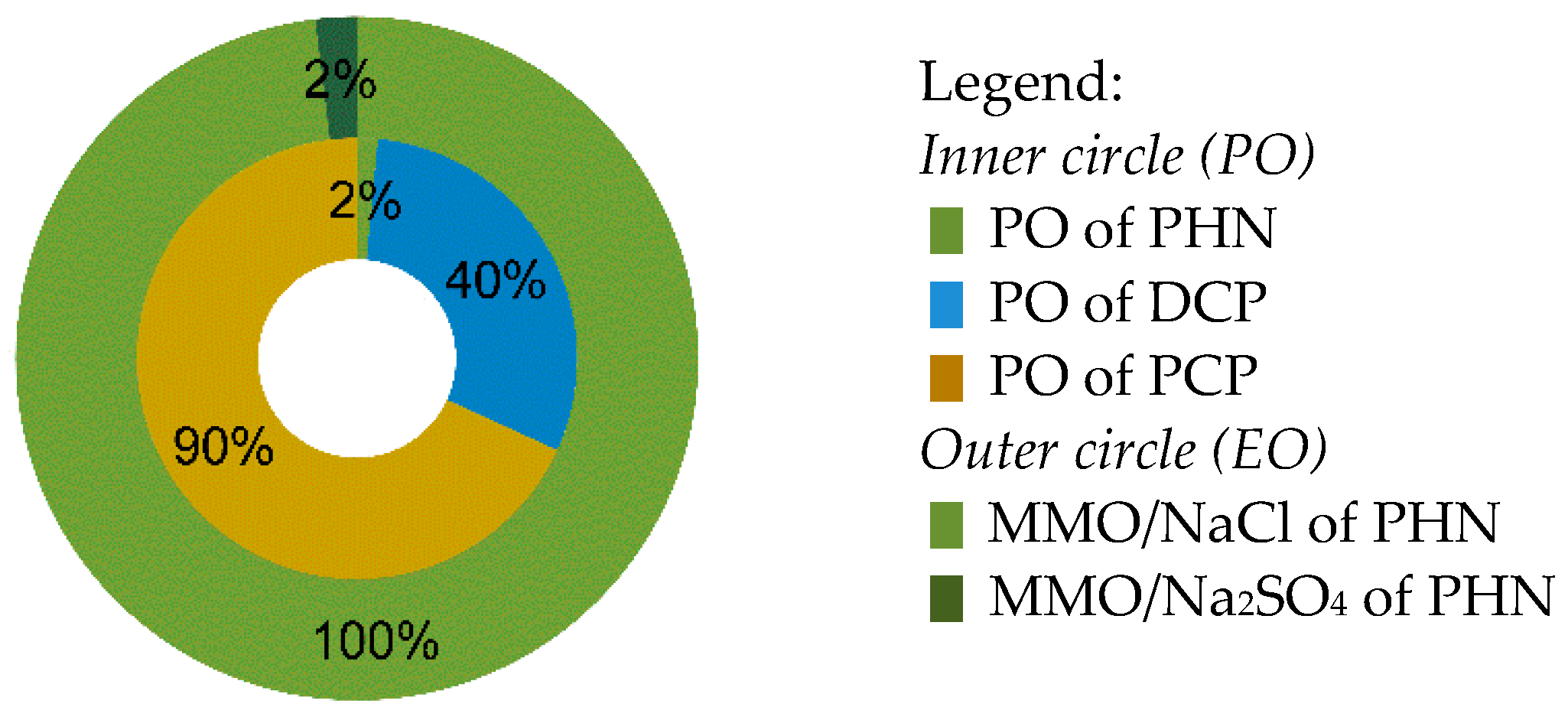
2.4. Numerical Evaluation
- Complete theoretical conversion ‘C6H5–xOClx → a chosen product’ (a is 3 and 6 for oxalic and formic acid, respectively) is assumed, regardless of other chemical transformations and changes in the volume of test mixture due to sampling during AOP treatment.
3. Results and Discussion
3.1. Removal of Target Phenols, Mineralization, and Progressivity
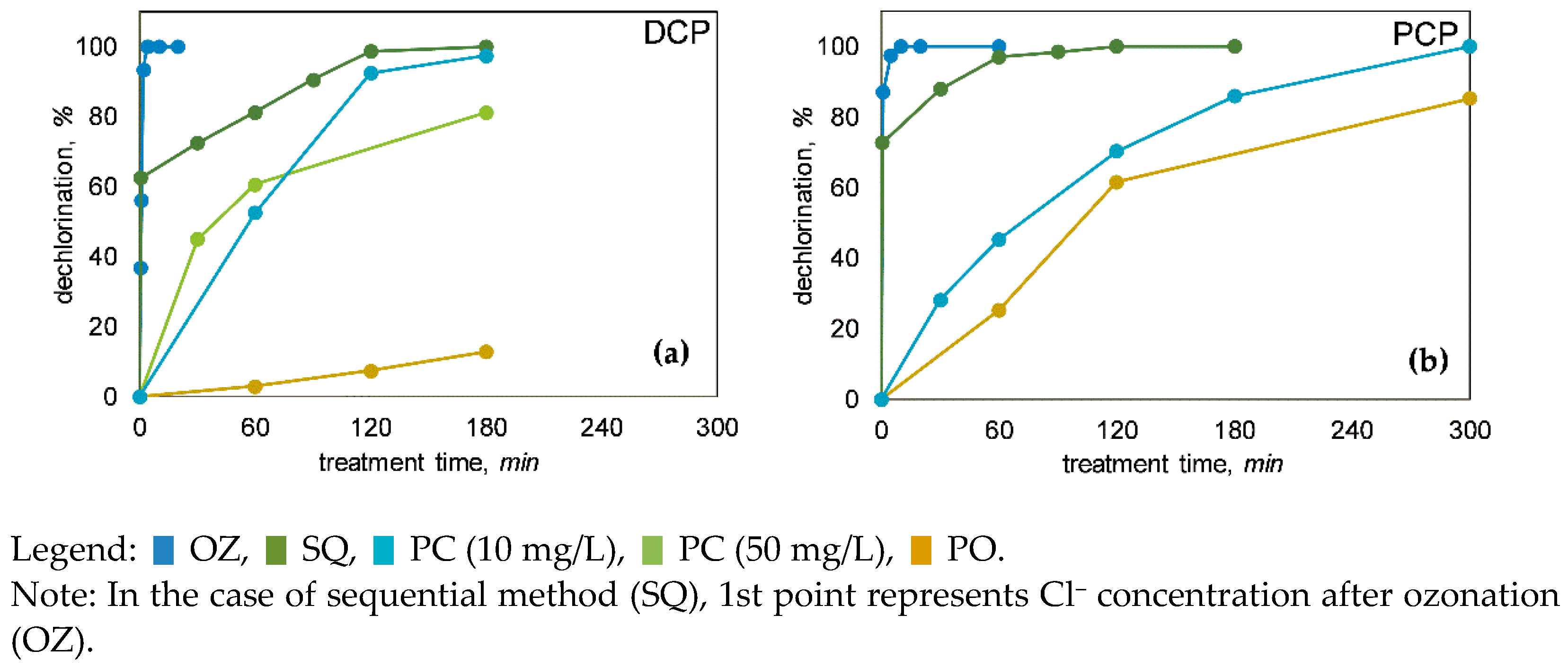
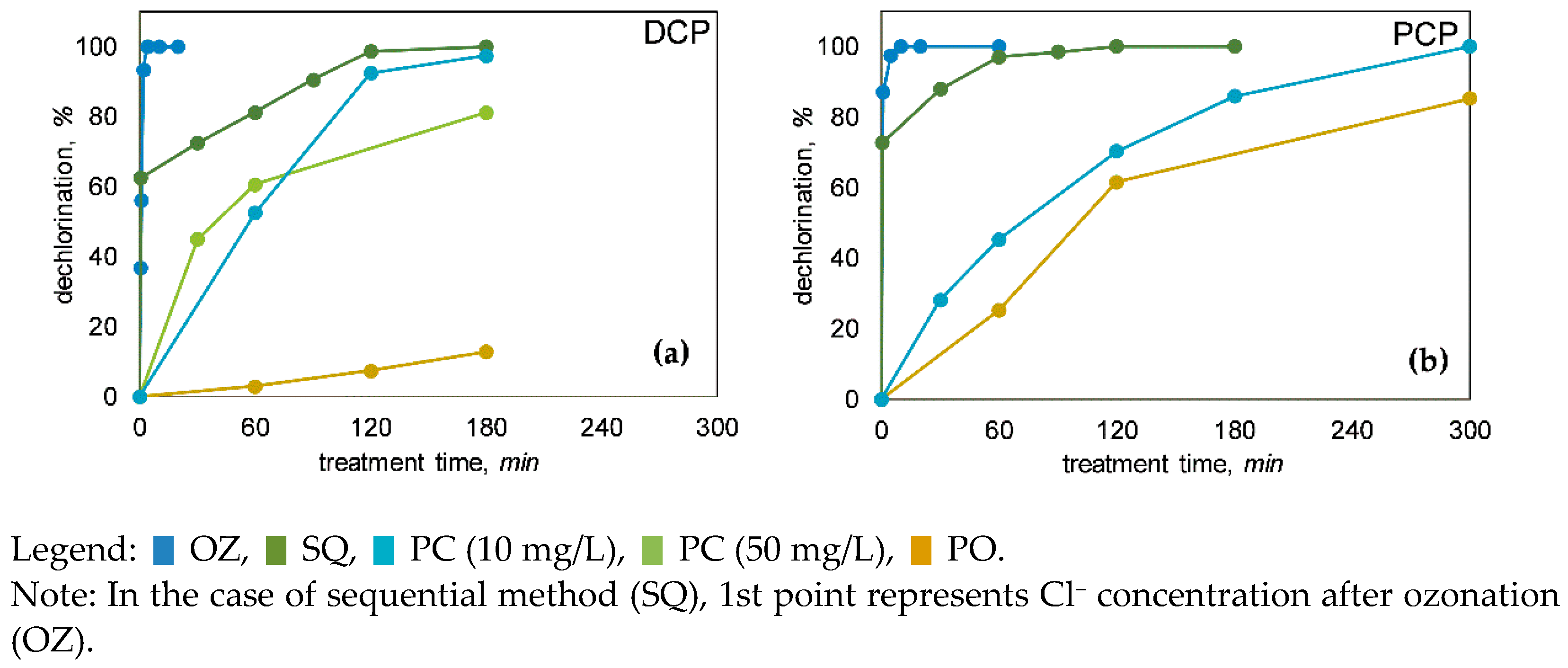
3.2. Dechlorination of Chlorophenols
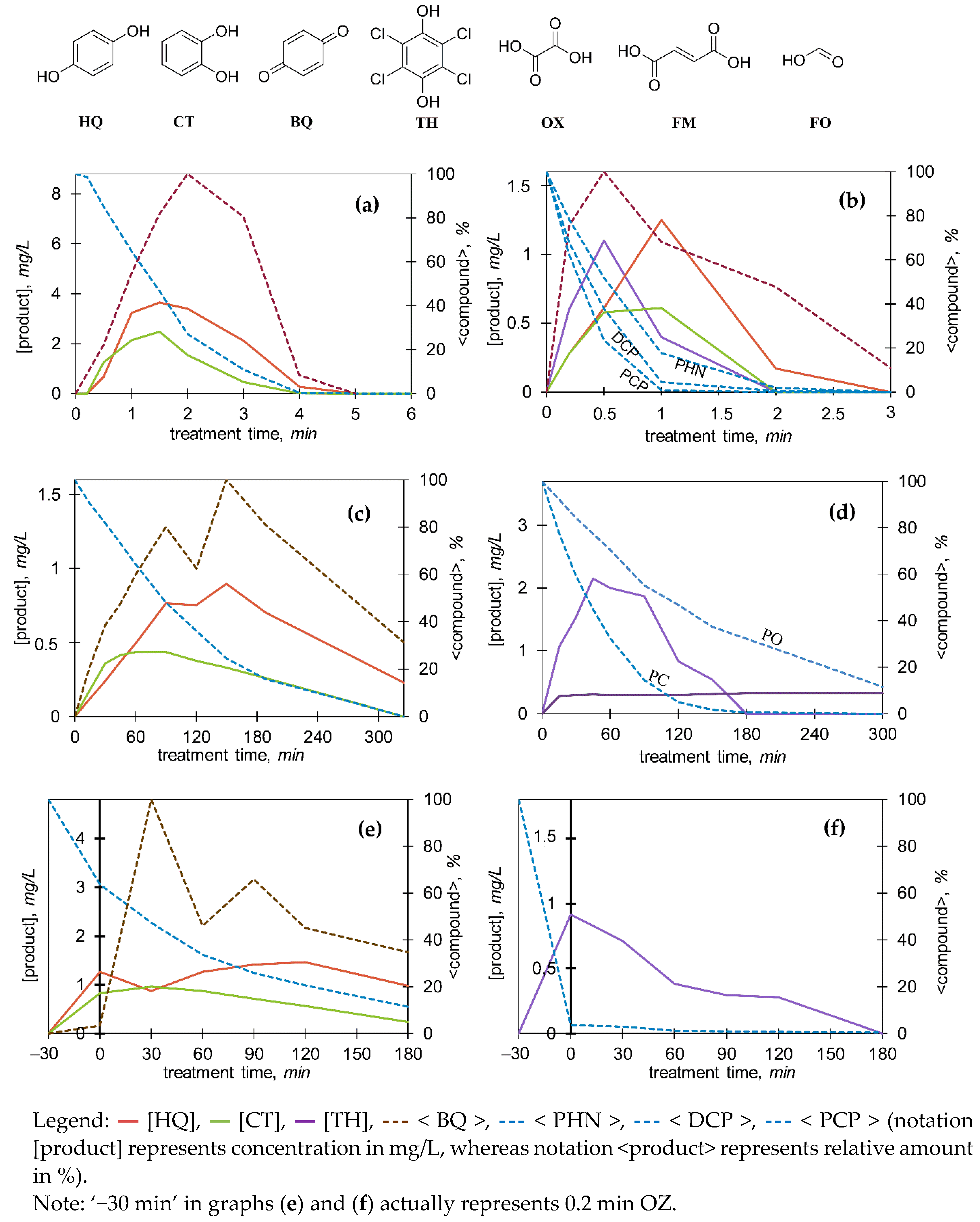
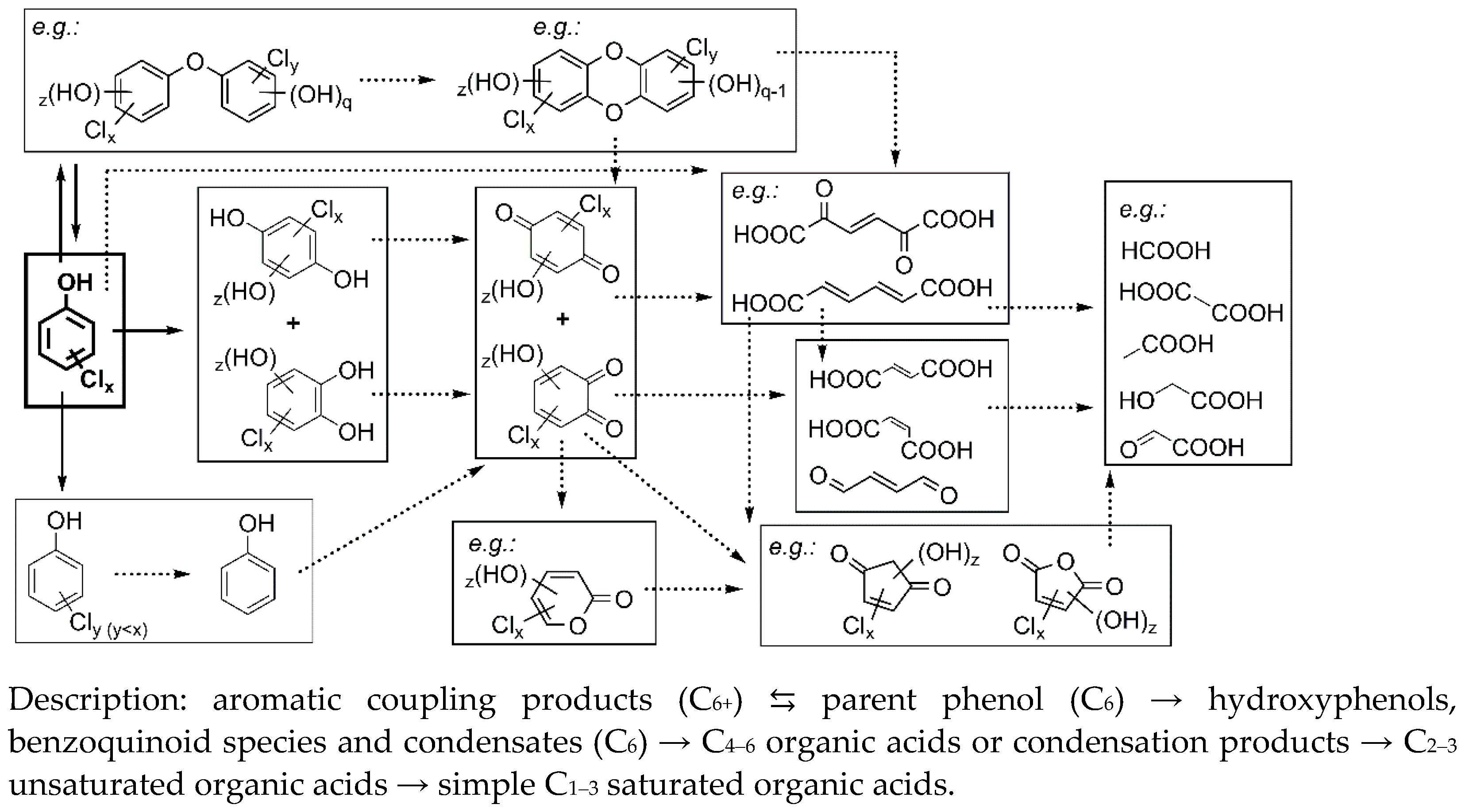
3.3. Transformation Products and Possible Degradation Pathways
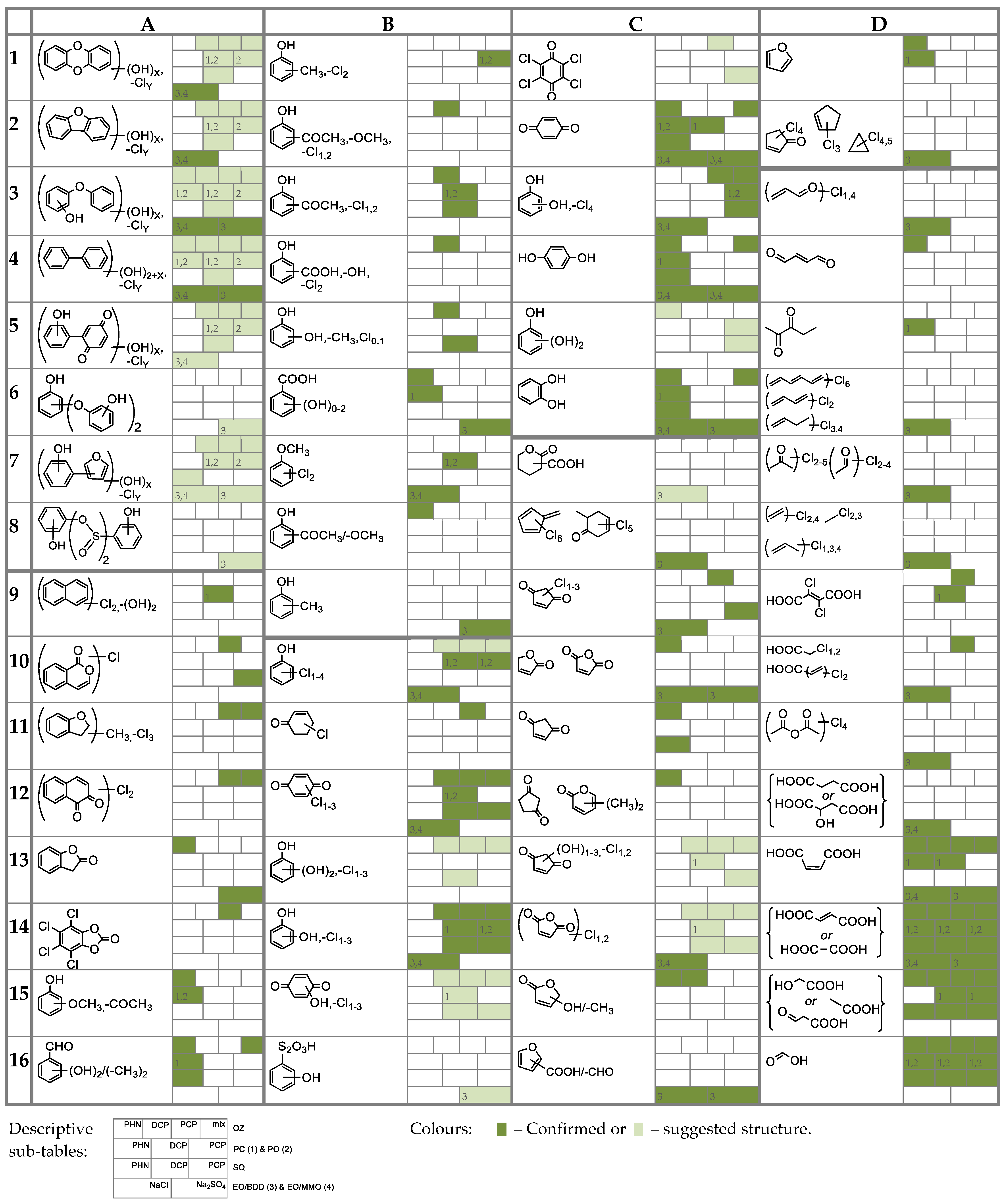
3.3.1. General Identification
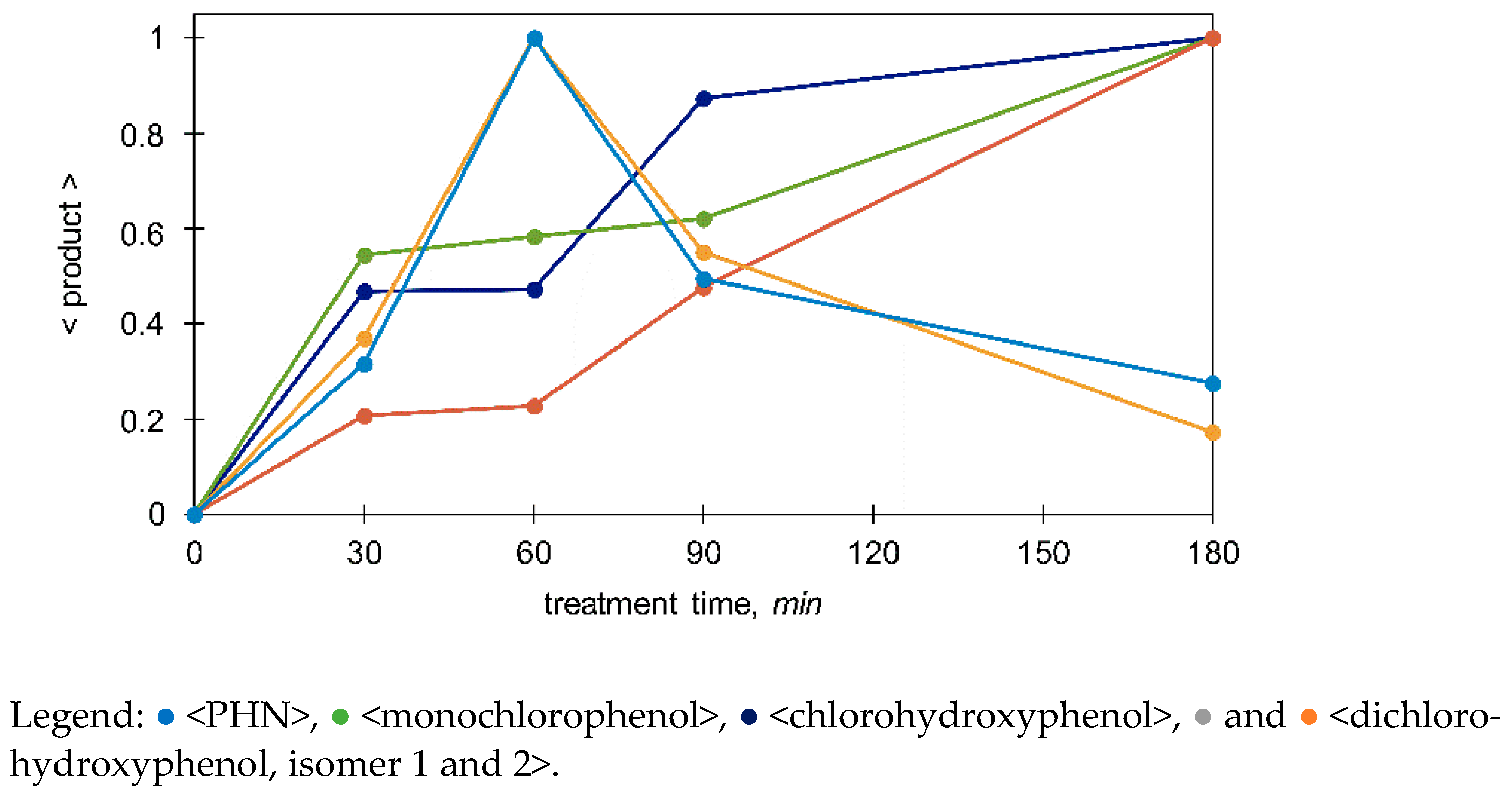
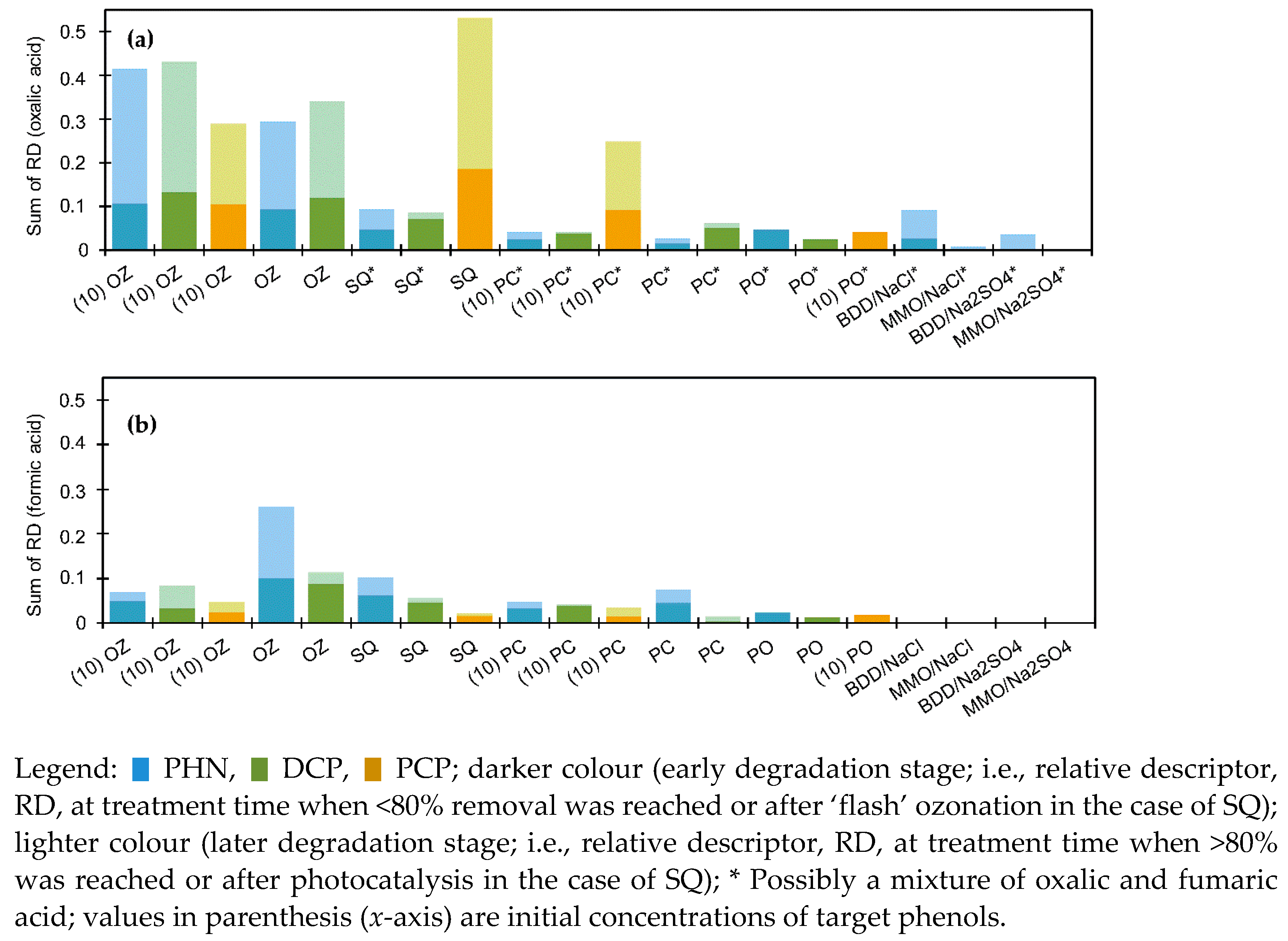
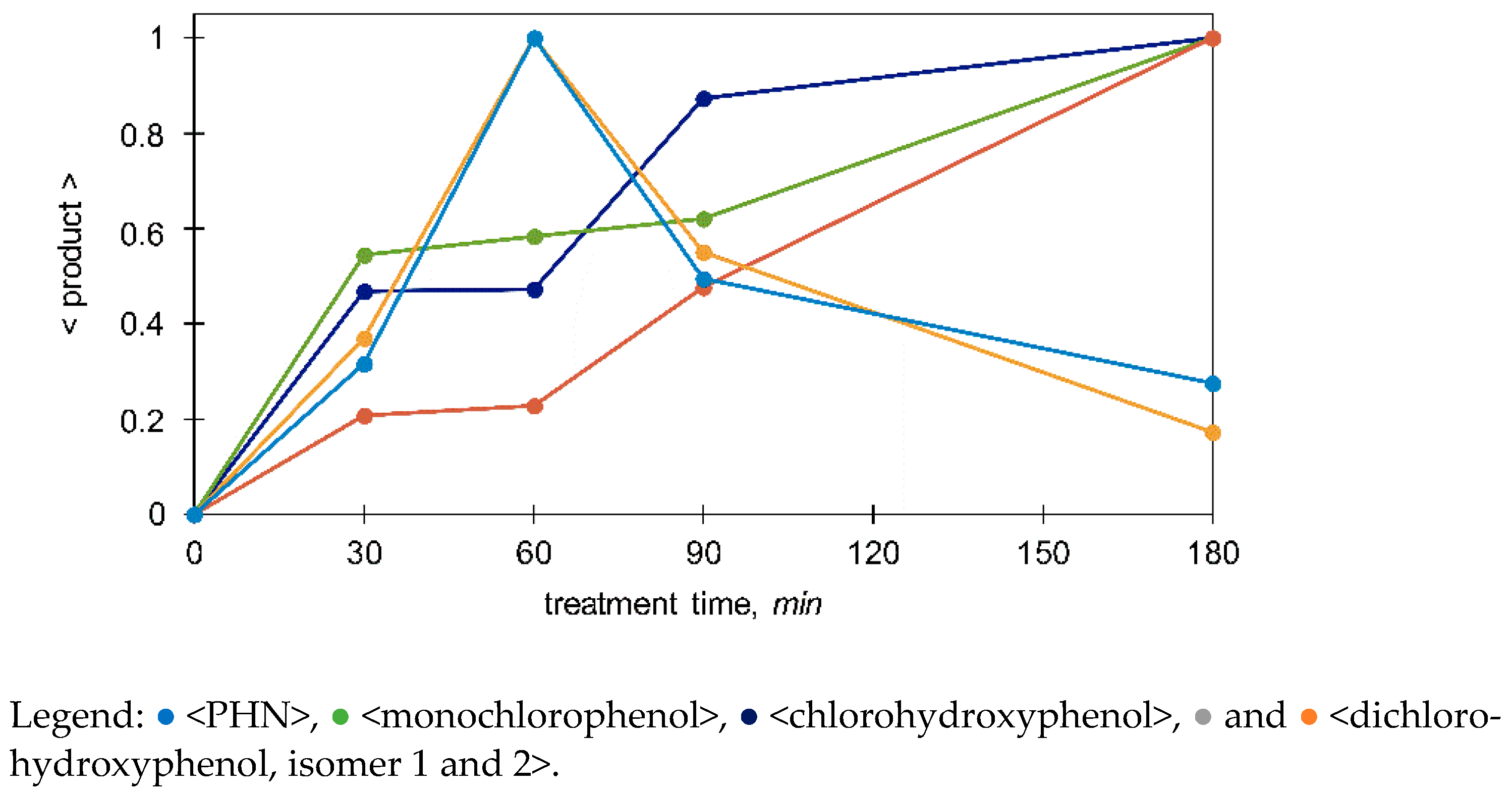
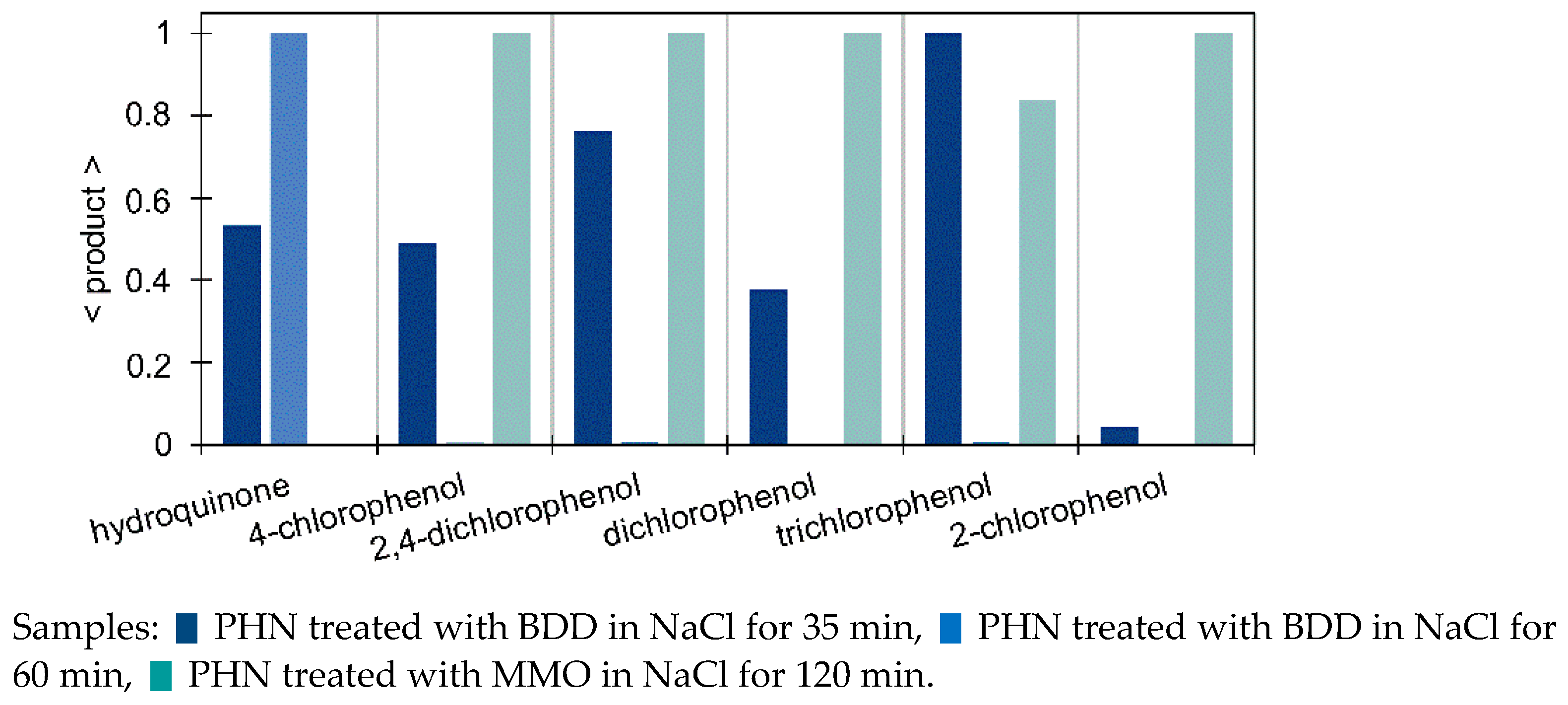
3.3.2. Monitoring of the Selected Products
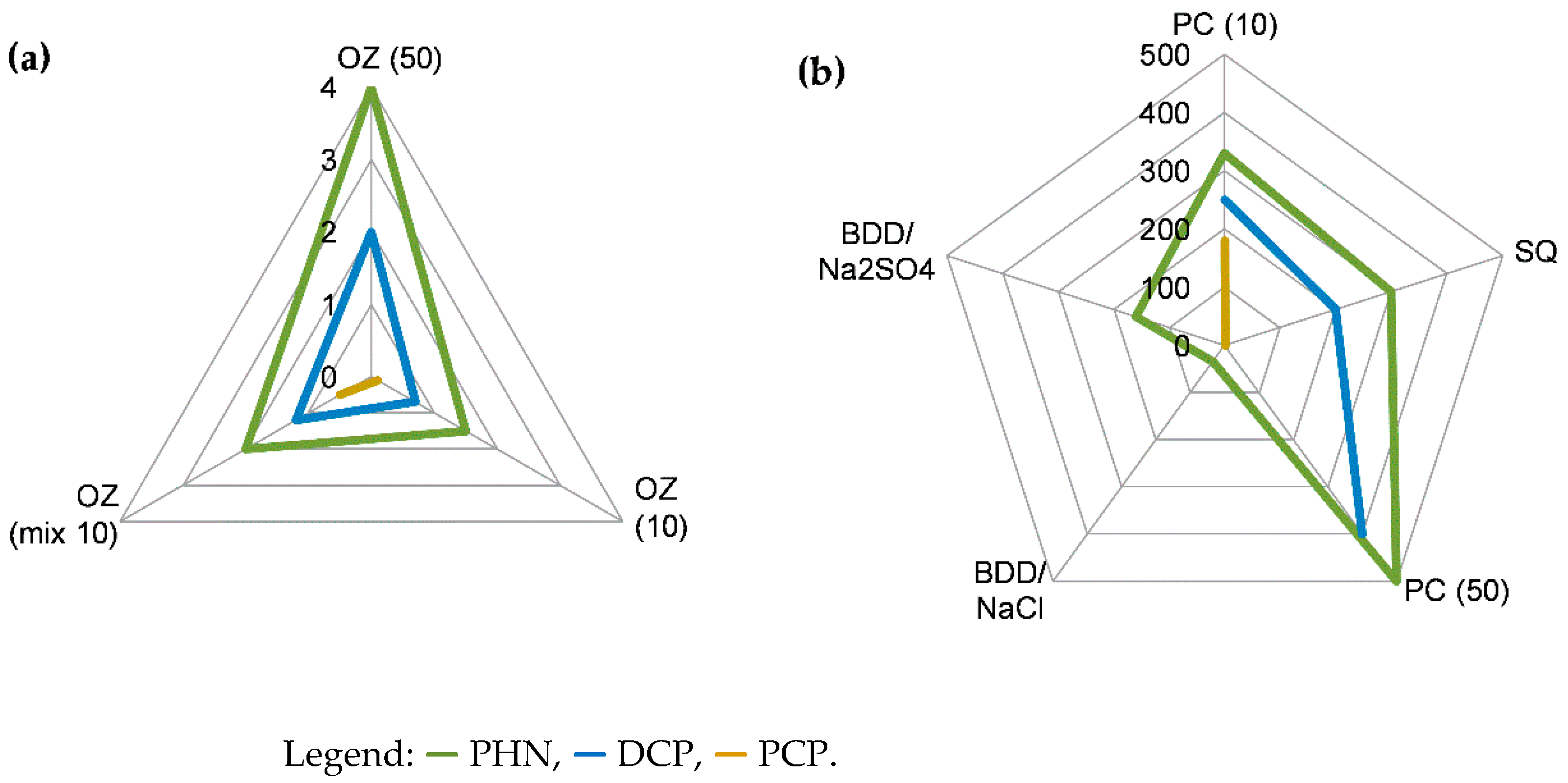
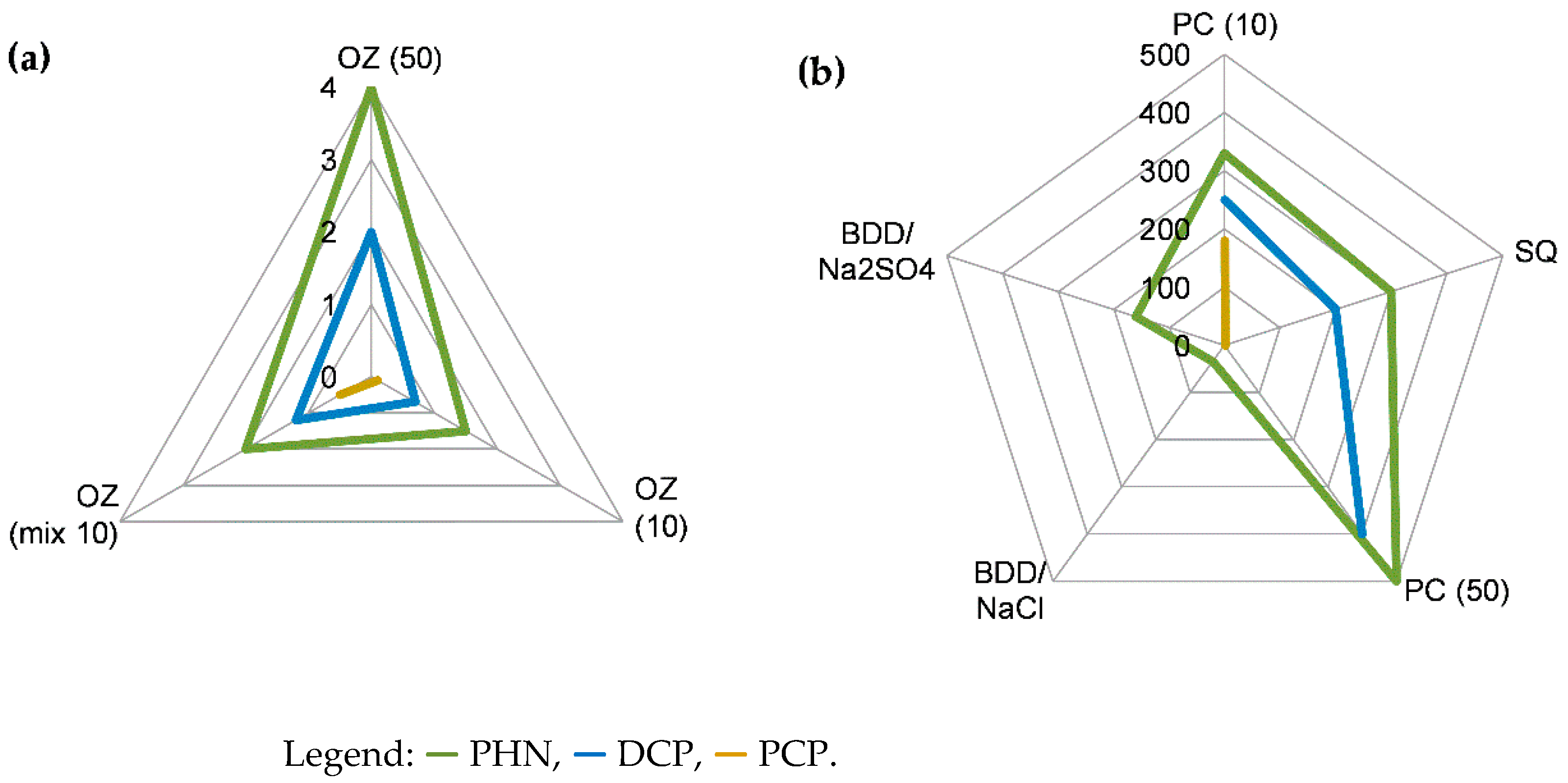
3.4. Detoxification Estimated from Acute Ecotoxicity Tests with Daphnia magna
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- New Indicator on Pesticides in European Waters (European Environment Agency, EEA News). Available online: https://www.eea.europa.eu/ims/pesticides-in-rivers-lakes-and (accessed on 1 January 2022).
- European Environment Agency. Contaminants in Europe’s Seas: Moving towards a Clean, Non-Toxic Marine Environment, 1st ed.; (Report No 25/2018); Reker, J., Ed.; Publications Office of the European Union: Copehnagen, Denmark, 2018; pp. 8, 49–55. [Google Scholar] [CrossRef]
- Waste Water Treatment Improves in Europe but Large Differences Remain (European Environment Agency, EEA News). Available online: https://www.eea.europa.eu/highlights/waste-water-treatment-improves-in (accessed on 1 January 2022).
- Garba, Z.N.; Zhou, W.; Lawan, I.; Xiao, W.; Zhang, M.; Wang, L.; Chen, L.; Yuan, Z. An overview of chlorophenols as contaminants and their removal from wastewater by adsorption: A review. J. Environ. Manag. 2019, 241, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Czaplicka, M. Sources and transformations of chlorophenols in the natural environment. Sci. Total Environ. 2004, 322, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Karci, A. Degradation of chlorophenols and alkylphenol ethoxylates, two representative textile chemicals, in water by advanced oxidation processes: The state of the art on transformation products and toxicity. Chemosphere 2014, 99, 1–18. [Google Scholar] [CrossRef]
- Villegas, L.G.C.; Mashhadi, N.; Chen, M.; Mukherjee, D.; Taylor, K.E.; Biswas, N. A Short Review of Techniques for Phenol Removal from Wastewater. Curr. Pollut. Rep. 2016, 2, 157–167. [Google Scholar] [CrossRef] [Green Version]
- European Environment Agency. Industrial Waste Water Treatment—Pressures on Europes’s Environment, 1st ed.; No 23/2018; Granger, M., Ed.; Publications Office of the European Union: Copehnagen, Denmark, 2019; p. 39. [Google Scholar] [CrossRef]
- Igbinosa, E.O.; Odjadjare, E.E.; Chigor, V.N.; Igbinosa, I.H.; Emoghene, A.O.; Ekhaise, F.O.; Igiehon, N.O.; Idemudia, O.G. Toxicological profile of chlorophenols and their derivatives in the environment: The public health perspective. Sci. World J. 2013, 2013, 460215. [Google Scholar] [CrossRef] [Green Version]
- Espinoza-Montero, P.J.; Vasquez-Medrano, R.; Ibanez, J.G.; Frontana-Uribe, B.A. Efficient Anodic Degradation of Phenol Paired to Improved Cathodic Production of H2O2 at BDD Electrodes. J. Electrochem. Soc. 2013, 160, G3171–G3177. [Google Scholar] [CrossRef]
- Ahmed, S.; Rasul, M.G.; Martens, W.N.; Brown, R.; Hashib, M.A. Heterogeneous photocatalytic degradation of phenols in wastewater: A review on current status and developments. Desalination 2010, 261, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Pera-Titus, M.; García-Molina, V.; Baños, M.A.; Giménez, J.; Esplugas, S. Degradation of chlorophenols by means of advanced oxidation processes: A general review. Appl. Catal. B Environ. 2004, 47, 219–256. [Google Scholar] [CrossRef]
- Teodosiu, C.; Gilca, A.F.; Barjoveanu, G.; Fiore, S. Emerging pollutants removal through advanced drinking water treatment: A review on processes and environmental performances assessment. J. Clean. Prod. 2018, 197, 1210–1221. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H. Catalytic ozonation for water and wastewater treatment: Recent advances and perspective. Sci. Total Environ. 2020, 704, 135249. [Google Scholar] [CrossRef]
- Mangindaan, D.; Lin, G.Y.; Kuo, C.J.; Chien, H.W. Biosynthesis of silver nanoparticles as catalyst by spent coffee ground/recycled poly(ethylene terephthalate) composites. Food Bioprod. Process. 2020, 121, 193–201. [Google Scholar] [CrossRef]
- Suzuki, H.; Araki, S.; Yamamoto, H. Evaluation of advanced oxidation processes (AOP) using O3, UV, and TiO2 for the degradation of phenol in water. J. Water Process Eng. 2015, 7, 54–60. [Google Scholar] [CrossRef]
- Amado-Piña, D.; Roa-Morales, G.; Barrera-Díaz, C.; Balderas-Hernandez, P.; Romero, R.; Martín del Campo, E.; Natividad, R. Synergic effect of ozonation and electrochemical methods on oxidation and toxicity reduction: Phenol degradation. Fuel 2017, 198, 82–90. [Google Scholar] [CrossRef]
- Poznyak, T.; Vivero, J. Degradation of aqueous phenol and chlorinated phenols by ozone. Ozone Sci. Eng. 2005, 27, 447–458. [Google Scholar] [CrossRef]
- Jaramillo-Sierra, B.; Mercado-Cabrera, A.; Peña-Eguiluz, R.; Hernández-Arias, A.N.; López-Callejas, R.; RodríGuez-Méndez, B.G.; Valencia-Alvarado, R. Assessing some advanced oxidation processes in the abatement of phenol aqueous solutions. Environ. Prot. Eng. 2019, 45, 23–38. [Google Scholar] [CrossRef]
- Oputu, O.U.; Fatoki, O.S.; Opeolu, B.O.; Akharame, M.O. Degradation Pathway of Ozone Oxidation of Phenols and Chlorophenols as Followed by LC-MS-TOF. Ozone Sci. Eng. 2020, 42, 294–318. [Google Scholar] [CrossRef]
- Van Aken, P.; Van den Broeck, R.; Degrève, J.; Dewil, R. The effect of ozonation on the toxicity and biodegradability of 2,4-dichlorophenol-containing wastewater. Chem. Eng. J. 2015, 280, 728–736. [Google Scholar] [CrossRef]
- Hirvonen, A.; Trapido, M.; Hentunen, J.; Tarhanen, J. Formation of hydroxylated and dimeric intermediates during oxidation of chlorinated phenols in aqueous solution. Chemosphere 2000, 41, 1211–1218. [Google Scholar] [CrossRef]
- Quispe, C.; Valdés, C.; Delgadillo, A.; Villaseñor, J.; Cheel, J.; Pecchi, G. Toxicity studies during the degradation of pentachlorophenol by ozonation in the presence of MnO2/TiO2. J. Chil. Chem. Soc. 2018, 63, 4090–4097. [Google Scholar] [CrossRef] [Green Version]
- Hong, P.K.A.; Zeng, Y. Degradation of pentachlorophenol by ozonation and biodegradability of intermediates. Water Res. 2002, 36, 4243–4254. [Google Scholar] [CrossRef]
- Anotai, J.; Wuttipong, R.; Visvanathan, C. Oxidation and detoxification of pentachlorophenol in aqueous phase by ozonation. J. Environ. Manag. 2007, 85, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Trapido, M.; Hirvonen, A.; Veressinina, Y.; Hentunen, J.; Munter, R. Ozonation, ozone/UV and UV/H2O2 degradation of chlorophenols. Ozone Sci. Eng. 1997, 19, 75–96. [Google Scholar] [CrossRef]
- Beltrán, F.J.; Rivas, F.J.; Gimeno, O. Comparison between photocatalytic ozonation and other oxidation processes for the removal of phenols from water. J. Chem. Technol. Biotechnol. 2005, 80, 973–984. [Google Scholar] [CrossRef]
- Turhan, K.; Uzman, S. Removal of phenol from water using ozone. Desalination 2008, 229, 257–263. [Google Scholar] [CrossRef]
- Zangeneh, H.; Zinatizadeh, A.A.L.; Habibi, M.; Akia, M.; Hasnain Isa, M. Photocatalytic oxidation of organic dyes and pollutants in wastewater using different modified titanium dioxides: A comparative review. J. Ind. Eng. Chem. 2015, 26, 1–36. [Google Scholar] [CrossRef]
- Scotti, R.; D’Arienzo, M.; Morazzoni, F.; Bellobono, I.R. Immobilization of hydrothermally produced TiO2 with different phase composition for photocatalytic degradation of phenol. Appl. Catal. B Environ. 2009, 88, 323–330. [Google Scholar] [CrossRef]
- Arana, J.; Dona-Rodriguez, J.M.; Portillo-Carrizo, D.; Fernández-Rodríguez, C.; Pérez-Pena, J.; Gonzalez Diaz, O.; Navio, J.A.; Macias, M. Applied Catalysis B: Environmental Photocatalytic degradation of phenolic compounds with new TiO2 catalysts. Appl. Catal. B Environ. 2010, 100, 346–354. [Google Scholar] [CrossRef]
- Nickheslat, A.; Amin, M.M.; Izanloo, H.; Fatehizadeh, A.; Mousavi, S.M. Phenol photocatalytic degradation by advanced oxidation process under ultraviolet radiation using titanium dioxide. J. Environ. Public Health 2013, 2013, 815310. [Google Scholar] [CrossRef] [Green Version]
- Dougna, A.A.; Gombert, B.; Kodom, T.; Djaneye-Boundjou, G.; Boukari, S.O.B.; Leitner, N.K.V.; Bawa, L.M. Photocatalytic removal of phenol using titanium dioxide deposited on different substrates: Effect of inorganic oxidants. J. Photochem. Photobiol. A Chem. 2015, 305, 67–77. [Google Scholar] [CrossRef]
- Sampaio, M.J.; Silva, C.G.; Silva, A.M.T.; Vilar, V.J.P.; Boaventura, R.A.R.; Faria, J.L. Photocatalytic activity of TiO2-coated glass raschig rings on the degradation of phenolic derivatives under simulated solar light irradiation. Chem. Eng. J. 2013, 224, 32–38. [Google Scholar] [CrossRef]
- Gar Alalm, M.; Samy, M.; Ookawara, S.; Ohno, T. Immobilization of S-TiO2 on reusable aluminum plates by polysiloxane for photocatalytic degradation of 2,4-dichlorophenol in water. J. Water Process Eng. 2018, 26, 329–335. [Google Scholar] [CrossRef]
- Yin, L.; Shen, Z.; Niu, J.; Chen, J.; Duan, Y. Degradation of Pentachlorophenol and 2,4-Dichlorophenol by Sequential Visible-Light Driven Photocatalysis and Laccase Catalysis. Environ. Sci. Technol. 2010, 44, 9117–9122. [Google Scholar] [CrossRef] [PubMed]
- Gunlazuardi, J.; Lindu, W.A. Photocatalytic degradation of pentachlorophenol in aqueous solution employing immobilized TiO2 supported on titanium metal. J. Photochem. Photobiol. A Chem. 2005, 173, 51–55. [Google Scholar] [CrossRef]
- Ratnawati, R.; Enjarlis, E.; Husnil, Y.A.; Christwardana, M.; Slamet, S. Degradation of Phenol in Pharmaceutical Wastewater using TiO2/Pumice and O3/Active Carbon. Bull. Chem. Reac. Eng. Cat. 2020, 15, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Chen, Y.; Yang, J.; Liu, C.; Zhao, H.; Cao, H. Distinct synergetic effects in the ozone enhanced photocatalytic degradation of phenol and oxalic acid with Fe3+/TiO2 catalyst. Chin. J. Chem. Eng. 2018, 26, 1528–1535. [Google Scholar] [CrossRef]
- Jiang, H.; Dang, C.; Liu, W.; Wang, T. Radical attack and mineralization mechanisms on electrochemical oxidation of p-substituted phenols at boron-doped diamond anodes. Chemosphere 2020, 248, 126033. [Google Scholar] [CrossRef]
- Xing, X.; Ni, J.; Zhu, X.; Jiang, Y.; Xia, J. Maximization of current efficiency for organic pollutants oxidation at BDD, Ti/SnO2-Sb/PbO2, and Ti/SnO2-Sb anodes. Chemosphere 2018, 205, 361–368. [Google Scholar] [CrossRef]
- Weiss, E.; Groenen-Serrano, K.; Savall, A. A comparison of electrochemical degradation of phenol on boron doped diamond and lead dioxide anodes. J. Appl. Electrochem. 2008, 38, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Chatzisymeon, E.; Fierro, S.; Karafyllis, I.; Mantzavinos, D.; Kalogerakis, N.; Katsaounis, A. Anodic oxidation of phenol on Ti/IrO2 electrode: Experimental studies. Catal. Today 2010, 151, 185–189. [Google Scholar] [CrossRef]
- Barışçı, S.; Turkay, O.; Öztürk, H.; Şeker, M.G. Anodic Oxidation of Phenol by Mixed-Metal Oxide Electrodes: Identification of Transformation By-Products and Toxicity Assessment. J. Electrochem. Soc. 2017, 164, E129–E137. [Google Scholar] [CrossRef]
- OECD. Test No. 202: Daphnia sp. Acute Immobilisation Test. In OECD Guidelines for the Testing of Chemicals; Section 2; OECD Publishing: Paris, France, 2004; pp. 1–12. [Google Scholar] [CrossRef]
- Results of Eco-Toxicity Tests of Chemicals Conducted by Ministry of the Environment in Japan. Available online: https://www.env.go.jp/chemi/sesaku/02e.pdf (accessed on 13 October 2020).
- Martins, J.; Teles, L.O.; Vasconcelos, V. Assays with Daphnia magna and Danio rerio as alert systems in aquatic toxicology. Environ. Int. 2007, 33, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Žener, B.; Matoh, L.; Carraro, G.; Miljević, B.; Korošec, R.C. Sulfur-, nitrogen- and platinum-doped titania thin films with high catalytic efficiency under visible-light illumination. Beilstein J. Nanotechnol. 2018, 9, 1629–1640, (in Supplementary Materials). [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | AOP | Phenols | Approx. conc. (mg/L) | Solvent; Initial pH | Max. TT (min) |
| 1 | OZ | PHN | 10 | MQ; 8 | 60 |
| DCP | 10 | ||||
| PCP | 10 | ||||
| 2 | OZ | PHN | 50 | MQ; 8 | 8 |
| DCP | 50 | ||||
| 3 | OZ | PHN, DCP, PCP | 10, 10, 10 (mix) | MQ; 8 | 3 |
| 4 | PC & PO | PHN | 50 | MQ, 8 | 180/300 |
| DCP | 50 | ||||
| PHN | 10 | ||||
| DCP | 10 | ||||
| PCP | 10 | ||||
| 5 | SQ | PHN | 20 | MQ; 8 | 0.2 (OZ); 180 (PC) |
| DCP | 20 | ||||
| PCP | 10 | ||||
| 6 | EO/BDD EO/MMO | PHN PHN | 50 50 | 2 g/L NaCl; 6 | 60 120 |
| 7 | EO/BDD EO/MMO | PHN PHN | 50 50 | 2 g/L Na2SO4; 6 | 160 160 |
| PHN | TT0% | TT0–25% | TT25–50% | TT50–100% | >TT100% |
|---|---|---|---|---|---|
| OZ (50 mg/L) | |||||
| OZ (10 mg/L) | |||||
| PC (50 mg/L) | |||||
| PC (10 mg/L) | |||||
| PO | |||||
| SQ | OZ | PC | PC | PC | |
| BDD/NaCl | |||||
| MMO/NaCl | |||||
| BDD/Na2SO4 | |||||
| MMO/Na2SO4 | |||||
| DCP | TT0% | TT0–25% | TT25–50% | TT50–100% | >TT>100% |
| OZ (50 mg/L) | |||||
| OZ (10 mg/L) | |||||
| PC (50 mg/L) | |||||
| PC (10 mg/L) | |||||
| PO | |||||
| SQ | OZ | OZ | OZ & PC | PC | |
| PCP | TT0% | TT0–25% | TT25–50% | TT50–100% | >TT>100% |
| OZ | |||||
| PC | |||||
| PO | |||||
| SQ | OZ | OZ | OZ & PC | PC | |
| Mixture | TT0% | TT0–25% | TT25–50% | TT50–100% | >TT>100% |
| OZ * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kravos, A.; Žgajnar Gotvajn, A.; Lavrenčič Štangar, U.; Malinović, B.N.; Prosen, H. Combined Analytical Study on Chemical Transformations and Detoxification of Model Phenolic Pollutants during Various Advanced Oxidation Treatment Processes. Molecules 2022, 27, 1935. https://doi.org/10.3390/molecules27061935
Kravos A, Žgajnar Gotvajn A, Lavrenčič Štangar U, Malinović BN, Prosen H. Combined Analytical Study on Chemical Transformations and Detoxification of Model Phenolic Pollutants during Various Advanced Oxidation Treatment Processes. Molecules. 2022; 27(6):1935. https://doi.org/10.3390/molecules27061935
Chicago/Turabian StyleKravos, Aleksander, Andreja Žgajnar Gotvajn, Urška Lavrenčič Štangar, Borislav N. Malinović, and Helena Prosen. 2022. "Combined Analytical Study on Chemical Transformations and Detoxification of Model Phenolic Pollutants during Various Advanced Oxidation Treatment Processes" Molecules 27, no. 6: 1935. https://doi.org/10.3390/molecules27061935
APA StyleKravos, A., Žgajnar Gotvajn, A., Lavrenčič Štangar, U., Malinović, B. N., & Prosen, H. (2022). Combined Analytical Study on Chemical Transformations and Detoxification of Model Phenolic Pollutants during Various Advanced Oxidation Treatment Processes. Molecules, 27(6), 1935. https://doi.org/10.3390/molecules27061935