
The Production and Evaluation of an Electrochemical Sensors for Strychnine and Its Main Metabolite Strychnine N-Oxide for Their Use in Biological Samples


Abstract
1. Introduction
2. Results and Discussion
2.1. Electrochemical Behaviour of Strychnine
2.2. Electrochemical Behaviour of Strychnine N-Oxide Strychnine N-Oxide (SNO) Voltammetry
2.3. Strychnine Molecularly Imprinted Sensor (STN-MIP Sensor)
2.3.1. Selection of Functional Monomers
2.3.2. Fabrication of the STN Imprinted Sensor
2.3.3. Optimization of the Imprinted Sensor
2.3.4. STN-MIP Sensor Voltammetry
2.3.5. Precision and Recovery
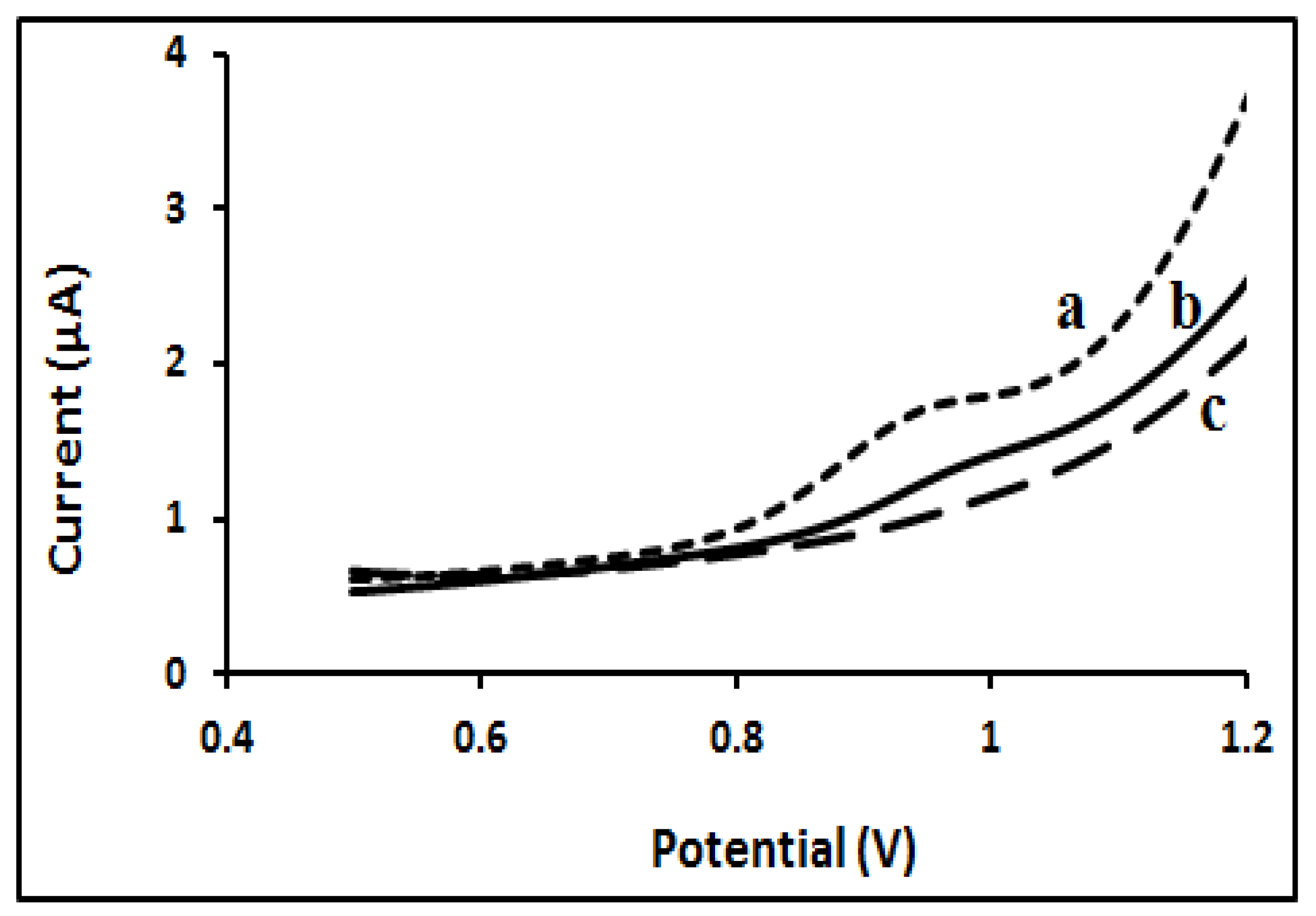
2.3.6. STN-MIP Sensor Selectivity
2.3.7. Application of Developed Electrochemical Methods in Biological Matrix Media
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Instruments and Apparatus
3.3. Solution Preparation
3.4. Experimental Procedures
3.5. Fabrication of GC Electrode with Imprinted Polymer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Philippe, G.; Angenot, L.; Tits, M.; Frederich, M. About the toxicity of some Strychnos species and their alkaloids. Toxicon Off. J. Int. Soc. Toxinology 2004, 44, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Makarovsky, I.; Markel, G.; Hoffman, A.; Schein, O.; Brosh-Nissimov, T.; Tashma, Z.; Dushnitsky, T.; Eisenkraft, A. Strychnine–a killer from the past. Isr. Med. Assoc. J. IMAJ 2008, 10, 142–145. [Google Scholar]
- Wood, D.M.; Webster, E.; Martinez, D.; Dargan, P.I.; Jones, A.L. Case report: Survival after deliberate strychnine self-poisoning, with toxicokinetic data. Crit. Care 2002, 6, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qu, Y.; Wang, D.; Peng, P.; Cai, H.; Gao, Y.; Chen, Z.; Cai, B. Pharmacological evaluation of total alkaloids from nux vomica: Effect of reducing strychnine contents. Molecules 2014, 19, 4395–4408. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, Y. Rapid and sensitive determination of strychnine and brucine in human urine by capillary electrophoresis with field-amplified sample stacking. Biomed. Chromatogr. BMC 2010, 24, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qi, X.; Yang, Y.-W.; Pan, Y.; Bian, H.-M. Toxic effects of strychnine and strychnine N-oxide on zebrafish embryos. Chin. J. Nat. Med. 2014, 12, 760–767. [Google Scholar] [CrossRef]
- Rao, P.S.; Ramanadham, M.; Prasad, M.N.V. Anti-proliferative and cytotoxic effects of Strychnos nux-vomica root extract on human multiple myeloma cell line—RPMI 8226. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2009, 47, 283–288. [Google Scholar] [CrossRef]
- Chen, X.; Lai, Y.; Cai, Z. Simultaneous analysis of strychnine and brucine and their major metabolites by liquid chromatography-electrospray ion trap mass spectrometry. J. Anal. Toxicol. 2012, 36, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, C.G. Handbook of Toxicology of Chemical Warfare Agents, 1st ed.; Elsevier Inc.: San Diego, CA, USA, 2009; Chapter 14; pp. 199–205. [Google Scholar]
- Liu, X.; Zheng, S.; Jiang, Z.; Liang, C.; Wang, R.; Zhou, Z.; Zhang, Y.; Yu, Y. Rapid separation and identification of Strychnos alkaloids metabolites in rats by ultra high performance liquid chromatography with linear ion trap Orbitrap mass spectrometry. J. Sep. Sci. 2014, 37, 764–774. [Google Scholar] [CrossRef]
- Tang, H.B.; Cai, H.L.; Li, H.D.; Zhang, L.J.; Li, X.L.; Tang, J.H.; Chen, M.L. HPLC-DAD method for comprehensive quality control of Semen Strychni. Pharm. Biol. 2013, 51, 1378–1383. [Google Scholar] [CrossRef]
- Tian, J.X.; Peng, C.; Xu, L.; Tian, Y.; Zhang, Z.J. In vitro metabolism study of Strychnos alkaloids using high-performance liquid chromatography combined with hybrid ion trap/time-of-flight mass spectrometry. Biomed. Chromatogr. BMC 2013, 27, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Frederich, M.; Choi, Y.H.; Verpoorte, R. Quantitative analysis of strychnine and Brucine in Strychnos nux-vomica using 1H-NMR. Planta Medica 2003, 69, 1169–1171. [Google Scholar] [PubMed]
- Oguri, K.; Tanimoto, Y.; Mishima, M.; Yoshimura, H. Metabolic fate of strychnine in rats. Xenobiotica Fate Foreign Compd. Biol. Syst. 1989, 19, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Dhalwal, K.; Shinde, V.M.; Namdeo, A.G.; Mahadik, K.R.; Kadam, S.S. Development and validation of a TLC-densitometric method for the simultaneous quantitation of strychnine and brucine from Strychnos spp.and its formulations. J. Chromatogr. Sci. 2007, 45, 706–709. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, J.; Wang, X.; Qu, Y.-G.; Chen, Z.-P.; Cai, H.; Liu, X.; Xu, F.; Lu, T.-L.; Cai, B.-C. Analgesic and anti-inflammatory activity and pharmacokinetics of alkaloids from seeds of Strychnos nux-vomica after transdermal administration: Effect of changes in alkaloid composition. J. Ethnopharmacol. 2012, 139, 181–188. [Google Scholar] [CrossRef]
- Ahmad, S.; Saleem, K.; Kamal, A.; Ahmad, F.J.; Kamal, Y.T. Simultaneous HPTLC determination of strychnine and brucine in strychnos nux-vomica seed. J. Pharm. Bioallied Sci. 2012, 4, 134–139. [Google Scholar] [CrossRef]
- Duverneuil, C.; de la Grandmaison, G.L.; de Mazancourt, P.; Alvarez, J.C. Liquid chromatography/photodiode array detection for determination of strychnine in blood: A fatal case report. Forensic Sci. Int. 2004, 141, 17–21. [Google Scholar] [CrossRef]
- Stahl, R.S.; Arjo, W.M.; Wagner, K.K.; Furcolow, C.; Nolte, D.L.; Johnston, J.J. Development of a high performance liquid chromatography/mass spectroscopy method for the determination of strychnine concentrations in insects used to assess potential risks to insectivores. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 811, 257–262. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, J.; Xing, J.; He, Y.; Guo, D. Analysis of strychnine and brucine in postmortem specimens by RP-HPLC: A case report of fatal intoxication. J. Anal. Toxicol. 2004, 28, 141–144. [Google Scholar] [CrossRef]
- Jablonski, J.E.; Schlesser, J.E.; Mariappagoudar, P. HPLC-UV method for nicotine, strychnine, and aconitine in dairy products. J. Agric. Food Chem. 2006, 54, 7460–7465. [Google Scholar] [CrossRef]
- Song, X.-Y.; Shi, Y.-P.; Chen, J. Carbon nanotubes reinforced hollow fiber solid phase microextraction for the determination of strychnine and brucine in urine. Talanta 2013, 116, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Van Eenoo, P.; Deventer, K.; Roels, K.; Delbeke, F.T. Quantitative LC-MS determination of strychnine in urine after ingestion of a Strychnos nux-vomica preparation and its consequences in doping control. Forensic Sci. Int. 2006, 164, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Si, D.; Liu, C. Determination of strychnine and brucine in rat plasma using liquid chromatography electrospray ionization mass spectrometry. J. Pharm. Biomed. Anal. 2009, 49, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, R.; Li, H.; Yan, M.; Lei, Y. Ultra-performance liquid chromatography-tandem mass spectrometric method for the determination of strychnine and brucine in mice plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2714–2719. [Google Scholar] [CrossRef] [PubMed]
- Marques, E.; Gil, F.; Proença, P.; Monsanto, P.; Oliveira, M.; Castanheira, A.; Vieira, D. Analytical method for the determination of strychnine in tissues by gas chromatography/mass spectrometry: Two case reports. Forensic Sci. Int. 2000, 110, 145–152. [Google Scholar] [CrossRef]
- Rosano, T.G.; Hubbard, J.D.; Meola, J.M.; Swift, T.A. Fatal strychnine poisoning: Application of gas chromatography and tandem mass spectrometry. J. Anal. Toxicol. 2000, 24, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Greene, R.; Meatherall, R. Dermal exposure to strychnine. J. Anal. Toxicol. 2001, 25, 344–347. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; He, X.; Qi, S.; Gao, W.; Chen, X.; Hu, Z. Separation and determination of strychnine and brucine in Strychnos nux-vomica L.and its preparation by nonaqueous capillary electrophoresis. J. Pharm. Biomed. Anal. 2006, 41, 400–407. [Google Scholar] [CrossRef]
- Zheng, X.; Xiao, H.; Hoshi, T.; Anzai, J.-I.; Li, G. Voltammetric Behavior of Strychnine, and its Determination in Strychno Nux-Vomica Seeds Extract. Microchim Acta 2005, 152, 69–74. [Google Scholar] [CrossRef]
- Behpour, M.; Ghoreishi, S.M.; Khayatkashani, M.; Motaghedifard, M. A new method for the simultaneous analysis of strychnine and brucine in Strychnos nux-vomica unprocessed and processed seeds using a carbon-paste electrode modified with multi-walled carbon nanotubes. Phytochem. Anal. PCA 2011, 23, 95–102. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Xu, J.J.; Lian, H.Z.; Li, X.Y.; Chen, H.Y. Rapid separation of strychnine and brucine on a dynamically modified poly(dimethylsiloxane) microchip followed by electrochemical detection. Anal. Bioanal. Chem 2006, 384, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Qiu, P.; Kokot, S. Study of the voltammetric behaviour of maleic hydrazide and its determination at a hanging mercury drop electrode. Talanta 2004, 63, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Guziejewski, D.; Skrzypek, S.; Ciesielski, W. Square wave adsorptive stripping voltammetric determination of diazinon in its insecticidal formulations. Environ. Monit. Assess. 2012, 184, 6575–6582. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhao, Y.-X.; Xiao, B.-L.; Moosavi-Movahedi, A.A.; Ghourchian, H.; Sheibani, N. Direct electrochemistry of hemoglobin immobilized on a functionalized multi-walled carbon nanotubes and gold nanoparticles nanocomplex-modified glassy carbon electrode. Sensors 2013, 13, 8595–8611. [Google Scholar] [CrossRef] [PubMed]
- Gowda, J.I.; Nandibewoor, S.T. Electrochemical behavior of paclitaxel and its determination at glassy carbon electrode. Asian J. Pharm. Sci. 2014, 9, 42–49. [Google Scholar] [CrossRef]
- Hrichi, H.; Louhaichi, M.R.; Monser, L.; Adhoum, N. Gliclazide voltammetric sensor based on electropolymerized molecularly imprinted polypyrrole film onto glassy carbon electrode. Sens. Actuators B 2014, 204, 42–49. [Google Scholar] [CrossRef]
- Ahuja, T.; Mir, I.A.; Kumar, D.; Rajesh. Biomolecular immobilization on conducting polymers for biosensing applications. Biomaterials 2007, 28, 791–805. [Google Scholar] [CrossRef]
- Mamo, S.K.; Gonzalez-Rodriguez, J. Development of a molecularly imprinted polymer-based sensor for the electrochemical determination of triacetone triperoxide (TATP). Sensors 2014, 14, 23269–23282. [Google Scholar] [CrossRef]
- Tahar, N.B.; Savall, A. Electropolymerization of phenol on a vitreous carbon electrode in acidic aqueous solution at different temperatures. J. Appl. Electrochem. 2011, 41, 983–989. [Google Scholar] [CrossRef][Green Version]
- Schweiger, B.; Kim, J.; Kim, Y.J.; Ulbricht, M. Electropolymerized molecularly imprinted polypyrrole film for sensing of clofibric acid. Sensors 2015, 15, 4870–4889. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interferent Molecules | Concentration (µM) a | Signal Change (%) b | RSD (%) (N = 3) |
|---|---|---|---|
| Brucine | 67.08 | 18.4 | 1.77 |
| SNO | 45.36 | 14.96 | 7.01 |
| Scopolamine | 45.98 | 14.0 | 11.35 |
| Interference Media | Analytical Technique | Concentration Spiked (µM) | Mean (µM) | Recovered Percentage (%) |
|---|---|---|---|---|
| Plasma | GC/CV | 25 | 20.2 | 80.8 |
| GC/DPV | 25 | 20.7 | 82.8 | |
| DPV/STN-MIP | 25 | 21.9 | 87.6 | |
| Urine | GC/CV | 25 | 22.1 | 88.4 |
| GC/DPV | 25 | 25.7 | 102.8 | |
| DPV/STN-MIP | 25 | 21.7 | 86.8 |
| Interference Media | Concentration Spiked (µM) | Mean (µM) | Recovered Percentage (%) |
|---|---|---|---|
| Plasma | 100 | 89.01 | 89.01% |
| 200 | 177.04 | 88.52% | |
| Urine | 100 | 93.6 | 93.6% |
| 200 | 183.2 | 91.6% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qader, B.; Hussain, I.; Baron, M.; Estevez-Brito, R.; Cassella, J.P.; Gonzalez-Rodriguez, J. The Production and Evaluation of an Electrochemical Sensors for Strychnine and Its Main Metabolite Strychnine N-Oxide for Their Use in Biological Samples. Molecules 2022, 27, 1826. https://doi.org/10.3390/molecules27061826
Qader B, Hussain I, Baron M, Estevez-Brito R, Cassella JP, Gonzalez-Rodriguez J. The Production and Evaluation of an Electrochemical Sensors for Strychnine and Its Main Metabolite Strychnine N-Oxide for Their Use in Biological Samples. Molecules. 2022; 27(6):1826. https://doi.org/10.3390/molecules27061826
Chicago/Turabian StyleQader, Bakhtiyar, Issam Hussain, Mark Baron, Rafael Estevez-Brito, John Paul Cassella, and Jose Gonzalez-Rodriguez. 2022. "The Production and Evaluation of an Electrochemical Sensors for Strychnine and Its Main Metabolite Strychnine N-Oxide for Their Use in Biological Samples" Molecules 27, no. 6: 1826. https://doi.org/10.3390/molecules27061826
APA StyleQader, B., Hussain, I., Baron, M., Estevez-Brito, R., Cassella, J. P., & Gonzalez-Rodriguez, J. (2022). The Production and Evaluation of an Electrochemical Sensors for Strychnine and Its Main Metabolite Strychnine N-Oxide for Their Use in Biological Samples. Molecules, 27(6), 1826. https://doi.org/10.3390/molecules27061826