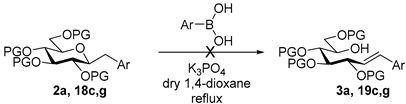
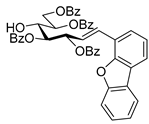
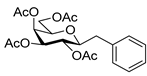
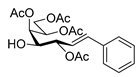
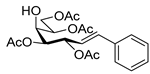
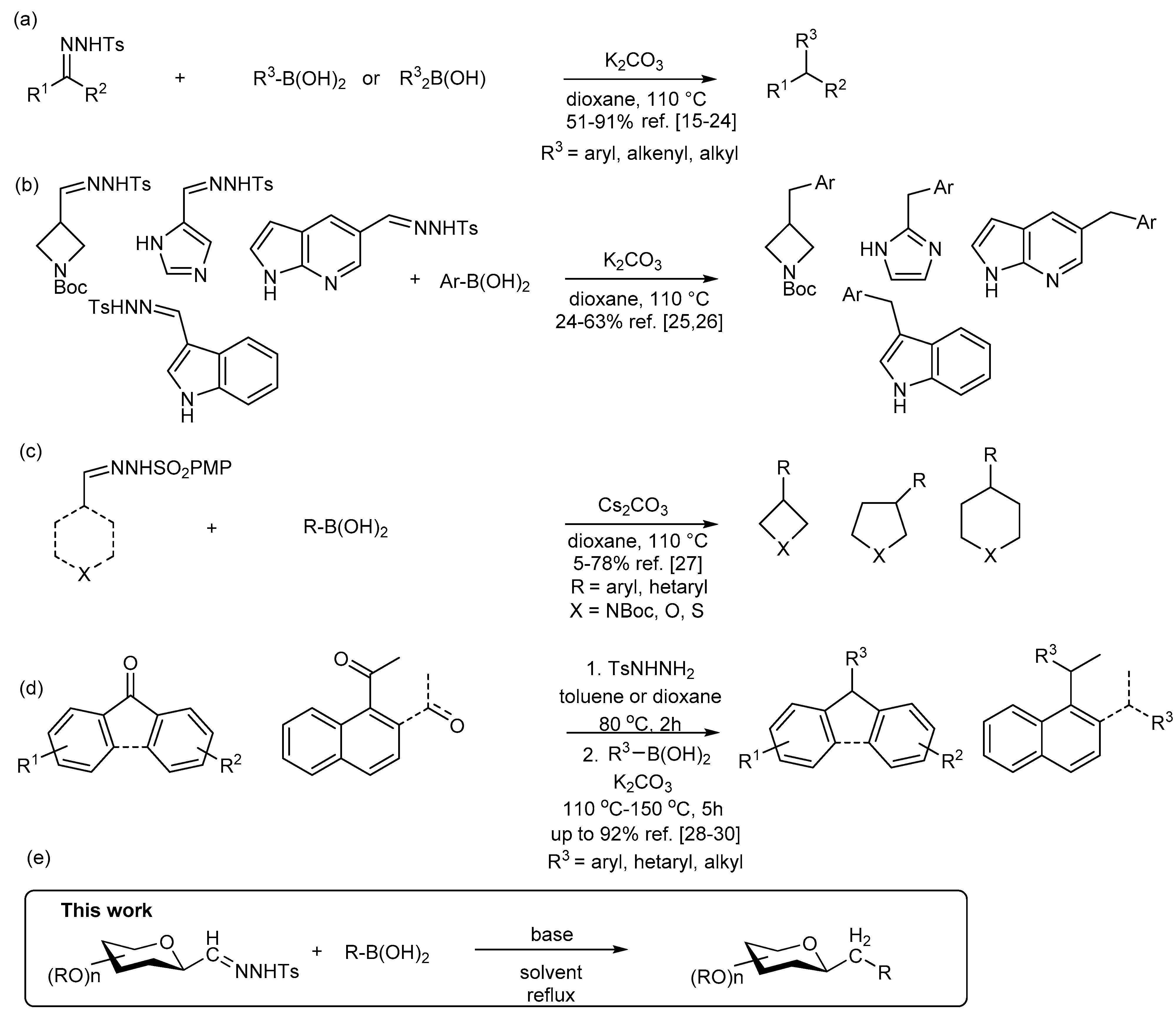
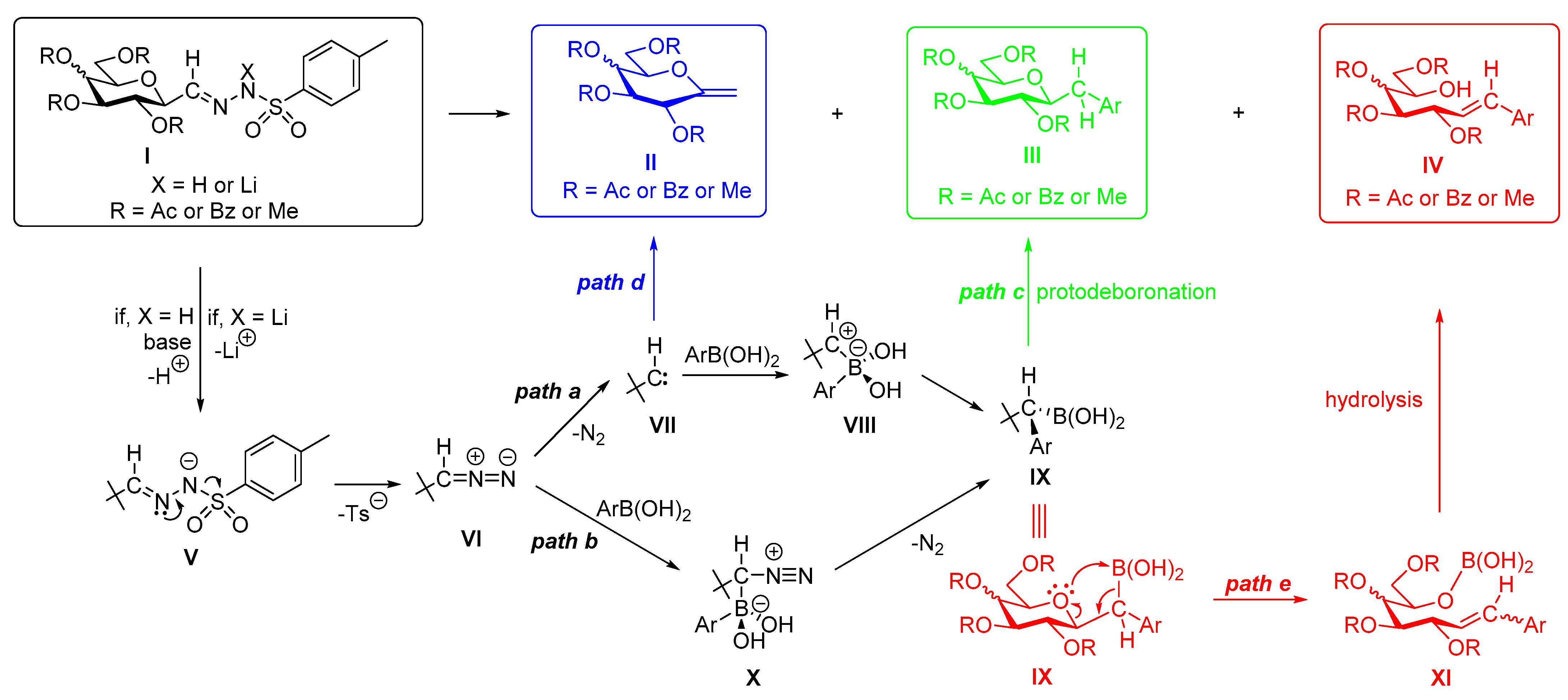
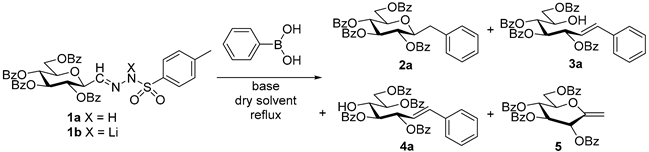
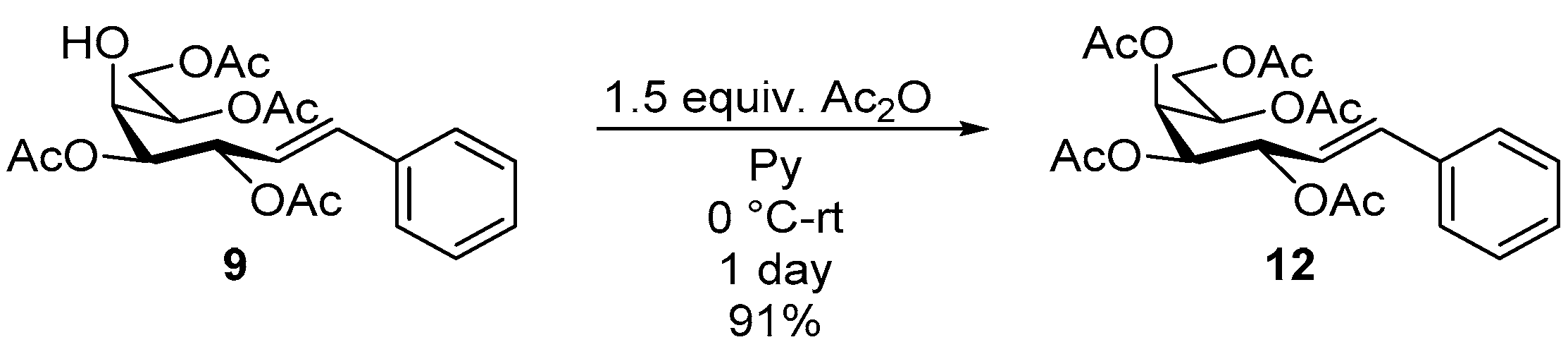4.3. Characterization of Anhydro-Heptitols 2
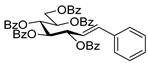
4.3.1. 2,6-Anhydro-3,4,5,7-Tetra-O-Benzoyl-1-Deoxy-1-Phenyl-d-glycero-d-gulo-Heptitol (2a)
Isolated from a reaction of tosylhydrazone
1a (0.10 g, 0.13 mmol), phenylboronic acid (1.5 equiv., 0.02 g, 0.19 mmol), and K
3PO
4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 3 mg (4%) of
2a as a white amorphous product. Optical rotation, NMR and MS spectra are identical with those reported [
13].
4.3.2. 2,6-Anhydro-3,4,5,7-Tetra-O-Benzoyl-1-Deoxy-1-(Naphth-2-yl)-d-glycero-d-gulo-Heptitol (2b)
Isolated from a reaction of tosylhydrazone 1a (0.10 g, 0.13 mmol), naphthalen-2-ylboronic acid (20 equiv., 0.44 g, 2.57 mmol), and K3PO4 (10 equiv., 0.27 g, 1.29 mmol) according to General procedure I by column chromatography (1:4 EtOAc–hexane) to yield 4 mg (4%) of 2b as a pale brown amorphous solid. Rf: 0.42 (1:2 EtOAc–hexane); [α]D + 6 (c 0.16, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.99–7.19 (27H, m, aromatics), 5.90 (1H, pszeudo t, J4,5 9.6 Hz, H-4), 5.62 (1H, pseudo t, J5,6 9.7 Hz, H-5), 5.52 (1H, pseudo t, J3,4 9.6 Hz, H-3), 4.57 (1H, dd, J7a,7b 12.0 Hz, H-7a), 4.41 (1H, dd, H-7b), 4.09 (1H, ddd, J1a,2 5.1, J1b,2 6.6, J2,3 9.8 Hz, H-2), 4.04 (1H, ddd, J6,7a 2.7, J6,7b 6.3 Hz, H-6), 3.12 (1H, dd, J1a,1b 14.8 Hz, H-1a), 3.08 (1H, dd, H-1b). 13C NMR (90 MHz, CDCl3) δ 166.3, 166.1, 165.6, 165.5 (4 × CO), 136.6–124.7 (aromatics), 79.2 (C-2), 76.3 (C-6), 74.7 (C-4), 72.6 (C-3), 70.1 (C-5), 63.6 (C-7), 38.3 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 743.2252, found: [M + Na]+ = 743.2253; C45H36O9 (720.24).
![Molecules 27 01795 i009]()
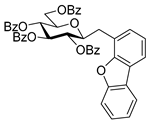
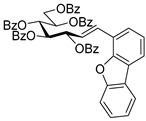
4.3.3. 2,6-Anhydro-3,4,5,7-Tetra-O-Benzoyl-1-(4-Dibenzo[b,d]furanyl)-1-Deoxy-d-glycero-d-gulo-Heptitol (2c)
Isolated from a reaction of tosylhydrazone 1a (0.10 g, 0.13 mmol), dibenzo[b,d]furan-4-ylboronic acid (20 equiv., 0.55 g, 2.57 mmol), and K3PO4 (10 equiv., 0.27 g, 1.29 mmol) according to General procedure I by column chromatography (1:3 EtOAc–hexane) to yield 30 mg pale brown amorphous solid containing 2c and 5 in 1:1 ratio. Rf: 0.50 (1:2 EtOAc–hexane). 1H NMR (400 MHz, CDCl3) δ 8.21–6.93 (27H, m, aromatics), 5.91 (1H, pseudo t, J4,5 9.5 Hz, H-4), 5.64 (1H, pseudo t, J5,6 9.8 Hz, H-5), 5.52 (1H, pseudo t, J3,4 9.8 Hz, H-3), 4.56 (1H, dd, J7a,7b 12.0 Hz, H-7a), 4.42 (1H, dd, H-7b), 4.33 (1H, ddd, J1a,2 3.2, J1b,2 8.0, J2,3 9.8 Hz, H-2), 4.07 (1H, ddd, J6,7a 2.9, J6,7b 5.9 Hz, H-6), 3.44 (1H, dd, J1a,1b 14.6 Hz, H-1a), 3.29 (1H, dd, H-1b). 13C NMR (100 MHz, CDCl3) δ 166.3, 166.1, 165.5 (4 × CO), 135.6–110.4 (aromatics), 77.9 (C-2), 76.2 (C-6), 74.8 (C-4), 72.5 (C-3), 70.1 (C-5), 63.5 (C-7), 32.1 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 761.2381, found: [M + H]+ = 761.2379; C47H36O10 (760.23).
![Molecules 27 01795 i010]()
4.3.4. 2,6-Anhydro-3,4,5,7-Tetra-O-Benzoyl-1-(3-Chlorophenyl)-1-Deoxy-d-glycero-d-gulo-Heptitol (2f)
Isolated from a reaction of tosylhydrazone 1a (0.10 g, 0.13 mmol), 3-chlorophenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:4 EtOAc–hexane) to yield 13 mg white amorphous solid containing 2f and 5 in 1:2 ratio. Rf: 0.48 (1:2 EtOAc–hexane). 1H NMR (400 MHz, CDCl3) δ 8.13–7.76 (12H, m, aromatics), 7.63–6.94 (12H, m, aromatics), 5.89 (1H, pseudo t, J4,5 9.7 Hz, H-4), 5.60 (1H, pseudo t, J5,6 9.7 Hz, H-5), 5.45 (1H, pseudo t, J3,4 9.5 Hz, H-3), 4.57 (1H, dd, J7a,7b 12.1 Hz, H-7a), 4.42 (1H, dd, H-7b), 4.05 (1H, ddd, J6,7a 2.8, J6,7b 6.2 Hz, H-6), 3.98 (1H, ddd, J1a,2 5.3, J1b,2 6.6, J2,3 9.7 Hz, H-2), 2.92 (1H, dd, J1a,1b 15.0 Hz, H-1a), 2.90 (1H, dd, H-1b). 13C NMR (90 MHz, CDCl3) δ 166.3, 166.1, 165.7, 165.6 (4 × CO), 156.3–125.7 (aromatics), 78.8 (C-2), 76.4 (C-6), 74.6 (C-4), 72.6 (C-3), 70.1 (C-5), 63.5 (C-7), 37.4 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 727.1705, found: [M + Na]+ = 727.1708; C41H33ClO9 (704.18).
![Molecules 27 01795 i011]()
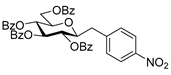
4.3.5. 2,6-Anhydro-3,4,5,7-Tetra-O-Benzoyl-1-Deoxy-1-(4-Nitrophenyl)-d-glycero-d-gulo-Heptitol (2h)
Isolated from a reaction of tosylhydrazone 1a (0.30 g, 0.39 mmol), 4-nitrophenylboronic acid (20 equiv., 1.30 g, 7.72 mmol), and K3PO4 (10 equiv., 0.82 g, 3.86 mmol) according to General procedure I by column chromatography (1:3 EtOAc–hexane) to yield 32 mg pale brown amorphous solid containing 2h and 5 in 4:1 ratio. Rf: 0.44 (1:2 EtOAc–hexane). 1H NMR (400 MHz, CDCl3) δ 8.32–7.72 (8H, m, aromatics), 7.69–7.16 (16H, m, aromatics), 5.90 (1H, pseudo t, J4,5 9.5 Hz, H-4), 5.58 (1H, pseudo t, J5,6 9.8 Hz, H-5), 5.45 (1H, pseudo t, J3,4 9.7 Hz, H-3), 4.53 (1H, dd, J7a,7b 12.2 Hz, H-7a), 4.48 (1H, dd, H-7b), 4.05 (1H, ddd, J6,7a 3.2, J6,7b 6.6 Hz, H-6), 3.99 (1H, ddd, J1a,2 5.1, J1b,2 7.0, J2,3 9.7 Hz, H-2), 3.03 (1H, dd, J1a,1b 14.3 Hz, H-1a), 3.02 (1H, dd, H-1b). 13C NMR (90 MHz, CDCl3) δ 166.4, 166.2, 165.7, 165.6 (4 × CO), 161.7–115.1 (aromatics), 78.3 (C-2), 76.4 (C-6), 74.5 (C-4), 72.5 (C-3), 70.0 (C-5), 63.3 (C-7), 37.8 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 738.1946, found: [M + Na]+ = 738.1950; C41H33NO11 (715.21).
![Molecules 27 01795 i012]()
4.4. Characterization of Hept-1-Enitols 3 and 4
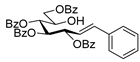
4.4.1. (E)-3,4,5,7-Tetra-O-Benzoyl-1,2-Dideoxy-1-Phenyl-d-gluco-Hept-1-Enitol (3a)
Prepared from tosylhydrazone 1a (0.80 g, 1.03 mmol), phenylboronic acid (20 equiv., 2.51 g, 20.60 mmol), and K3PO4 (10 equiv., 2.19 g, 10.30 mmol) according to General procedure I. Purified by column chromatography (1:4 EtOAc–hexane) to yield 484 mg (70%) of 3a as a white amorphous solid. Rf: 0.36 (1:2 EtOAc–hexane); [α]D + 21 (c 0.20, CH2Cl2). 1H NMR (500 MHz, CDCl3) δ 8.18–7.82 (8H, m, aromatics), 7.64–7.15 (17H, m, aromatics), 6.78 (1H, d, J1,2 15.9 Hz, H-1), 6.32 (1H, dd, J2,3 6.9 Hz, H-2), 6.14–6.02 (2H, m, H-3, H-4), 5.76 (1H, dd, J4,5 0.8, J5,6 8.9 Hz, H-5), 4.53 (1H, dd, J6,7a 2.6, J7a,7b 11.9 Hz, H-7a), 4.34 (1H, dd, J6,7b 5.7 Hz, H-7b), 4.21–4.11 (1H, m, H-6), 3.58 (1H, d, J6,OH 4.3 Hz, OH). 13C NMR (125 MHz, CDCl3) δ 167.3, 166.7, 165.6, 165.4 (4 × CO), 136.7 (C-1), 136.3–125.9 (aromatics), 122.1 (C-2), 73.9 (C-3), 73.3 (C-4), 71.3 (C-5), 68.6 (C-6), 65.5 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 693.2095, found: [M + Na]+ = 693.2095; C41H34O9 (670.22).
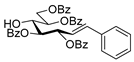
4.4.2. (E)-3,4,6,7-Tetra-O-Benzoyl-1,2-Dideoxy-1-Phenyl-d-gluco-Hept-1-Enitol (4a)
Isolated from a reaction of tosylhydrazone 1a (0.30 g, 0.39 mmol), phenylboronic acid (20 equiv., 0.94 g, 7.72 mmol), and K3PO4 (10 equiv., 8.20 g, 3.86 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 24 mg yellow amorphous solid containing 4a and 3a in 10:2 ratio. Rf: 0.37 (1:2 EtOAc–hexane). 1H NMR (400 MHz, CDCl3) δ 8.22–7.77 (8H, m, aromatics), 7.63–7.06 (17H, m, aromatics), 6.99 (1H, d, J1,2 15.6 Hz, H-1), 6.31 (1H, dd, J2,3 8.0 Hz, H-2), 6.23 (1H, pseudo t, J3,4 8.6 Hz, H-3), 5.82 (1H, dd, J4,5 1.3 Hz, H-4), 5.44 (1H, ddd, J6,7a 3.3, J6,7b 4.4, J5,6 8.0 Hz, H-6), 4.81 (1H, dd, J7a,7b 12.4 Hz, H-7a), 4.74 (1H, dd, H-7b), 4.39 (1H, pseudo t, H-5), 3.25 (1H, d, J5,OH 8.4 Hz, OH). 13C NMR (100 MHz, CDCl3) δ 167.3, 166.7, 165.7, 165.4 (4 × CO), 136.9 (C-1), 136.3–124.1 (aromatics), 122.7 (C-2), 74.6 (C-3), 72.4 (C-4), 71.7 (C-6), 68.5 (C-5), 63.4 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 693.2095, found: [M + Na]+ = 693.2096; C41H34O9 (670.22).
![Molecules 27 01795 i014]()
4.4.3. (E)-3,4,5,7-Tetra-O-Benzoyl-1,2-Dideoxy-1-Naphth-2-yl-d-gluco-Hept-1-Enitol (3b) and (E)-3,4,6,7-Tetra-O-Benzoyl-1,2-Dideoxy-1-Naphth-2-yl-d-gluco-Hept-1-Enitol (4b)
Isolated from a reaction of tosylhydrazone 1a (0.10 g, 0.13 mmol), naphthalen-2-ylboronic acid (20 equiv., 0.44 g, 2.57 mmol), and K3PO4 (10 equiv., 0.27 g, 1.29 mmol) according to General procedure I by column chromatography (1:4 EtOAc–hexane) to yield 70 mg pale brown amorphous solid containing 3b and 4b in 1.5:1 ratio. Rf: 0.25 (1:2 EtOAc–hexane).
3b: 1H NMR (400 MHz, CDCl3) δ 8.20–7.03 (27H, m, aromatics), 6.94 (1H, d, J1,2 15.9 Hz, H-1), 6.45 (1H, dd, J2,3 6.7 Hz, H-2), 6.19–6.09 (2H, m, H-3, H-4), 5.82 (1H, dd, J4,5 1.2, J5,6 8.9 Hz, H-5), 4.54 (1H, dd, J6,7a 3.0, J7a,7b 11.9 Hz, H-7a), 4.35 (1H, dd, J6,7b 5.7 Hz, H-7b), 4.24–4.13 (1H, m, H-6), 3.66 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 167.2, 166.7, 165.6, 165.4 (4 × CO), 136.6 (C-1), 136.4–123.3 (aromatics), 122.4 (C-2), 73.9 (C-3), 73.2 (C-4), 71.3 (C-5), 68.5 (C-6), 65.5 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 743.2252, found: [M + Na]+ = 743.2250; C45H36O9 (720.24).
4b: 1H NMR (400 MHz, CDCl3) δ 8.20–7.03 (28H, m, aromatics, H-1), 6.44 (1H, dd, J1,2 15.8, J2,3 8.4 Hz, H-2), 6.31 (1H, pseudo t, J3,4 8.9 Hz, H-3), 5.88 (1H, dd, J4,5 1.4 Hz, H-4), 5.48 (1H, ddd, J6,7a 3.3, J6,7b 4.4, J5,6 8.0 Hz, H-6), 4.82 (1H, dd, J7a,7b 12.4 Hz, H-7a), 4.75 (1H, dd, H-7b), 4.44 (1H, d, H-5), 3.57 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 166.9, 166.3, 165.8, 165.4 (4 × CO), 136.9 (C-1), 136.4–123.0 (aromatics), 123.1 (C-2), 74.8 (C-3), 72.5 (C-4), 71.6 (C-6), 68.4 (C-5), 63.4 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 743.2252, found: [M + Na]+ = 743.2254; C45H36O9 (720.24).
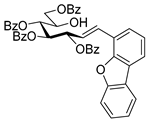
4.4.4. (E)-3,4,5,7-Tetra-O-Benzoyl-1-(4-Dibenzo[b,d]furanyl)-1,2-Dideoxy-d-gluco-Hept-1-Enitol (3c)
Prepared from tosylhydrazone 1a (0.10 g, 0.13 mmol), dibenzo[b,d]furan-4-ylboronic acid (1.5 equiv., 0.04 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I. Purified by column chromatography (1:2 EtOAc–hexane) to yield 16 mg (16%) of 3c as a pale brown amorphous solid. Rf: 0.32 (1:2 EtOAc–hexane); [α]D + 5 (c 0.11, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.38–7.68 (12H, m, aromatics), 7.64–7.16 (15H, m, aromatics), 7.15–6.92 (2H, m, H-1, H-2), 6.22 (1H, dd, J2,3 5.5, J3,4 8.0 Hz, H-3), 6.16 (1H, dd, J4,5 1.7 Hz, H-4), 5.87 (1H, dd, J5,6 8.9 Hz, H-5), 4.54 (1H, dd, J6,7a 3.0, J7a,7b 11.9 Hz, H-7a), 4.35 (1H, dd, J6,7b 5.7 Hz, H-7b), 4.23–4.15 (1H, m, H-6), 3.60 (1H, d, J6,OH 5.3 Hz, OH). 13C NMR (100 MHz, CDCl3) δ 167.2, 166.7, 165.6, 165.4 (4 × CO), 130.8 (C-1), 156.5–111.9 (aromatics), 125.9 (C-2), 74.1 (C-3), 73.3 (C-4), 71.3 (C-5), 68.6 (C-6), 65.5 (C-7). C47H36O10 (760.23). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 783.2201, found: [M + Na]+ = 783.2202; C47H36O10 (760.23).
![Molecules 27 01795 i016]()
4.4.5. (E)-3,4,6,7-Tetra-O-Benzoyl-1-(4-Dibenzo[b,d]furanyl)-1,2-Dideoxy-d-gluco-Hept-1-Enitol (4c)
Prepared from tosylhydrazone 1a (0.10 g, 0.13 mmol), dibenzo[b,d]furan-4-ylboronic acid (20 equiv., 0.55 g, 2.57 mmol), and K3PO4 (10 equiv., 0.27 g, 1.29 mmol) according to General procedure I. Purified by column chromatography (1:3 EtOAc–hexane) to yield 29 mg (30%) of 4c as a yellow amorphous solid. Rf: 0.32 (1:2 EtOAc–hexane); [α]D + 5 (c 0.11, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.23–6.76 (27H, m, aromatics), 7.02 (1H, d, J1,2 16.2 Hz, H-1), 6.97 (1H, dd, J2,3 8.2 Hz, H-2), 6.29 (1H, pseudo t, J3,4 9.0 Hz, H-3), 5.91 (1H, dd, J4,5 1.5 Hz, H-4), 5.44 (1H, ddd, J6,7a 3.5, J6,7b 4.4, J5,6 8.0 Hz, H-6), 4.77 (1H, dd, J7a,7b 12.4 Hz, H-7a), 4.68 (1H, dd, H-7b), 4.50–4.41 (1H, m, H-5), 3.28 (1H, d, J5,OH 6.0 Hz, OH). 13C NMR (100 MHz, CDCl3) δ 167.1, 166.9, 165.9, 165.8 (4 × CO), 131.7 (C-1), 156.3–111.0 (aromatics), 120.6 (C-2), 75.1 (C-3), 72.5 (C-4), 71.7 (C-6), 68.4 (C-5), 63.3 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 783.2201, found: [M + Na]+ = 783.2202; C47H36O10 (760.23).
![Molecules 27 01795 i017]()
4.4.6. (E)-3,4,5,7-Tetra-O-Benzoyl-1,2-Dideoxy-1-(4-Methylphenyl)-d-gluco-Hept-1-Enitol (3d) and (E)-3,4,6,7-Tetra-O-Benzoyl-1,2-Dideoxy-1-(4-Methylphenyl)-d-gluco-Hept-1-enitol (4d)
Isolated from a reaction of tosylhydrazone 1a (0.10 g, 0.13 mmol), 4-methylphenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 44 mg pale yellow amorphous solid containing 3d and 4d in 2:1 ratio. Rf: 0.38 (1:2 EtOAc–hexane).
![Molecules 27 01795 i018]()
3d: 1H NMR (400 MHz, CDCl3) δ 8.20–7.81 (8H, m, aromatics), 7.64–7.01 (16H, m, aromatics), 6.74 (1H, d, J1,2 15.9 Hz, H-1), 6.26 (1H, dd, J2,3 6.7 Hz, H-2), 6.12–6.02 (2H, m, H-3, H-4), 5.75 (1H, dd, J4,5 1.1, J5,6 8.9 Hz, H-5), 4.52 (1H, dd, J6,7a 2.9, J7a,7b 11.9 Hz, H-7a), 4.34 (1H, dd, J6,7b 5.9 Hz, H-7b), 4.20–4.10 (1H, m, H-6), 3.60 (1H, d, J6,OH 5.2 Hz, OH), 2.33 (3H, s, CH3). 13C NMR (100 MHz, CDCl3) δ 167.3, 166.7, 165.6, 165.4 (4 × CO), 136.8 (C-1), 139.5–126.1 (aromatics), 120.9 (C-2), 74.0 (C-3), 73.4 (C-4), 71.4 (C-5), 68.6 (C-6), 65.5 (C-7), 21.4 (CH3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 707.2252, found: [M + Na]+ = 707.2251; C42H36O9 (684.24).
4d: 1H NMR (400 MHz, CDCl3) δ 8.21–7.73 (8H, m, aromatics), 7.72–7.01 (16H, m, aromatics), 6.96 (1H, d, J1,2 14.9 Hz, H-1), 6.25 (1H, dd, J2,3 6.6 Hz, H-2), 6.21 (1H, pseudo t, J3,4 8.6 Hz, H-3), 5.81 (1H, dd, J4,5 1.3 Hz, H-4), 5.44 (1H, ddd, J6,7a 3.4, J6,7b 4.5, J5,6 8.5 Hz, H-6), 4.79 (1H, dd, J7a,7b 12.4 Hz, H-7a), 4.74 (1H, dd, H-7b), 4.39 (1H, pseudo t, J5,OH 8.8 Hz, H-5), 3.16 (1H, bs, OH), 2.35 (3H, s, CH3). 13C NMR (100 MHz, CDCl3) δ 167.1, 166.9, 165.9, 165.3 (4 × CO), 135.9 (C-1), 139.5–115.0 (aromatics), 121.6 (C-2), 74.7 (C-3), 72.5 (C-4), 71.7 (C-6), 68.5 (C-5), 63.4 (C-7), 21.4 (CH3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 707.2252, found: [M + Na]+ = 707.2254; C42H36O9 (684.24).
4.4.7. (E)-3,4,5,7-Tetra-O-Benzoyl-1,2-Dideoxy-1-(4-Methoxyphenyl)-d-gluco-Hept-1-Enitol (3e) and (E)-3,4,6,7-Tetra-O-Benzoyl-1,2-Dideoxy-1-(4-Methoxyphenyl)-d-gluco-Hept-1-Enitol (4e)
Isolated from a reaction of tosylhydrazone 1a (0.10 g, 0.13 mmol), 4-methoxyphenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 43 mg yellow amorphous solid containing 3e and 4e in 2.5:1 ratio. Rf: 0.31 (1:2 EtOAc–hexane).
![Molecules 27 01795 i019]()
3e: 1H NMR (400 MHz, CDCl3) δ 8.28–7.67 (8H, m, aromatics), 7.65–7.09 (14H, m, aromatics), 6.81 (2H, d, J 8.8 Hz, aromatics), 6.71 (1H, d, J1,2 15.7 Hz, H-1), 6.25–6.11 (1H, m, H-2), 6.10–6.02 (2H, m, H-3, H-4), 5.75 (1H, dd, J4,5 1.0, J5,6 8.9 Hz, H-5), 4.52 (1H, dd, J6,7a 3.0, J7a,7b 11.9 Hz, H-7a), 4.34 (1H, dd, J6,7b 5.9 Hz, H-7b), 4.21–4.09 (1H, m, H-6), 3.80 (3H, s, OCH3), 3.63 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 167.3, 166.7, 165.6, 165.4 (4 × CO), 136.5 (C-1), 160.9–112.7 (aromatics), 119.6 (C-2), 74.2 (C-3), 73.4 (C-4), 71.4 (C-5), 68.5 (C-6), 65.5 (C-7), 55.4 (OCH3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 723.2201, found: [M + Na]+ = 723.2204; C42H36O10 (700.23).
4e: 1H NMR (400 MHz, CD3OD) δ 8.19–7.74 (8H, m, aromatics), 7.65–7.12 (14H, m, aromatics), 6.96 (1H, d, J1,2 15.8 Hz, H-1), 6.90 (2H, d, J 8.7 Hz, aromatics), 6.32 (1H, dd, J2,3 8.2 Hz, H-2), 6.21 (1H, pseudo t, J3,4 9.1 Hz, H-3), 5.82 (1H, dd, J4,5 1.5 Hz, H-4), 5.45 (1H, ddd, J6,7a 2.5, J6,7b 5.1 Hz, H-6), 4.93 (1H, dd, J7a,7b 12.2 Hz, H-7a), 4.57 (1H, dd, H-7b), 4.52 (1H, dd, J5,6 9.1 Hz, H-5), 3.80 (3H, s, OCH3), 3.58 (1H, bs, OH). 13C NMR (90 MHz, CDCl3) δ 167.0, 166.9, 165.8, 165.4 (4 × CO), 136.6 (C-1), 160.9–112.7 (aromatics), 120.3 (C-2), 74.5 (C-3), 72.5, 71.7 (C-4, C-6), 68.8 (C-5), 63.4 (C-7), 55.5 (OCH3). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 701.2381, found: [M + H]+ = 701.2381; C42H36O10 (700.23).
4.4.8. (E)-3,4,5,7-Tetra-O-Benzoyl-1-(3-Chlorophenyl)-1,2-Dideoxy-d-gluco-Hept-1-Enitol (3f) and (E)-3,4,6,7-Tetra-O-Benzoyl-1-(3-Chlorophenyl)-1,2-Dideoxy-d-gluco-Hept-1-Enitol (4f)
Isolated from a reaction of tosylhydrazone 1a (0.10 g, 0.13 mmol), 3-chlorophenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:4 EtOAc–hexane) to yield 60 mg white amorphous solid containing 3f and 4f in 2:1 ratio with two unidentified species. Rf: 0.35 (1:2 EtOAc–hexane).
![Molecules 27 01795 i020]()
3f: 1H NMR (400 MHz, CDCl3) δ 8.18–7.82 (8H, m, aromatics), 7.64–7.02 (16H, m, aromatics), 6.70 (1H, d, J1,2 15.9 Hz, H-1), 6.34 (1H, dd, J2,3 6.9 Hz, H-2), 6.12–6.04 (2H, m, H-3, H-4), 5.76 (1H, dd, J4,5 0.6, J5,6 8.9 Hz, H-5), 4.54 (1H, dd, J6,7a 3.0, J7a,7b 11.9 Hz, H-7a), 4.34 (1H, dd, J6,7b 5.7 Hz, H-7b), 4.23–4.14 (1H, m, H-6), 3.64 (1H, d, J6,OH 3.9 Hz, OH). 13C NMR (90 MHz, CDCl3) δ 167.1, 166.7, 165.7, 165.5 (4 × CO), 134.9 (C-1), 138.4–123.4 (aromatics), 123.7 (C-2), 73.5 (C-3), 73.0 (C-4), 71.2 (C-5), 68.5 (C-6), 65.5 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 727.1705, found: [M + Na]+ = 727.1706; C41H33ClO9 (704.18).
4f: 1H NMR (400 MHz, CDCl3) δ 8.18–7.79 (8H, m, aromatics), 7.69–7.06 (16H, m, aromatics), 6.91 (1H, d, J1,2 15.8 Hz, H-1), 6.32 (1H, dd, J2,3 8.0 Hz, H-2), 6.21 (1H, pseudo t, J3,4 8.4 Hz, H-3), 5.81 (1H, dd, J4,5 1.5 Hz, H-4), 5.43 (1H, ddd, J6,7a 3.3, J6,7b 4.3, J5,6 8.0 Hz, H-6), 4.84 (1H, dd, J7a,7b 12.4 Hz, H-7a), 4.73 (1H, dd, H-7b), 4.38–4.27 (1H, m, H-5), 3.34 (1H, d, J5,OH 8.4 Hz, OH). 13C NMR (90 MHz, CDCl3) δ 167.1, 166.8, 165.8, 165.4 (4 × CO), 135.2 (C-1), 153.1–123.4 (aromatics), 124.4 (C-2), 74.4 (C-3), 72.4 (C-4), 71.7 (C-6), 68.4 (C-5), 63.4 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 727.1705, found: [M + Na]+ = 727.1706; C41H33ClO9 (704.18).
4.4.9. 3,4,5,7-Tetra-O-Benzoyl-1-(4-Chlorophenyl)-1,2-Dideoxy-d-gluco-Hept-1-Enitol (3g)
Prepared from tosylhydrazone 1a (0.10 g, 0.13 mmol), 4-chlorophenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I. Purified by column chromatography (1:2 acetone–hexane) to yield 62 mg (68%) of 3g as a white amorphous solid. Rf: 0.36 (1:2 EtOAc–hexane); [α]D + 9 (c 0.57, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.16–7.85 (8H, m, aromatics), 7.64–7.12 (16H, m, aromatics), 6.71 (1H, d, J1,2 16.0 Hz, H-1), 6.34–6.24 (1H, m, H-2), 6.10–6.02 (2H, m, H-3, H-4), 5.74 (1H, dd, J4,5 0.9, J5,6 8.9 Hz, H-5), 4.53 (1H, dd, J6,7a 2.9, J7a,7b 11.9 Hz, H-7a), 4.34 (1H, dd, J6,7b 5.7 Hz, H-7b), 4.21–4.10 (1H, m, H-6), 3.60 (1H, d, J6,OH 5.1 Hz, OH). 13C NMR (100 MHz, CDCl3) δ 167.2, 166.8, 165.5, 165.4 (4 × CO), 135.2 (C-1), 134.7–127.2 (aromatics), 122.7 (C-2), 73.7 (C-3), 73.1 (C-4), 71.2 (C-5), 68.5 (C-6), 65.5 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 727.1705, found: [M + Na]+ = 727.1703; C41H33ClO9 (704.18).
4.6. General Procedure II for the Synthesis of 1-Aryl-3,4,5,6,7-Penta-O-Benzoyl-1,2-Dideoxy-d-gluco-Hept-1-Enitols 11 and 12
A mixture of 1-aryl-tetra-O-benzoyl-1,2-dideoxy-d-gluco-hept-1-enitol (3 and 4, 1 mmol) and dry pyridine (6.3 mmol) were dissolved in dry chloroform (3 mL). Then, benzoyl–chloride (7 mmol) was added dropwise to the solution. The reaction mixture was stirred and heated at 80 °C. When TLC (1:2 EtOAc–hexane) showed complete consumption of the starting compound (~2 h), the mixture was cooled down. The organic layer was washed with 2M aqueous hydrogen chloride solution (1 × 3 mL), cold, saturated sodium hydrogen carbonate solution (1 × 3 mL), water (1 × 3 mL), and then dried on anhydrous magnesium sulfate. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (1:2 EtOAc–hexane) to give hept-1-enitols.
4.6.1. (E)-3,4,5,6,7-Penta-O-Benzoyl-1,2-Dideoxy-1-Phenyl-d-gluco-Hept-1-Enitol (11a)
Prepared from (E)-3,4,5,7-tetra-O-benzoyl-1,2-dideoxy-1-phenyl-d-gluco-hept-1-enitol 3a and (E)-3,4,6,7-tetra-O-benzoyl-1,2-dideoxy-1-phenyl-d-gluco-hept-1-enitol 4a (0.10 g, 0.15 mmol) according to General procedure II. Purified by column chromatography (1:2 EtOAc–hexane) to yield 104 mg (90%) of 11a as a white amorphous solid. Rf: 0.41 (1:2 EtOAc–hexane); [α]D − 2 (c 0.50, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.24–7.84 (8H, m, aromatics), 7.66–7.17 (17H, m, aromatics), 6.80 (1H, d, J1,2 15.9 Hz, H-1), 6.40–6.29 (1H, m, H-2), 6.18 (1H, dd, J4,5 2.0 Hz, H-5), 6.12–6.04 (2H, m, H-3, H-4), 5.91 (1H, ddd, J6,7a 3.6, J6,7b 5.9, J5,6 7.2 Hz, H-6), 4.82 (1H, dd, J7a,7b 12.3 Hz, H-7a), 4.55 (1H, dd, H-7b). 13C NMR (100 MHz, CDCl3) δ 166.1, 165.7, 165.5, 165.4, 165.3 (5 × CO), 136.7 (C-1), 135.8–127.0 (aromatics), 122.0 (C-2), 73.8 (C-3), 71.8 (C-4), 69.9, 69.7 (C-5, C-6), 62.8 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 797.2357, found: [M + Na]+ = 797.2355; C48H38O10 (774.25).
4.6.2. (E)-3,4,5,6,7-Penta-O-Benzoyl-1-(4-Dibenzo[b,d]furanyl)-1,2-Dideoxy-d-gluco-Hept-1-Enitol (11b)
Prepared from (E)-3,4,5,7-tetra-O-benzoyl-1-(4-dibenzo[b,d]furanyl)-1,2-dideoxy-d-gluco-hept-1-enitol 3c and (E)-3,4,6,7-tetra-O-benzoyl-1-(4-dibenzo[b,d]furanyl)-1,2-dideoxy-d-gluco-hept-1-enitol 4c (0.05 g, 0.06 mmol), according to General procedure II. Purified by column chromatography (1:2 EtOAc–hexane) to yield 28 mg (55%) of 11b as a yellow amorphous solid. Rf: 0.39 (1:2 EtOAc–hexane); [α]D − 1 (c 0.48, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.44–6.81 (32H, m, aromatics), 7.08–7.04 (2H, m, H-1, H-2), 6.28 (1H, dd, J4,5 2.0 Hz, H-5), 6.20–6.14 (2H, m, H-3, H-4), 5.92 (1H, ddd, J6,7a 3.8, J6,7b 5.8, J5,6 7.1 Hz, H-6), 4.82 (1H, dd, J7a,7b 12.2 Hz, H-7a), 4.53 (1H, dd, H-7b). 13C NMR (100 MHz, CDCl3) δ 166.1, 165.7, 165.5, 165.4, 165.3 (5 × CO), 130.9 (C-1), 162.8–110.9 (aromatics), 125.8 (C-2), 74.0 (C-3), 71.8 (C-4), 69.9 (C-6), 69.6 (C-5), 62.8 (C-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 887.2463, found: [M + Na]+ = 887.2460; C54H40O11 (864.26).
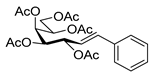
4.6.3. (E)-3,4,5,6,7-Penta-O-Acetyl-1,2-Dideoxy-1-Phenyl-d-galacto-Hept-1-Enitol (12)
3,4,6,7-Tetra-O-acetyl-1,2-dideoxy-1-phenyl-d-galacto-hept-1-enitol (9, 0.12 g, 0.29 mmol) was dissolved in dry pyridine (1 mL) and cooled to 0 °C. Then, acetic anhydride (1.5 equiv., 0.04 mL, 0.04 g, 0.43 mmol) was added dropwise to the solution. The reaction mixture was stirred for a day at room temperature and the pyridine was evaporated. The residue was dissolved in dichloromethane and washed with water (1 × 2 mL), then dried on anhydrous magnesium sulfate. The solution was concentrated under reduced pressure and traces of pyridine were removed by repeated co-evaporations with toluene to yield 122 mg (91%) of 12 as a white amorphous solid. Rf: 0.36 (1:2 EtOAc–hexane); [α]D + 115 (c 0.02, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.57–7.06 (5H, m, aromatics), 6.58 (1H, dd, JAr,1 0.7, J1,2 15.9 Hz, H-1), 5.97 (1H, dd, J2,3 6.1 Hz, H-2), 5.67–5.59 (1H, m, H-3), 5.45 (1H, dd, J5,6 1.8 Hz, H-5), 5.41–5.31 (1H, m, H-6), 5.37 (1H, dd, J3,4 2.5, J4,5 10.0 Hz, H-4), 4.29 (1H, dd, J6,7a 5.0, J7a,7b 11.6 Hz, H-7a), 3.88 (1H, dd, J6,7a 7.5 Hz, H-7b), 2.14, 2.10, 2.08, 2.04, 202 (15H, 5s, 5 × CH3). 13C NMR (90 MHz, CDCl3) δ 170.5, 170.3, 170.1, 169.8 (5 × CO), 133.5 (C-1), 136.5–122.2 (aromatics), 122.9 (C-2), 71.1 (C-3), 69.5 (C-4), 68.1 (C-5), 68.0 (C-6), 62.3 (C-7), 21.0, 20.8, 20.7 (5 × CH3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 487.1575, found: [M + Na]+ = 487.11577; C23H28O10 (464.17).
![Molecules 27 01795 i027]()
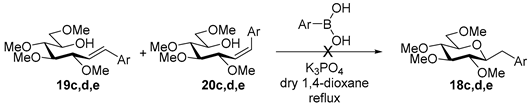
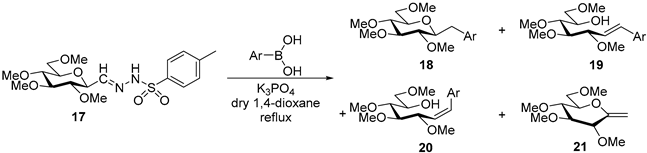
4.8. Characterization of Anhydro-Heptitols 18
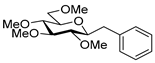
4.8.1. 2,6-Anhydro-1-Deoxy-3,4,5,7-Tetra-O-Methyl-1-Phenyl-d-glycero-d-gulo-Heptitol (18a)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), phenylboronic acid (1.5 equiv., 0.02 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 18 mg amorphous solid containing 18a and 21 in 1.3:1 ratio. Rf: 0.50 (1:2 EtOAc–hexane). 1H NMR (400 MHz, CDCl3) δ 8.34–6.74 (5H, m, aromatics), 3.65 (3H, s, CH3OC-4), 3.59 (3H, s, CH3OC-3), 3.53 (3H, s, CH3OC-5), 3.55–3.50 (2H, m, H-7a, H-7b), 3.36 (3H, s, CH3OC-7), 3.30 (1H, ddd, J1a,2 2.4, J1b,2 8.8, J2,3 8.9 Hz, H-2), 3.23–3.15 (2H, m, H-4, H-5), 3.12 (1H, ddd, J6,7a 2.0, J6,7b 4.0, J5,6 9.8 Hz, H-6), 3.07 (1H, dd, J1a,1b 14.3 Hz, H-1a), 2.90 (1H, pseudo t, J3,4 9.0 Hz, strongly coupled, H-3), 2.74 (1H, dd, H-1b). 13C NMR (90 MHz, CDCl3) δ 139.4–126.1 (aromatics), 89.2 (C-4), 83.7 (C-3), 80.3 (C-2), 80.1 (C-5), 78.8 (C-6), 71.5 (C-7), 60.8 (CH3OC-4) 60.7 (CH3OC-3), 60.4 (CH3OC-5), 59.5 (CH3OC-7), 37.9 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 333.1672, found: [M + Na]+ = 333.1672; C17H26O5 (310.39).
![Molecules 27 01795 i029]()
4.8.2. 2,6-Anhydro-1-Deoxy-1-(4-Dibenzo[b,d]furanyl)-3,4,5,7-Tetra-O-Methyl-d-glycero-d-gulo-Heptitol (18b)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), dibenzo[b,d]furan-4-ylboronic acid (1.5 equiv., 0.04 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:4 EtOAc–hexane) to yield 4 mg (8%) of 18b as a white amorphous solid. Rf: 0.47 (1:2 EtOAc–hexane). 1H NMR (500 MHz, CDCl3) δ 7.93 (1H, d, J 7.7 Hz, aromatic), 7.80 (1H, dd, J 1.1, 7.7 Hz, aromatic), 7.58 (1H, d, J 8.2 Hz, aromatic), 7.47–7.39 (2H, m, aromatics), 7.35–7.30 (1H, m, aromatic), 7.29–7.23 (1H, m, aromatics), 3.67 (3H, s, CH3OC-4), 3.62 (3H, s, CH3OC-3), 3.58 (1H, ddd, J1a,2 2.9, J1b,2 8.9, J2,3 9.2 Hz, H-2), 3.54 (1H, dd, H-1a), 3.53 (3H, s, CH3OC-5), 3.48 (1H, dd, H-7a), 3.46 (1H, dd, J7a,7b 11.2 Hz, H-7b), 3.28 (3H, s, CH3OC-7), 3.26 (1H, pseudo t, J3,4 8.7 Hz, H-4), 3.21 (1H, pseudo t, J4,5 8.8 Hz, H-5), 3.14 (1H, ddd, J6,7a 2.5, J6,7b 3.4, J5,6 9.5 Hz, H-6), 3.09 (1H, dd, J1a,1b 14.4 Hz, H-1b) 3.02 (1H, pseudo t, J3,4 8.9 Hz, H-3). 13C NMR (90 MHz, CDCl3) δ 129.4–110.9 (aromatics), 89.2 (C-4), 84.2 (C-3), 80.2 (C-5), 79.0, 78.9 (C-2, C-6), 71.5 (C-7), 60.9 (CH3OC-4), 60.8 (CH3OC-3), 60.5 (CH3OC-5), 59.5 (CH3OC-7), 32.0 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 423.1778, found: [M + Na]+ = 423.1777; C23H28O6 (400.19).
![Molecules 27 01795 i030]()
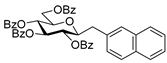
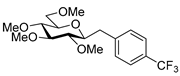
4.8.3. 2,6-Anhydro-1-Deoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Trifluoromethylphenyl)-d-glycero-d-gulo-Heptitol (18c)
Prepared from tosylhydrazone 17 (0.05 g, 0.13 mmol), (4-trifluoromethly)phenylboronic acid (1.5 equiv., 0.04 g, 0.39 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I. Purified by column chromatography (1:3 EtOAc–hexane) to yield 22 mg (45%) of 18c as a white amorphous solid. Rf: 0.50 (1:2 EtOAc–hexane); [α]D − 6 (c 0.30, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.52 (2H, d, J 8.1 Hz, aromatics), 7.38 (2H, d, J 8.1 Hz, aromatics), 3.65 (3H, s, CH3OC-4), 3.59 (3H, s, CH3OC-3), 3.54 (1H, dd, H-7a), 3.53 (3H, s, CH3OC-5), 3.50 (1H, dd, J7a,7b 11.1 Hz, H-7b), 3.36 (3H, s, CH3OC-7), 3.29 (1H, ddd, J1a,2 2.3, J1b,2 8.9, J2,3 9.2 Hz, H-2), 3.24–3.08 (3H, m, H-1a, H-4, H-5), 3.13 (1H, ddd, J6,7a 2.0, J6,7b 3.9, J5,6 9.8 Hz, H-6), 2.88 (1H, pseudo t, J3,4 8.9 Hz, strongly coupled, H-3), 2.80 (1H, dd, J1a,1b 14.2 Hz, H-1b). 13C NMR (100 MHz, CDCl3) δ 143.7–124.1 (aromatics), 89.2 (C-4), 83.6 (C-3), 80.0 (C-2), 79.8 (C-5), 78.8 (C-6), 71.4 (C-7), 60.8 (CH3OC-4), 60.8 (CH3OC-3), 60.5 (CH3OC-5), 59.5 (CH3OC-7), 37.7 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 379.1727, found: [M + H]+ = 379.1727; C18H25F3O5 (378.17).
![Molecules 27 01795 i031]()
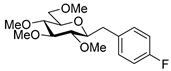
4.8.4. 2,6-Anhydro-1-Deoxy-1-(4-Fluorophenyl)-3,4,5,7-Tetra-O-Methyl-d-glycero-d-gulo-Heptitol (18d)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-fluorophenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:4 EtOAc–hexane) to yield 6 mg (14%) of 18d as a white amorphous solid. Rf: 0.41 (1:2 EtOAc–hexane); [α]D + 0.5 (c 0.20, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.22 (2H, dd, J 5.6, 8.6 Hz, aromatics), 6.94 (2H, t, J 8.8 Hz, aromatics), 3.65 (3H, s, CH3OC-4), 3.58 (3H, s, CH3OC-3), 3.54 (1H, dd, H-7a), 3.53 (3H, s, CH3OC-5), 3.50 (1H, dd, J7a,7b 10.8 Hz, H-7b), 3.37 (3H, s, CH3OC-7), 3.24 (1H, ddd, J1a,2 2.1, J1b,2 8.8, J2,3 9.1 Hz, H-2), 3.21–3.13 (2H, m, H-4, H-5), 3.12 (1H, ddd, J6,7a 1.9, J6,7b 3.6, J5,6 8.7 Hz, H-6), 3.04 (1H, dd, J1a,1b 14.3 Hz, H-1a), 2.87 (1H, pseudo t, J3,4 9.0 Hz, strongly coupled, H-3), 2.71 (1H, dd, H-1b). 13C NMR (90 MHz, CDCl3) δ 131.4–109.0 (aromatics), 89.3 (C-4), 83.6 (C-3), 80.2, (C-2), 80.1 (C-5), 78.8 (C-6), 71.5 (C-7), 60.8 (CH3OC-3, CH3OC-4), 60.5 (CH3OC-5), 59.5 (CH3OC-7), 37.1 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 329.1759, found: [M + Na]+ = 329.1759; C17H25FO5 (328.17).
![Molecules 27 01795 i032]()
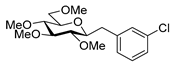
4.8.5. 2,6-Anhydro-1-(3-Chlorophenyl)-1-Deoxy-3,4,5,7-Tetra-O-Methyl-d-glycero-d-gulo-Heptitol (18e)
Prepared from tosylhydrazone 17 (0.05 g, 0.13 mmol), 3-chorophenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I. Purified by column chromatography (1:2 EtOAc–hexane) to yield 13 mg (29%) of 18e as a pale-yellow amorphous solid. Rf: 0.48 (1:2 EtOAc–hexane); [α]D − 3 (c 0.24, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.32–7.24 (1H, m, aromatic), 7.23–7.08 (3H, m, aromatics), 3.65 (3H, s, CH3OC-4), 3.59 (3H, s, CH3OC-3), 3.56 (1H, dd, J7a,7b 11.0 Hz, H-7a), 3.53 (3H, s, CH3OC-5), 3.51 (1H, dd, H-7b), 3.37 (3H, s, CH3OC-7), 3.27 (1H, ddd, J1a,2 2.3, J1b,2 8.8, J2,3 9.1 Hz, H-2), 3.23–3.14 (2H, m, H-4, H-5), 3.13 (1H, ddd, J6,7a 1.6, J6,7b 3.4, J5,6 8.6 Hz, H-6), 3.04 (1H, dd, J1a,1b 14.3 Hz, H-1a), 2.87 (1H, pseudo t, J3,4 8.8 Hz, H-3), 2.71 (1H, dd, H-1b). 13C NMR (90 MHz, CDCl3) δ 141.3–126.0 (aromatics), 89.2 (C-4), 83.5 (C-3), 80.1 (C-2), 79.9 (C-5), 78.8 (C-6), 71.5 (C-7), 60.9 (CH3OC-4), 60.8 (CH3OC-3), 60.5 (CH3OC-5), 59.6 (CH3OC-7), 37.6 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 345.1463, found: [M + H]+ = 345.1460; C17H25ClO5 (344.14).
![Molecules 27 01795 i033]()
4.8.6. 2,6-Anhydro-1-(4-Bromophenyl)-1-Deoxy-3,4,5,7-Tetra-O-Methyl-d-glycero-d-gulo-Heptitol (18f)
Prepared from tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-bromophenylboronic acid (1.5 equiv., 0.04 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I. Purified by column chromatography (1:4 EtOAc–hexane) to yield 11 mg (22%) of 18f as a white amorphous solid. Rf: 0.53 (1:2 EtOAc–hexane); [α]D − 6 (c 0.21, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.38 (2H, d, J 8.4 Hz, aromatics), 7.14 (2H, d, J 8.4 Hz, aromatics), 3.65 (3H, s, CH3OC-4), 3.58 (3H, s, CH3OC-3), 3.53 (3H, s, CH3OC-5), 3.53 (1H, dd, H-7a), 3.49 (1H, dd, J7a,7b 10.8 Hz, H-7b), 3.37 (3H, s, CH3OC-7), 3.24 (1H, ddd, J1a,2 2.2, J1b,2 8.9, J2,3 9.1 Hz, H-2), 3.21–3.13 (2H, m, H-4, H-5), 3.11 (1H, ddd, J6,7a 2.1, J6,7b 3.5, J5,6 9.8 Hz, H-6), 3.02 (1H, dd, J1a,1b 14.3 Hz, H-1a), 2.87 (1H, pseudo t, J3,4 9.0 Hz, strongly coupled, H-3), 2.69 (1H, dd, H-1b). 13C NMR (100 MHz, CDCl3) δ 138.4–119.3 (aromatics), 89.2 (C-4), 83.5 (C-3), 80.0 (C-2, C-5), 78.8 (C-6), 71.4 (C-7), 60.9 (CH3OC-4), 60.8 (CH3OC-3), 60.5 (CH3OC-5), 59.5 (CH3OC-7), 37.3 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 389.0958, found: [M + H]+ = 389.0959; C17H25BrO5 (389.29).
![Molecules 27 01795 i034]()
4.8.7. 2,6-Anhydro-1-Deoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Nitrophenyl)-d-glycero-d-gulo-Heptitol (18g)
Prepared from tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-nitrophenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I. Purified by column chromatography (1:2 EtOAc–hexane) to yield 21 mg (46%) of 18g as a yellow amorphous solid. Rf: 0.26 (1:2 EtOAc–hexane); [α]D + 3 (c 0.22, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 8.13 (2H, d, J 8.7 Hz, aromatics), 7.43 (2H, d, J 8.7 Hz, aromatics), 3.66 (3H, s, CH3OC-4), 3.60 (3H, s, CH3OC-3), 3.53 (3H, s, CH3OC-5), 3.53 (1H, dd, H-7a), 3.49 (1H, dd, J7a,7b 10.8 Hz, H-7b), 3.37 (3H, s, CH3OC-7), 3.29 (1H, ddd, J1a,2 2.4, J1b,2 9.0, J2,3 9.2 Hz, H-2), 3.23–3.14 (3H, m, H-1a, H-4, H-5), 3.12 (1H, ddd, J6,7a 1.3, J6,7b 3.1, J5,6 9.6 Hz, H-6), 2.89 (1H, pseudo t, J3,4 8.8 Hz, strongly coupled, H-3), 2.85 (1H, dd, H-1b). 13C NMR (90 MHz, CDCl3) δ 147.6–121.1 (aromatics), 89.2 (C-4), 83.5 (C-3), 80.0 (C-2), 79.6 (C-5), 78.8 (C-6), 71.4 (C-7), 60.9 (CH3OC-3, CH3OC-4), 60.5 (CH3OC-5), 59.5 (CH3OC-7), 37.8 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 356.1704, found: [M + H]+ = 356.1704; C17H25NO7 (355.16).
![Molecules 27 01795 i035]()
4.8.8. 2,6-Anhydro-1-Deoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Methoxyphenyl)-d-glycero-d-gulo-Heptitol (18h)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-methoxyphenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 4 mg (9%) of 18h as a pale-yellow amorphous solid. Rf: 0.41 (1:2 EtOAc–hexane); [α]D + 5 (c 0.57, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.19 (2H, d, J 8.6 Hz, aromatics), 6.81 (2H, d, J 8.7 Hz, aromatics), 3.79 (3H, s, OCH3), 3.65 (3H, s, CH3OC-4), 3.59 (3H, s, CH3OC-3), 3.55 (1H, dd, J7a,7b 10.9 Hz, H-7a), 3.53 (3H, s, CH3OC-5), 3.50 (1H, dd, H-7b), 3.38 (3H, s, CH3OC-7), 3.24 (1H, ddd, J1a,2 2.3, J1b,2 8.8, J2,3 9.1 Hz, H-2), 3.21–3.14 (2H, m, H-4, H-5), 3.12 (1H, ddd, J6,7a 2.0, J6,7b 3.9, J5,6 9.8 Hz, H-6), 3.01 (1H, dd, J1a,1b 14.3 Hz, H-1a), 2.88 (1H, pseudo t, J3,4 9.0 Hz, strongly coupled, H-3), 2.69 (1H, dd, H-1b). 13C NMR (90 MHz, CDCl3) δ 162.4–109.6 (aromatics), 89.3 (C-4), 83.7 (C-3), 80.5 (C-2), 80.1 (C-5), 78.8 (C-6), 71.6 (C-7), 60.8 (CH3OC-3, CH3OC-4), 60.5 (CH3OC-5), 59.6 (CH3OC-7), 55.4 (OCH3), 37.0 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 341.1959, found: [M + H]+ = 341.1957; C18H28O5 (340.42).
![Molecules 27 01795 i036]()
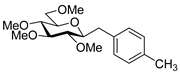
4.8.9. 2,6-Anhydro-1-Deoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Methylphenyl)-d-glycero-d-gulo-Heptitol (18i)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-methylphenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 11 mg white amorphous solid containing 18i and 21 in 3:1 ratio. Rf: 0.48 (1:2 EtOAc–hexane); [α]D + 0.5 (c 0.08, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 7.16 (2H, d, J 7.9 Hz, aromatics), 7.07 (2H, d, J 7.9 Hz, aromatics), 3.65 (3H, s, CH3OC-4), 3.59 (3H, s, CH3OC-3), 3.53 (3H, s, CH3OC-5), 3.55–3.50 (2H, m, H-7a, H-7b), 3.37 (3H, s, CH3OC-7), 3.26 (1H, ddd, J1a,2 2.1, J1b,2 8.8, J2,3 9.1 Hz, H-2), 3.23–3.14 (2H, m, H-4, H-5), 3.11 (1H, ddd, J6,7a 1.9, J6,7b 3.6, J5,6 9.7 Hz, H-6), 3.03 (1H, dd, J1a,1b 14.3 Hz, H-1a), 2.88 (1H, pseudo t, J3,4 9.0 Hz, strongly coupled, H-3), 2.70 (1H, dd, H-1b), 2.31 (3H, s, CH3). 13C NMR (100 MHz, CDCl3) δ 136.2–128.6 (aromatics), 89.3 (C-4), 83.7 (C-3), 80.4 (C-2), 80.1 (C-5), 78.8 (C-6), 71.5 (C-7), 60.8 (CH3OC-3, CH3OC-4), 60.5 (CH3OC-5), 59.6 (CH3OC-7), 37.5 (C-1), 21.2 (CH3). HR-ESI-MS positive mode (m/z): calc. for [M + H]+ = 325.2010, found: [M + H]+ = 325.2008; C18H28O5 (324.42).
![Molecules 27 01795 i037]()
4.9. Characterization of Heptenitols 19 and 20
4.9.1. (E)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-1-Phenyl-d-gluco-Hept-1-Enitol (19a) and (Z)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-1-Phenyl-d-gluco-Hept-1-Enitol (20a)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), phenylboronic acid (1.5 equiv., 0.02 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 29 mg white amorphous solid containing 19a and 20a in 9:1 ratio. Rf: 0.16 (1:2 EtOAc–hexane), [α]D + 28 (c 0.16, CH2Cl2).
![Molecules 27 01795 i038]()
19a: 1H NMR (500 MHz, CDCl3) δ 7.42 (2H, d, J 7.6 Hz, aromatics), 7.38–7.30 (2H, m, aromatics), 7.29–7.23 (1H, m, aromatic), 6.63 (1H, d, J1,2 16.0 Hz, H-1), 6.16 (1H, dd, J2,3 8.2 Hz, H-2), 4.05 (1H, dd, J3,4 6.0 Hz, H-3), 3.96 (1H, ddd, J6,7a 3.9, J6,7b 5.5, J5,6 6.7 Hz, H-6), 3.60 (3H, s, CH3OC-4), 3.59–3.50 (3H, m, H-4, H-7a, H-7b), 3.40 (6H, 2s, CH3OC-5, CH3OC-7), 3.40–3.37 (1H, m, H-5), 3.37 (3H, s, CH3OC-3), 3.32 (1H, bs, OH). 13C NMR (125 MHz, CDCl3) δ 134.0 (C-1), 137.0–126.3 (aromatics), 126.7 (C-2), 83.8 (C-4), 83.4 (C-3), 79.8 (C-5), 73.8 (C-7), 70.2 (C-6), 60.8 (CH3OC-4), 59.4 (CH3OC-5), 59.2 (CH3OC-7), 56.8 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 333.1672, found: [M + Na]+ = 333.1679; C17H26O5 (310.39).
20a: 1H NMR (500 MHz, CDCl3) δ 7.45–7.38 (2H, m, aromatics), 7.38–7.30 (2H, m, aromatics), 7.29–7.23 (1H, m, aromatic), 6.77 (1H, d, J1,2 12.0 Hz, H-1), 5.58 (1H, dd, J2,3 10.0 Hz, H-2), 4.59 (1H, dd, J3,4 4.6 Hz, H-3), 3.99–3.91 (1H, m, H-6), 3.57 (3H, s, CH3OC-4), 3.57–3.49 (3H, m, H-4, H-7a, H-7b), 3.45 (1H, dd, J4,5 3.6, J5,6 6.4 Hz, H-5), 3.40 (3H, s, CH3OC-7), 3.32 (3H, s, CH3OC-5), 3.23 (3H, s, CH3OC-3), 3.02 (1H, bs, OH). 13C NMR (125 MHz, CDCl3) δ 133.9 (C-1), 137.0–126.3 (aromatics), 129.5 (C-2), 84.1 (C-4), 79.6 (C-5), 76.8 (C-3), 73.9 (C-7), 70.4 (C-6), 60.7 (CH3OC-4), 59.2 (CH3OC-7), 59.1 (CH3OC-5), 56.4 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 333.1672, found: [M + Na]+ = 333.1669; C17H26O5 (310.39).
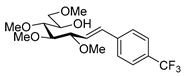
4.9.2. (E)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Trifluoromethylphenyl)-d-gluco-Hept-1-Enitol (19c)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), (4-trifluoromethly)phenylboronic acid (1.5 equiv., 0.04 g, 0.39 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:3 EtOAc–hexane) to yield 11 mg white amorphous solid containing 19c and an unidentified impurity in 3:1 ratio. Rf: 0.41 (1:2 EtOAc–hexane). 1H NMR (500 MHz, CDCl3) δ 8.40–7.40 (4H, m, aromatics), 6.68 (1H, d, J1,2 16.0 Hz, H-1), 6.29 (1H, dd, J2,3 7.7 Hz, H-2), 4.09 (1H, dd, J3,4 5.9 Hz, H-3), 4.00–3.91 (1H, m, H-6), 3.60 (3H, s, CH3OC-4), 3.60–3.53 (3H, m, H-4, H-7a, H-7b), 3.41 (3H, s, CH3OC-5), 3.40 (3H, s, CH3OC-7), 3.39 (3H, s, CH3OC-3), 3.40–3.36 (1H, m, H-5), 3.07 (1H, bs, OH). 13C NMR (90 MHz, CDCl3) δ 132.1 (C-1), 140.0–120.5 (aromatics), 129.5 (C-2), 83.5 (C-4), 82.7 (C-3), 79.7 (C-5), 73.6 (C-7), 70.2 (C-6), 60.8 (CH3OC-4), 59.5 (CH3OC-7), 59.2 (CH3OC-5), 57.1 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 401.1546, found: [M + Na]+ = 401.1542; C18H25F3O5 (378.17).
![Molecules 27 01795 i039]()
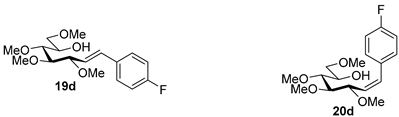
4.9.3. (E)-1,2-Dideoxy-1-(4-Fluorophenyl)-3,4,5,7-Tetra-O-Methyl-d-gluco-Hept-1-Enitol (19d) and (Z)-1,2-Dideoxy-(4-Fluorophenyl)-3,4,5,7-Tetra-O-Methyl-1-d-gluco-Hept-1-Enitol (20d)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-fluorophenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:4 EtOAc–hexane) to yield 31 mg white amorphous solid containing 19d and 20d in 3:1 ratio. Rf: 0.11 (1:2 EtOAc–hexane).
![Molecules 27 01795 i040]()
19d: 1H NMR (400 MHz, CDCl3) δ 7.39 (2H, dd, J 5.4, 8.7 Hz, aromatics), 7.03 (2H, t, J 8.7 Hz, aromatics), 6.60 (1H, d, J1,2 16.0 Hz, H-1), 6.09 (1H, dd, J2,3 8.1 Hz, H-2), 4.04 (1H, dd, J3,4 5.9 Hz, H-3), 4.00–3.91 (1H, m, H-6), 3.59 (3H, s, CH3OC-4), 3.59–3.49 (3H, m, H-4, H-7a, H-7b), 3.40 (6H, 2s, CH3OC-5, CH3OC-7), 3.38 (1H, dd, J4,5 3.1, J5,6 7.3 Hz, H-5), 3.36 (3H, s, CH3OC-3), 3.03 (1H, bs, OH). 13C NMR (90 MHz, CDCl3) δ 132.7 (C-1), 129.7–110.2 (aromatics), 126.4 (C-2), 83.7 (C-4), 83.2 (C-3), 79.8 (C-5), 73.7 (C-7), 70.2 (C-6), 60.8 (CH3OC-4), 59.4 (CH3OC-5), 59.2 (CH3OC-7), 56.9 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 351.1578, found: [M + Na]+ = 351.1579; C17H25FO5 (328.17).
20d: 1H NMR (500 MHz, CDCl3) δ 7.34 (2H, dd, J 5.5, 8.5 Hz, aromatics), 7.03 (2H, t, J 8.7 Hz, aromatics), 6.72 (1H, d, J1,2 11.9 Hz, H-1), 5.66 (1H, dd, J2,3 10.1 Hz, H-2), 4.54 (1H, dd, J3,4 4.8 Hz, H-3), 3.98–3.91 (1H, m, H-6), 3.57 (3H, s, CH3OC-4), 3.57–3.50 (3H, m, H-4, H-7a, H-7b), 3.45 (1H, dd, J4,5 3.3, J5,6 6.6 Hz, H-5), 3.40 (3H, s, CH3OC-7), 3.33 (3H, s, CH3OC-5), 3.22 (3H, s, CH3OC-3), 3.17 (1H, bs, OH). 13C NMR (125 MHz, CDCl3) δ 132.7 (C-1), 131.2–114.5 (aromatics), 129.4 (C-2), 84.1 (C-4), 79.6 (C-5), 76.8 (C-3), 73.9 (C-7), 70.4 (C-6), 60.7 (CH3OC-4), 59.3 (CH3OC-7), 59.0 (CH3OC-5), 56.4 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 351.1578, found: [M + Na]+ = 351.1579; C17H25FO5 (328.17).
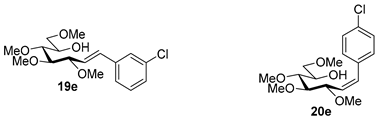
4.9.4. (E)-1-(3-Chlorophenyl)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-d-gluco-Hept-1-Enitol (19e) and (Z)-1-(3-Chlorophenyl)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-d-gluco-Hept-1-Enitol (20e)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), 3-chorophenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 18 mg pale yellow amorphous solid containing 19e and 20e in 9:1 ratio. Rf: 0.13 (1:2 EtOAc–hexane).
![Molecules 27 01795 i041]()
19e: 1H NMR (400 MHz, CDCl3) δ 7.50–7.37 (1H, m, aromatic), 7.36–7.19 (3H, m, aromatics), 6.58 (1H, d, J1,2 16.0 Hz, H-1), 6.19 (1H, dd, J2,3 7.9 Hz, H-2), 4.06 (1H, dd, J3,4 6.2 Hz, H-3), 4.01–3.90 (1H, m, H-6), 3.60 (3H, s, CH3OC-4), 3.59–3.44 (3H, m, H-4, H-7a, H-7b), 3.40 (6H, 2s, CH3OC-5, CH3OC-7), 3.38 (1H, dd, J4,5 2.6, J5,6 7.4 Hz, H-5), 3.37 (3H, s, CH3OC-3), 3.11 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 132.5 (C-1), 138.8–120.5 (aromatics), 124.9 (C-2), 83.5 (C-4), 82.9 (C-3), 79.7 (C-5), 73.6 (C-7), 70.2 (C-6), 60.8 (CH3OC-4), 59.4 (CH3OC-5), 59.2 (CH3OC-7), 57.0 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 367.1283, found: [M + Na]+ = 367.1282; C17H25ClO5 (344.14).
20e: 1H NMR (400 MHz, CDCl3) δ 7.84–6.06 (5H, m, H-1, aromatics), 5.75 (1H, dd, J1,2 11.9, J2,3 10.01 Hz, H-2), 4.52 (1H, dd, J3,4 4.3 Hz, H-3), 4.12–3.71 (1H, m, H-6), 3.57 (3H, s, CH3OC-4), 3.58–3.42 (4H, m, H-4, H-5, H-7a, H-7b), 3.40 (3H, s, CH3OC-7), 3.40–3.23 (3H, m, CH3OC-5), 3.23 (3H, s, CH3OC-3), 3.11 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 132.4 (C-1), 138.8–120.5 (aromatics), 129.3 (C-2), 83.9 (C-4), 83.0 (C-5), 81.4 (C-3), 74.5 (C-7), 73.9 (C-6), 60.4 (CH3OC-4), 59.3 (CH3OC-7), 59.1 (CH3OC-5), 57.0 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 367.1283, found: [M + Na]+ = 367.1282; C17H25ClO5 (344.14).
4.9.5. (E)-1-(4-Bromophenyl)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-d-gluco-Hept-1-Enitol (19f) and (Z)-1-(4-Bromophenyl)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-d-gluco-Hept-1-Enitol (20f)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-bromophenylboronic acid (1.5 equiv., 0.04 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:4 EtOAc–hexane) to yield 20 mg white amorphous solid containing 19f and 20f in 9:1 ratio. Rf: 0.10 (1:2 EtOAc–hexane).
![Molecules 27 01795 i042]()
19f: 1H NMR (400 MHz, CDCl3) δ 7.46 (2H, d, J 8.5 Hz, aromatics), 7.28 (2H, d, J 8.5 Hz, aromatics), 6.57 (1H, d, J1,2 16.0 Hz, H-1), 6.18 (1H, dd, J2,3 7.9 Hz, H-2), 4.04 (1H, dd, J3,4 5.9 Hz, H-3), 3.99–3.90 (1H, m, H-6), 3.59 (3H, s, CH3OC-4), 3.58–3.50 (3H, m, H-4, H-7a, H-7b), 3.40 (3H, s, CH3OC-5), 3.39 (3H, s, CH3OC-7), 3.37 (3H, s, CH3OC-3), 3.37 (1H, dd, J4,5 2.8, J5,6 6.7 Hz, H-5), 3.00 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 132.4 (C-1), 136.1–117.1 (aromatics), 127.6 (C-2), 83.6 (C-4), 83.0 (C-3), 79.7 (C-5), 73.7 (C-7), 70.2 (C-6), 60.7 (CH3OC-4), 59.4 (CH3OC-5), 59.2 (CH3OC-7), 57.0 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 411.0778, found: [M + Na]+ = 411.0777; C17H25BrO5 (389.29).
20f: 1H NMR (400 MHz, CDCl3) δ 7.47 (2H, d, J 8.4 Hz, aromatics), 7.25 (2H, d, J 8.4 Hz, aromatics), 6.69 (1H, d, J1,2 11.9 Hz, H-1), 5.71 (1H, dd, J2,3 10.1 Hz, H-2), 4.53 (1H, dd, J3,4 4.7 Hz, H-3), 3.99–3.90 (1H, m, H-6), 3.57 (3H, s, CH3OC-4), 3.56–3.47 (3H, m, H-4, H-7a, H-7b), 3.45 (1H, dd, J4,5 3.3, J5,6 6.5 Hz, H-5), 3.40 (3H, s, CH3OC-7), 3.34 (3H, s, CH3OC-5), 3.21 (3H, s, CH3OC-3), 3.00 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 132.6 (C-1), 136.1–117.1 (aromatics), 130.2 (C-2), 84.0 (C-4), 79.5 (C-5), 76.8 (C-3), 73.8 (C-7), 70.4 (C-6), 60.7 (CH3OC-4), 59.3 (CH3OC-7), 59.0 (CH3OC-5), 56.4 (CH3OC-3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 411.0778, found: [M + Na]+ = 411.0777; C17H25BrO5 (389.29).
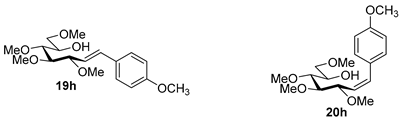
4.9.6. (E)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Methoxyphenyl)-d-gluco-Hept-1-Enitol (19h) and (Z)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Methoxyphenyl)-d-gluco-Hept-1-Enitol (20h)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-methoxyphenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 24 mg pale yellow amorphous solid containing 19h and 20h in 23:1 ratio. Rf: 0.13 (1:2 EtOAc–hexane).
![Molecules 27 01795 i043]()
19h: 1H NMR (400 MHz, CDCl3) δ 7.36 (2H, d, J 8.7 Hz, aromatics), 6.88 (2H, d, J 8.7 Hz, aromatics), 6.57 (1H, d, J1,2 16.0 Hz, H-1), 6.01 (1H, dd, J2,3 8.3 Hz, H-2), 4.02 (1H, dd, J3,4 6.0 Hz, H-3), 3.98–3.91 (1H, m, H-6), 3.82 (3H, s, OCH3), 3.60 (3H, s, CH3OC-4), 3.59–3.53 (2H, m, H-7a, H-7b), 3.54–3.49 (1H, m, H-4), 3.40 (3H, s, CH3OC-5), 3.39 (3H, s, CH3OC-7), 3.38 (1H, dd, J4,5 2.9, J5,6 6.9 Hz, H-5), 3.35 (3H, s, CH3OC-3), 3.02 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 133.6 (C-1), 159.9–112.7 (aromatics), 124.3 (C-2), 83.9 (C-4), 83.6 (C-3), 79.8 (C-5), 73.8 (C-7), 70.3 (C-6), 60.8 (CH3OC-4), 59.4 (CH3OC-5), 59.2 (CH3OC-7), 56.7 (CH3OC-3), 55.5 (OCH3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 363.1778, found: [M + Na]+ = 363.1779; C18H28O5 (340.42).
20h: 1H NMR (400 MHz, CDCl3) δ 7.32 (2H, d, J 8.7 Hz, aromatics), 6.88 (2H, d, J 8.7 Hz, aromatics), 6.69 (1H, d, J1,2 12.0 Hz, H-1), 5.56 (1H, dd, J2,3 10.0 Hz, H-2), 4.63 (1H, dd, J3,4 4.7 Hz, H-3), 3.99–3.90 (1H, m, H-6), 3.82 (3H, s, OCH3), 3.58 (3H, s, CH3OC-4), 3.57–3.49 (3H, m, H-4, H-7a, H-7b), 3.46 (1H, dd, J4,5 3.3, J5,6 6.6 Hz, H-5), 3.40 (3H, s, CH3OC-7), 3.33 (3H, s, CH3OC-5), 3.23 (3H, s, CH3OC-3), 3.02 (1H, bs, OH). 13C NMR (100 MHz, CDCl3) δ 133.3 (C-1), 161.0–112.7 (aromatics), 130.4 (C-2), 84.2 (C-4), 79.6 (C-5), 77.0 (C-3), 73.9 (C-7), 70.4 (C-6), 60.7 (CH3OC-4), 59.3 (CH3OC-7), 59.0 (CH3OC-5), 56.3 (CH3OC-3), 55.4 (OCH3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 363.1778, found: [M + Na]+ = 363.1776; C18H28O5 (340.42).
4.9.7. (E)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Methylphenyl)-d-gluco-Hept-1-Enitol (19i) and (Z)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methyl-1-(4-Methylphenyl)-d-gluco-Hept-1-Enitol (20i)
Isolated from a reaction of tosylhydrazone 17 (0.05 g, 0.13 mmol), 4-methylphenylboronic acid (1.5 equiv., 0.03 g, 0.19 mmol), and K3PO4 (3 equiv., 0.08 g, 0.39 mmol) according to General procedure I by column chromatography (1:2 EtOAc–hexane) to yield 24 mg pale white amorphous solid containing 19i and 20i in 8:1 ratio. Rf: 0.13 (1:2 EtOAc–hexane), [α]D + 28 (c 0.36, CH2Cl2).
![Molecules 27 01795 i044]()
19i: 1H NMR (400 MHz, CDCl3) δ 7.32 (2H, d, J 8.1 Hz, aromatics),7.15 (2H, d, J 7.9 Hz, aromatics), 6.60 (1H, d, J1,2 16.0 Hz, H-1), 6.10 (1H, dd, J2,3 8.3 Hz, H-2), 4.03 (1H, dd, J3,4 6.0 Hz, H-3), 3.98–3.90 (1H, m, H-6), 3.60 (3H, s, CH3OC-4), 3.58–3.49 (3H, m, H-4, H-7a, H-7b), 3.39 (6H, 2s, CH3OC-5, CH3OC-7), 3.39–3.36 (1H, m, H-5), 3.35 (3H, s, CH3OC-3), 3.03 (1H, bs, OH), 2.35 (3H, s, CH3). 13C NMR (100 MHz, CDCl3) δ 134.0 (C-1), 138.3–125.3 (aromatics), 125.5 (C-2), 83.8 (C-4), 83.6 (C-3), 79.8 (C-5), 73.8 (C-7), 70.2 (C-6), 60.8 (CH3OC-4), 59.4 (CH3OC-5), 59.2 (CH3OC-7), 56.7 (CH3OC-3), 21.3 (CH3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 347.1829, found: [M + Na]+ = 347.1828; C18H28O5 (324.42).
20i: 1H NMR (400 MHz, CDCl3) δ 7.28–7.23 (4H, m, aromatics), 6.73 (1H, d, J1,2 12.1 Hz, H-1), 5.62 (1H, dd, J2,3 10.0 Hz, H-2), 4.61 (1H, dd, J3,4 4.6 Hz, H-3), 3.98–3.90 (1H, m, H-6), 3.57 (3H, s, CH3OC-4), 3.58–3.49 (3H, m, H-4, H-7a, H-7b), 3.46 (1H, dd, J4,5 3.4, J5,6 6.5 Hz, H-5), 3.40 (3H, s, CH3OC-7), 3.33 (3H, s, CH3OC-5), 3.23 (3H, s, CH3OC-3), 3.03 (1H, bs, OH), 2.36 (3H, s, CH3). 13C NMR δ 133.7 (C-1), 138.2–125.3 (aromatics), 128.7 (C-2), 84.1 (C-4), 79.6 (C-5), 76.7 (C-3), 73.9 (C-7), 70.5 (C-6), 60.7 (CH3OC-4), 59.2 (CH3OC-7), 59.1 (CH3OC-5), 56.4 (CH3OC-3), 21.3 (CH3). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 347.1829, found: [M + Na]+ = 347.1828; C18H28O5 (324.42).
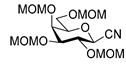
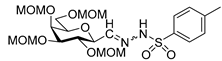
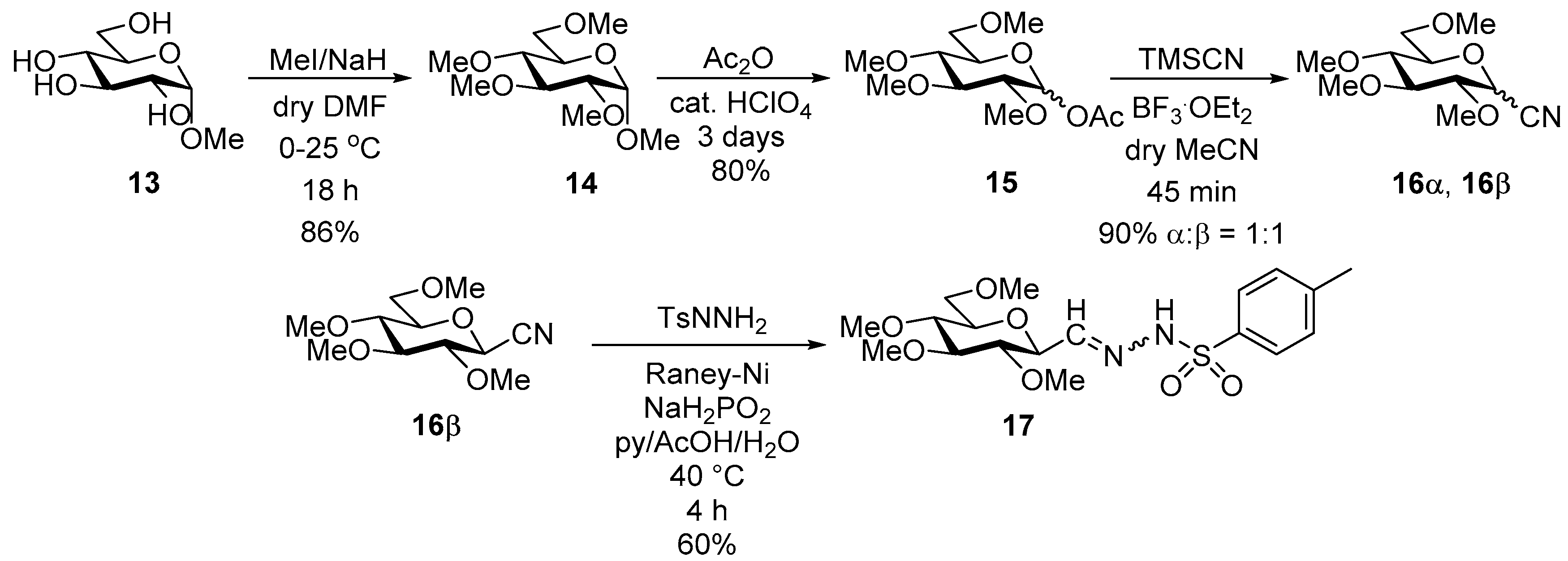
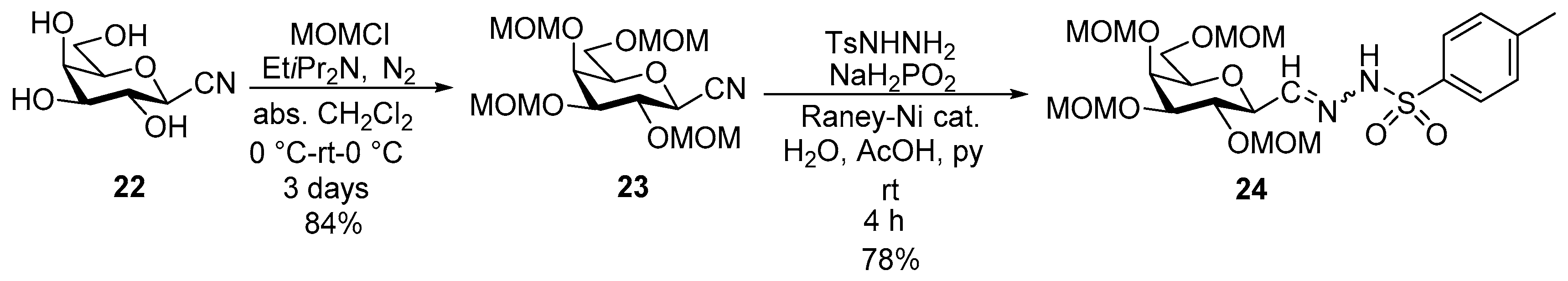
4.11. 2,6-Anhydro-3,4,5,7-Tetra-O-Methoxymethyl-d-glycero-L-manno-Heptose Tosylhydrazone (C-(2,3,4,6-Tetra-O-Methoxymethyl-β-d-Galactopyranosyl) Formaldehyde Tosylhydrazone) (24)
Prepared from cyanide 23 (0.10 g, 0.27 mmol) according to General procedure III. Purified by column chromatography (2:1 EtOAc–hexane) to get two unidentified isomers 24-1 and 24-2.
24-1 yellow oil, 19 mg (13%); Rf: 0.33 (2:1 EtOAc–hexane). 1H NMR (360 MHz, CDCl3) δ 9.50 (1H, s, NH), 7.84–7.75 (2H, m, aromatics), 7.33–7.22 (2H, m, aromatics), 4.89 (1H, d, J 6.8 Hz, CH2), 4.85 (1H, d, J 6.5 Hz, CH2), 4.79 (1H, d, J 6.8 Hz, CH2), 4.73–4.59 (4H, m, CH2), 4.57 (1H, d, J 6.5 Hz, CH2), 4.03 (1H, dd, J4,5 2.4, J5,6 0.6 Hz, H-5), 4.03–3.99 (1H, m, H-2 or H-4), 3.98 (1H, pseudo t, J2,3 9.9, J3,4 9.9 Hz, H-3), 3.78–3.65 (4H, m, H-2 or H-4, H-6, H-7a, H-7b), 3.41, 3.39, 3.21 (12H, 4s, 4 × CH3), 2.42 (3H, s, CH3-Ts). HR-ESI-MS positive mode (m/z): calcd. for [M + H]+ = 537.2113, found: [M + H]+ = 537.2111; C22H36N2O11S (536.20).
24-2 yellow oil, 96 mg (65%); Rf: 0.19 (2:1 EtOAc–hexane).1H NMR (360 MHz, CDCl3) δ 8.25 (1H, s, NH), 7.86–7.73 (2H, m, aromatics), 7.35–7.23 (2H, m, aromatics), 7.05 (1H, d, J1,2 4.4 Hz, H-1), 4.87 (1H, d, J 6.7 Hz, CH2), 4.77 (1H, d, J 6.6 Hz, CH2), 4.72–4.67 (2H, m, CH2), 4.65 (1H, d, J 6.7 Hz, CH2), 4.60 (2H, s, CH2), 4.42 (1H, d, J 6.7 Hz, CH2), 4.02 (1H, dd, J4,5 2.6, J5,6 0.6 Hz, H-5), 3.88–3.78 (2H, m) and 3.75–3.55 (4H, m) and 3.46–3.19 (1H, m): (H-2, H-3, H-4, H-6, H-7a, H-7b), 3.39, 3.32, 3.05 (12H, 4s, 4 × CH3), 2.42 (3H, s, CH3-Ts). 13C NMR (90 MHz, CDCl3) δ 146.6 (C-1), 144.8–127.4 (aromatics), 98.2, 97.6, 96.9, 95.7 (4 × CH2), 79.1, 78.8, 77.3, 74.6, 72.9 (C-2–C-6), 66.9 (C-7), 56.2, 55.9, 55.6 (4 × CH3-Ts). HR-ESI-MS positive mode (m/z): calcd. for [M + H]+ = 537.2113, found: [M + H]+ = 537.2111; C22H36N2O11S (536.20).
4.12. Characterization of Anhydro-Heptitol 25 and Heptenitols 26 and 27
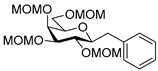
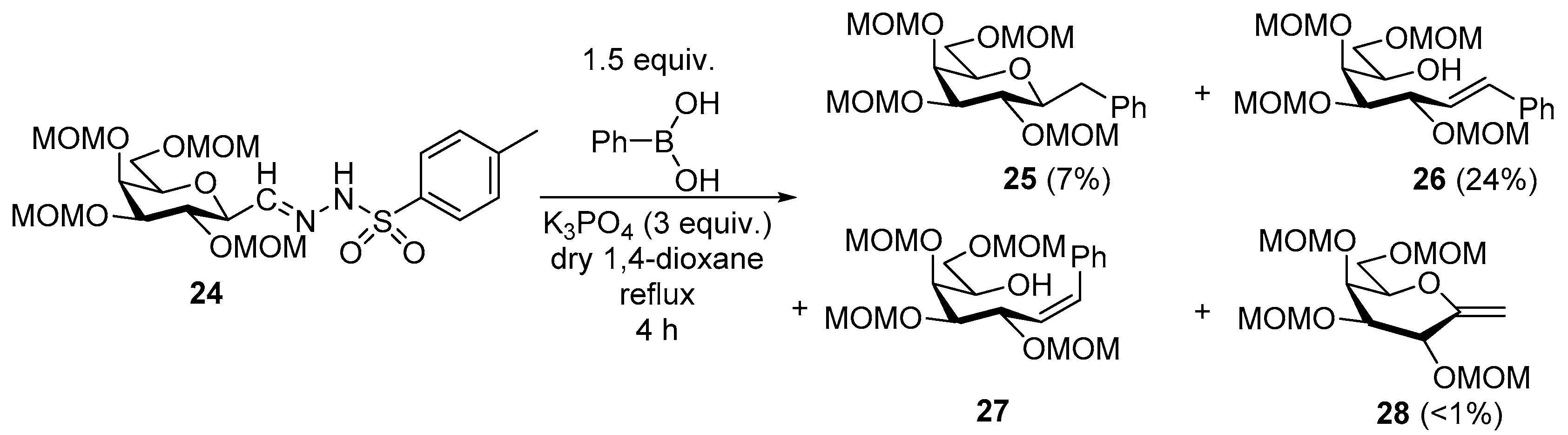
4.12.1. 2,6-Anhydro-1-Deoxy-3,4,5,7-Tetra-O-Methoxymethyl-1-Phenyl-d-glycero-d-gulo-Heptitol (25)
Isolated from a reaction of tosylhydrazone 24 (0.10 g, 0.19 mmol), phenylboronic acid (1.5 equiv., 0.03 g, 0.28 mmol), and K3PO4 (3 equiv., 0.12 g, 0.56 mmol) according to General procedure I by column chromatography (1:6 EtOAc–hexane) to yield 7 mg white amorphous solid containing 25 and 28 in 2.6:1 ratio. Rf: 0.35 (1:2 EtOAc–hexane). 1H NMR (400 MHz, CDCl3) δ 7.32–7.16 (5H, m, aromatics), 4.98 (1H, d, J 6.5 Hz, CH3OCH2OC-3), 4.92 (1H, d, J 6.8 Hz, CH3OCH2OC-5), 4.83 (1H, d, J 6.8 Hz, CH3OCH2OC-4), 4.76 (H, d, J 6.5 Hz, CH3OCH2OC-3), 4.70 (1H, d, J 6.8 Hz, CH3OCH2OC-4), 4.69 (1H, d, J 7.0 Hz, CH3OCH2OC-5), 4.55 (1H, d, J 6.5 Hz, CH3OCH2OC-7), 4.50 (1H, d, J 6.5 Hz, CH3OCH2OC-7), 4.05 (1H, dd, J4,5 2.0, J5,6 0.6 Hz, H-5), 3.76–3.67 (2H, m, H-3, H-4), 3.67 (1H, dd, J6,7a 6.3, J7a,7b 10.2 Hz, H-7a), 3.58 (1H, dd, J6,7b 6.5 Hz, H-7b), 3.48 (3H, s, CH3OCH2OC-3), 3.50–3.44 (1H, m, H-6), 3.43 (3H, s CH3OCH2OC-4), 3.42 (3H, s CH3OCH2OC-5), 3.42–3.39 (1H, m, H-2), 3.27 (3H, s, CH3OCH2OC-7), 3.23 (1H, dd, J1a,1b 14.2, J1a,2 1.5 Hz, H-1a), 2.77 (1H, dd, J1b,2 10.0 Hz, H-1b). 13C NMR (100 MHz, CDCl3) δ 139.7–125.1 (aromatics), 98.9 (CH3OCH2OC-3), 97.5 (CH3OCH2OC-5), 96.9 (CH3OCH2OC-7), 95.4 (CH3OCH2OC-4), 80.9 (C-2), 80.2 (C-4), 77.5 (C-3), 77.2 (C-6), 72.9 (C-5), 66.7 (C-7), 56.7 (CH3OCH2OC-3), 56.1 (CH3OCH2OC-5), 56.0 (CH3OCH2OC-4), 55.5 (CH3OCH2OC-7), 38.1 (C-1). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 453.2095, found: [M + Na]+ = 453.2093; C21H34O9 (430.49).
![Molecules 27 01795 i047]()
4.12.2. (E)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methoxymethyl-1-Phenyl-d-gluco-Hept-1-Enitol (26) and (Z)-1,2-Dideoxy-3,4,5,7-Tetra-O-Methoxymethyl-1-Phenyl-d-gluco-Hept-1-Enitol (27)
Isolated from a reaction of tosylhydrazone 24 (0.10 g, 0.19 mmol), phenylboronic acid (1.5 equiv., 0.03 g, 0.28 mmol), and K3PO4 (3 equiv., 0.12 g, 0.56 mmol) according to General procedure I by column chromatography (1:6 EtOAc–hexane) to yield 19 mg white amorphous solid containing 26 and 27 in 100:1 ratio. Rf: 0.29 (1:2 EtOAc–hexane), [α]D + 1 (c 0.30, CH2Cl2).
![Molecules 27 01795 i048]()
26: 1H NMR (500 MHz, CDCl3) δ 7.39 (2H, d, J 8.7 Hz, aromatics), 7.35–7.29 (2H, m, aromatics), 7.29–7.23 (1H, m, aromatic), 6.65 (1H, d, J1,2 16.0 Hz, H-1), 6.15 (1H, dd, J2,3 8.1 Hz, H-2), 4.86 (1H, d, J 6.6 Hz, CH3OCH2OC-4), 4.84 (2H, d, J 6.7 Hz, CH3OCH2OC-4, CH3OCH2OC-5), 4.78 (1H, d, J 6.7 Hz, CH3OCH2OC-3), 4.71 (1H, d, J 6.8 Hz, CH3OCH2OC-5), 4.64 (1H, d, J 6.7 Hz, CH3OCH2OC-3), 4.62 (1H, d, J 6.5 Hz, CH3OCH2OC-7), 4.60 (1H, d, J 6.5 Hz, CH3OCH2OC-7), 4.47 (1H, dd, J3,4 5.4 Hz, H-3), 4.20–4.12 (1H, m, H-6), 4.00 (1H, pseudo t, J4,5 4.6 Hz, H-4), 3.89 (1H, dd, J5,6 2.1 Hz, H-5), 3.66 (1H, dd, J6,7a 6.4, J7a,7b 10.3 Hz, H-7a), 3.64 (1H, dd, J6,7b 6.1 Hz, H-7b), 3.49 (1H, dd, J6,OH 3.9 Hz, OH), 3.46 (3H, s CH3OCH2OC-4), 3.44 (3H, s, CH3OCH2OC-5), 3.41 (3H, s, CH3OCH2OC-3), 3.31 (3H, s, CH3OCH2OC-7). 13C NMR (125 MHz, CDCl3) δ 134.6 (C-1), 136.4–125.5 (aromatics), 125.9 (C-2), 98.5 (CH3OCH2OC-4), 97.5 (CH3OCH2OC-5), 96.9 (CH3OCH2OC-7), 94.3 (CH3OCH2OC-3), 81.3 (C-4), 77.2 (C-3), 76.9 (C-5), 69.8 (C-6), 69.1 (C-7), 56.4 (CH3OCH2OC-4, CH3OCH2OC-5), 56.0 (CH3OCH2OC-3), 55.4 (CH3OCH2OC-7). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 453.2095, found: [M + Na]+ = 453.2099; C21H34O9 (430.49).
27: 1H NMR (500 MHz, CDCl3) δ 7.43–7.36 (2H, m, aromatics), 7.35–7.29 (2H, m, aromatics), 7.29–7.23 (1H, m, aromatic), 6.75 (1H, d, J1,2 11.4 Hz, H-1), 5.70 (1H, dd, J2,3 9.9 Hz, H-2), 4.93–4.22 (11H, m, H-3, H-4, H-5, 4 × CH3OCH2), 4.20–4.12 (1H, m, H-6), 3.96–3.83 (2H, m, H-7a, H-7b), 3.44, 3.35, 3.34 (12H, 4s, 4 × CH3OCH2). 13C NMR (125 MHz, CDCl3) δ 133.9 (C-1), 136.4–125.5 (aromatics), 129.2 (C-2), 98.9, 97.6, 97.0, 94.6 (4 × CH3OCH2), 81.6 (C-4), 76.9 (C-5), 71.7 (C-3), 69.5 (C-6), 65.7 (C-7), 56.6, 56.5, 55.9, 55.7 (4 × CH3OCH2). HR-ESI-MS positive mode (m/z): calc. for [M + Na]+ = 453.2095, found: [M + Na]+ = 453.2099; C21H34O9 (430.49).


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}