
Synthesis and Investigation of Flavanone Derivatives as Potential New Anti-Inflammatory Agents

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
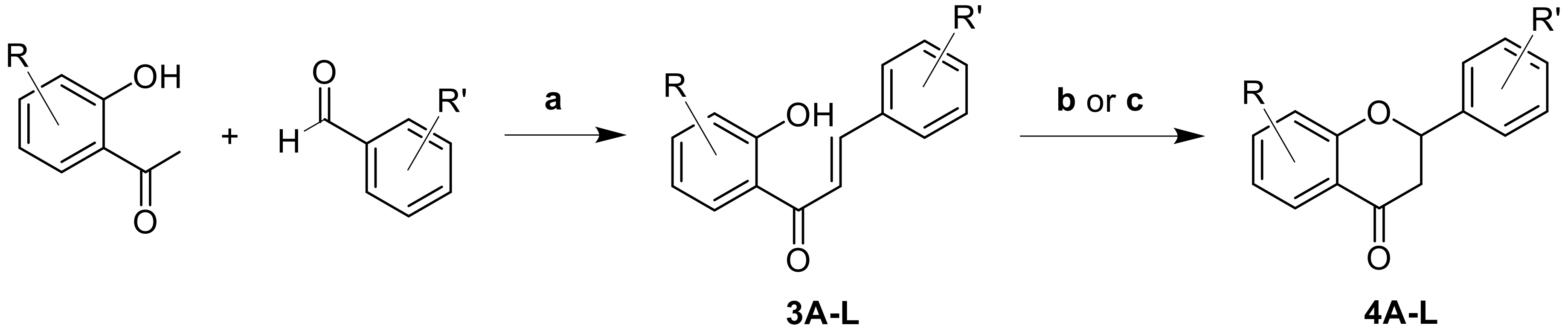
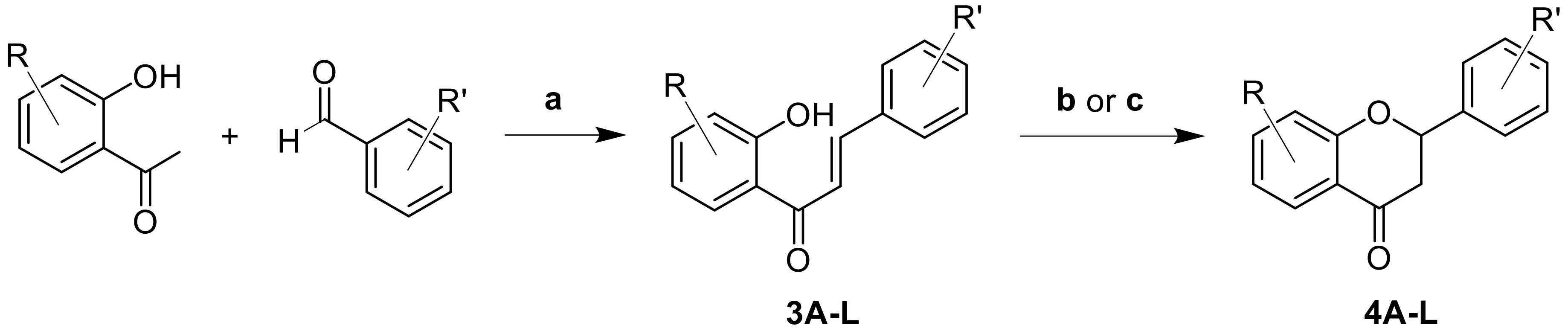
2.1. Synthesis
2.2. Evaluation of Biological Activities
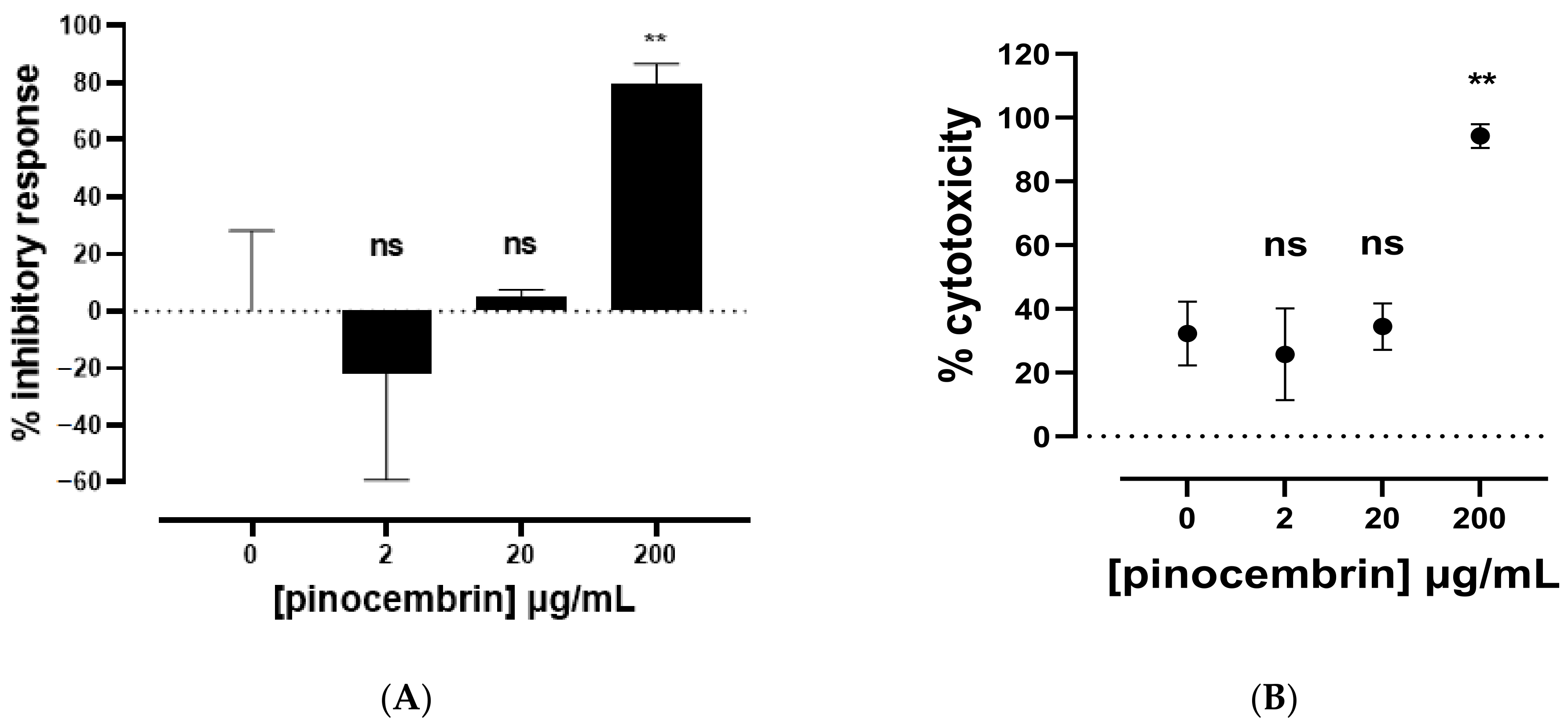
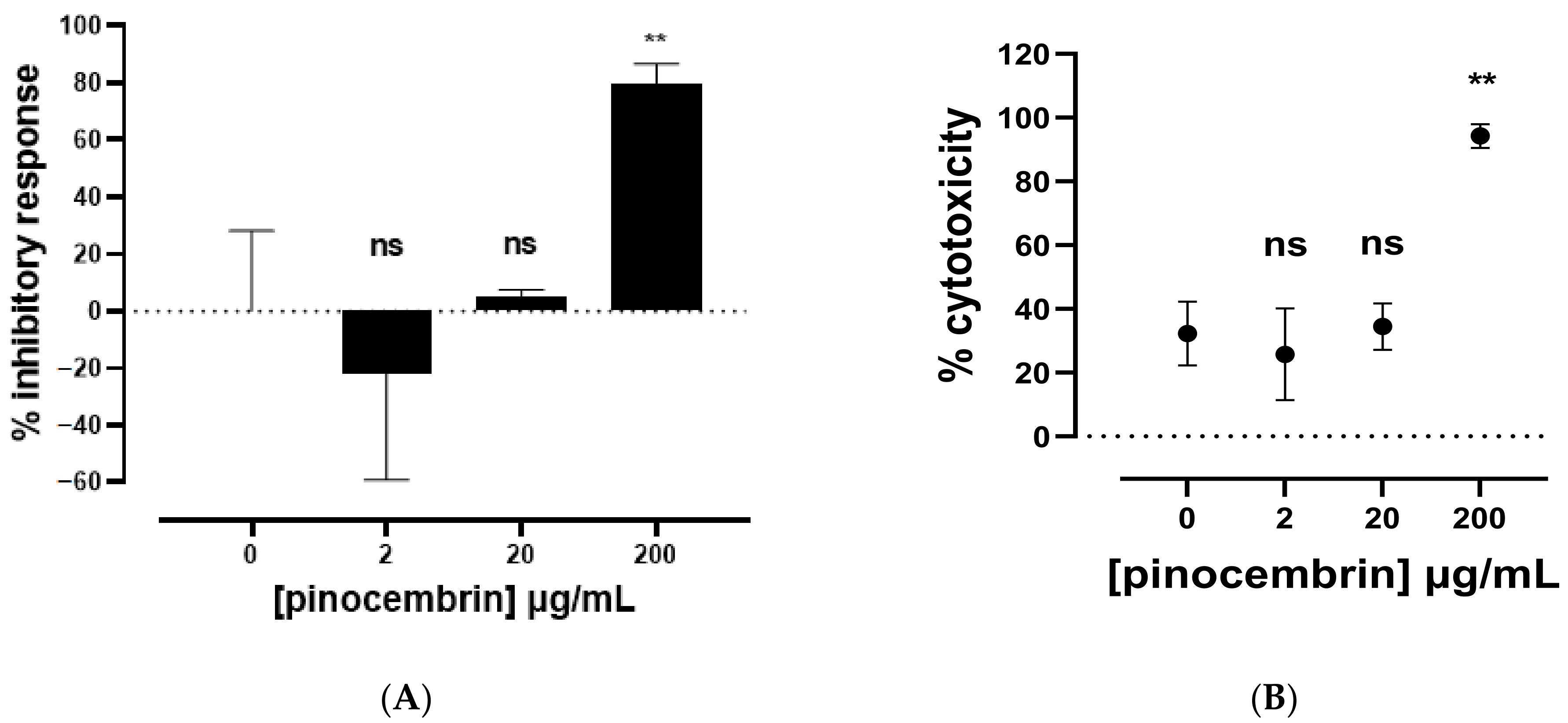
2.2.1. Biological Activities of Commercial Pinocembrin
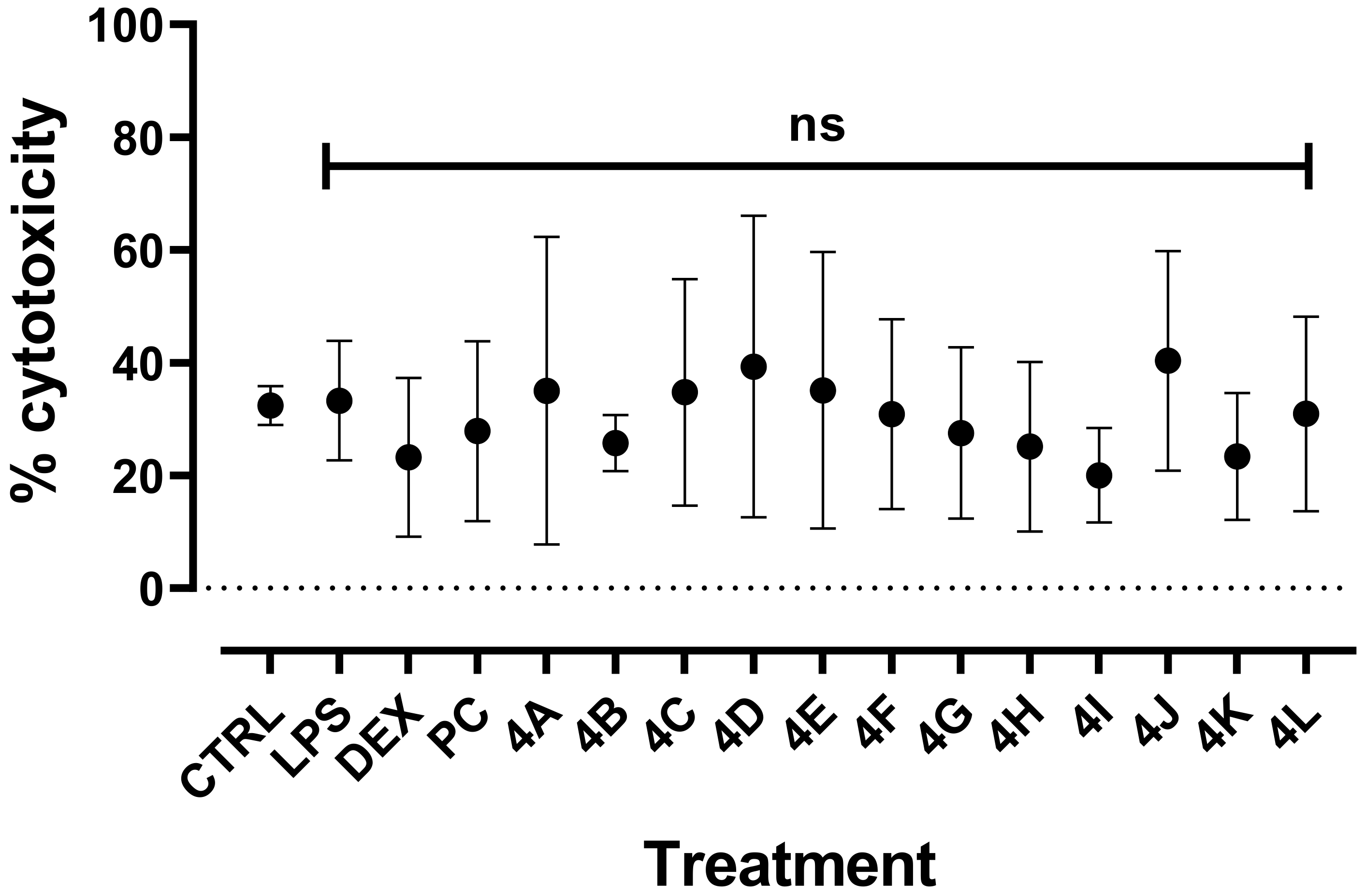
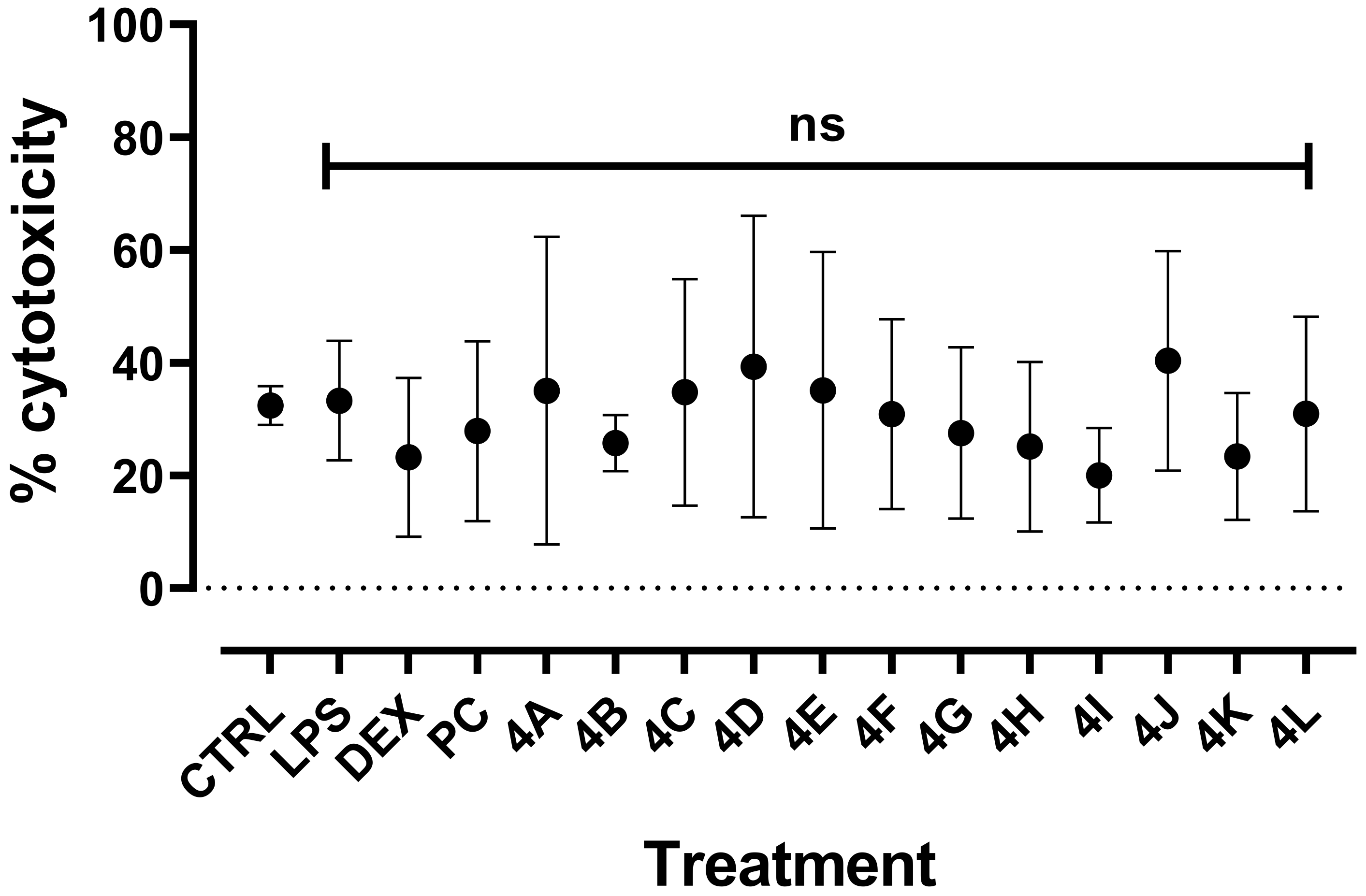
2.2.2. Cytotoxicity of Flavanone Derivatives
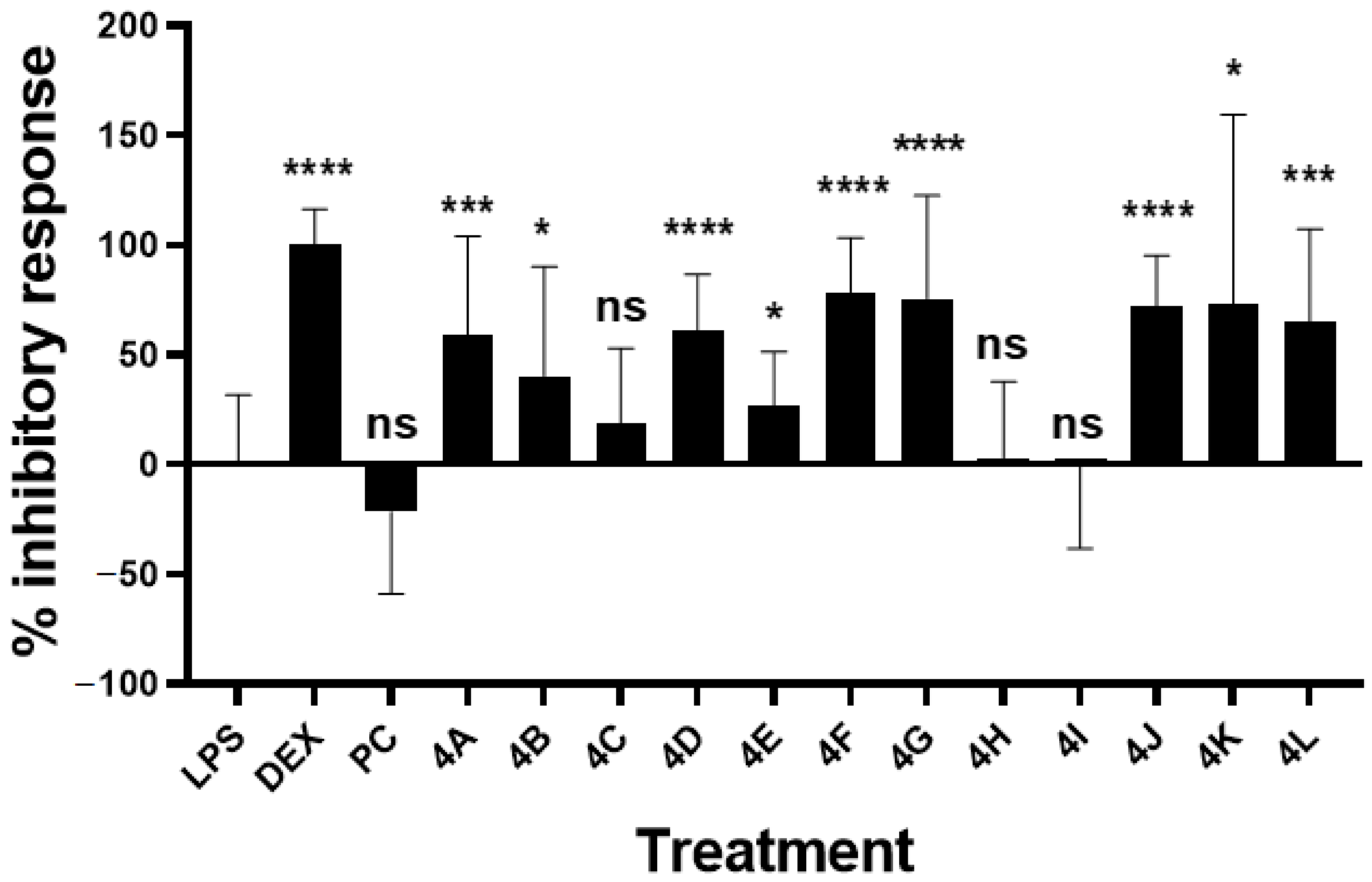
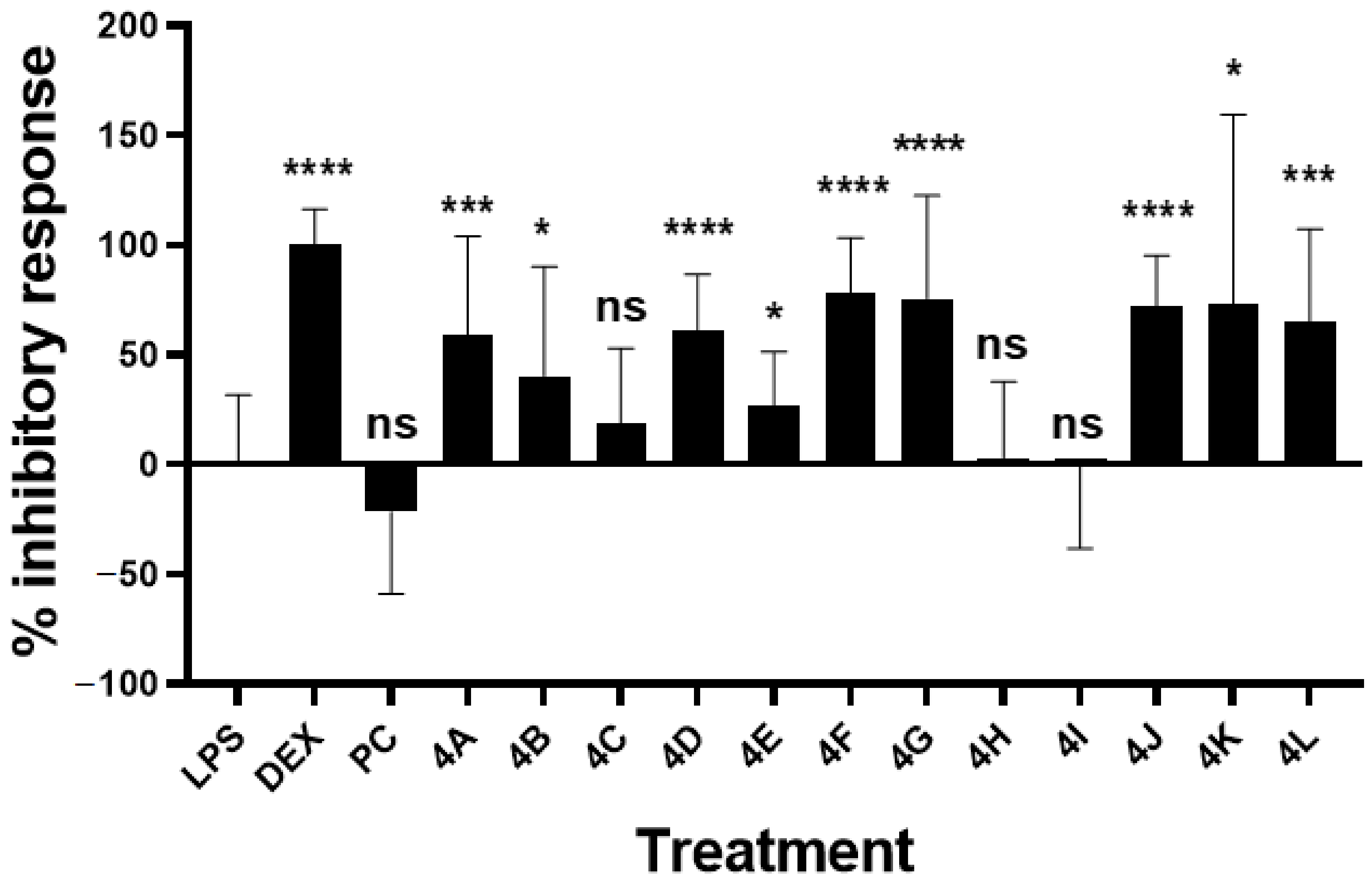
2.2.3. Inhibitory Activity on LPS-Induced NO Production
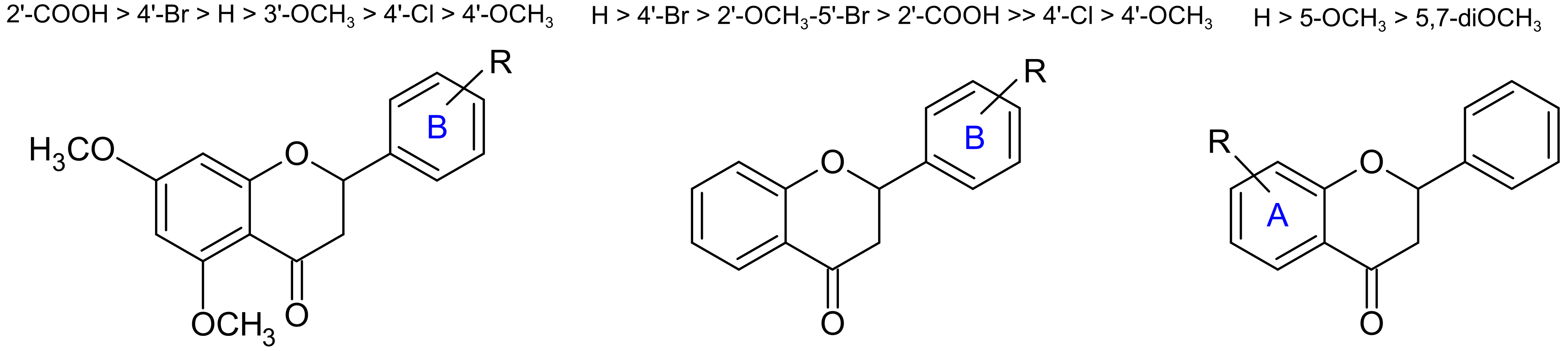
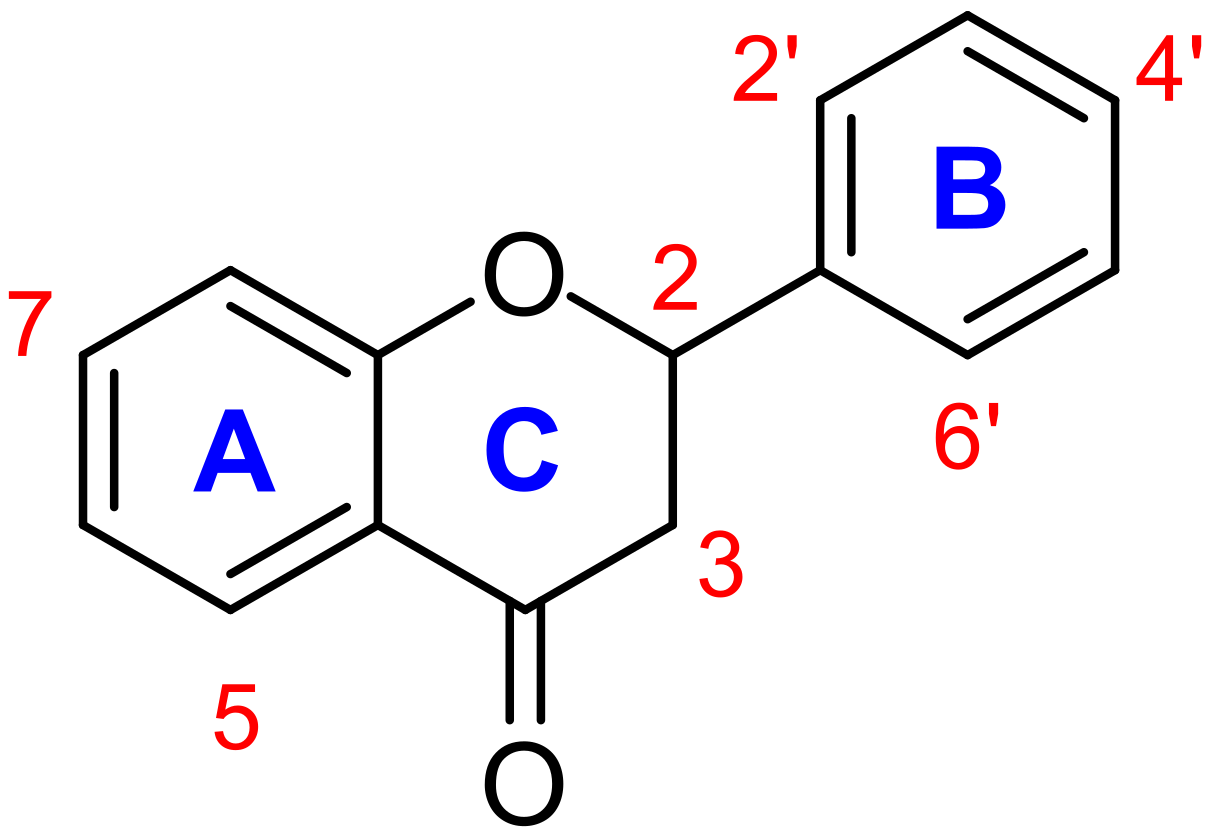
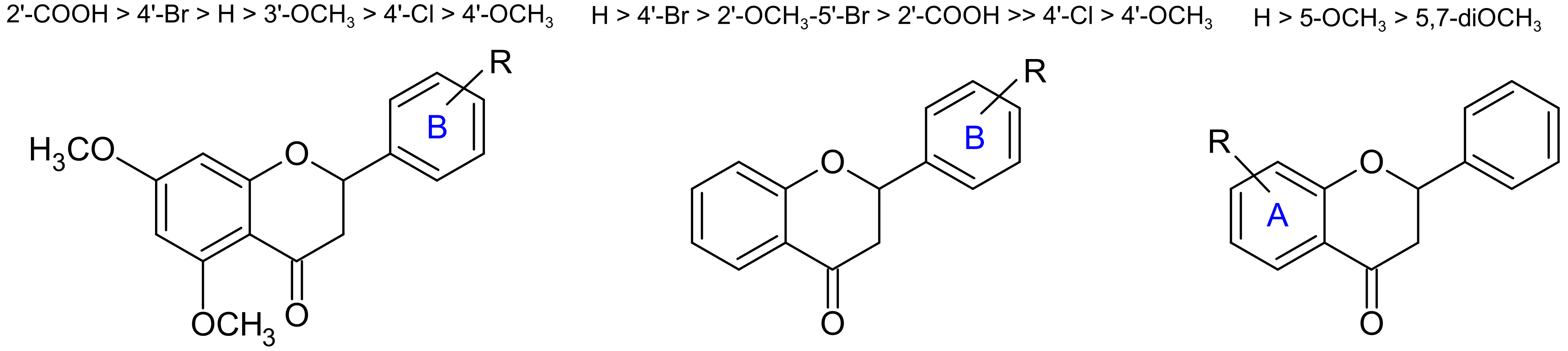
2.3. Structure–Activity Relationship Study
3. Materials and Methods
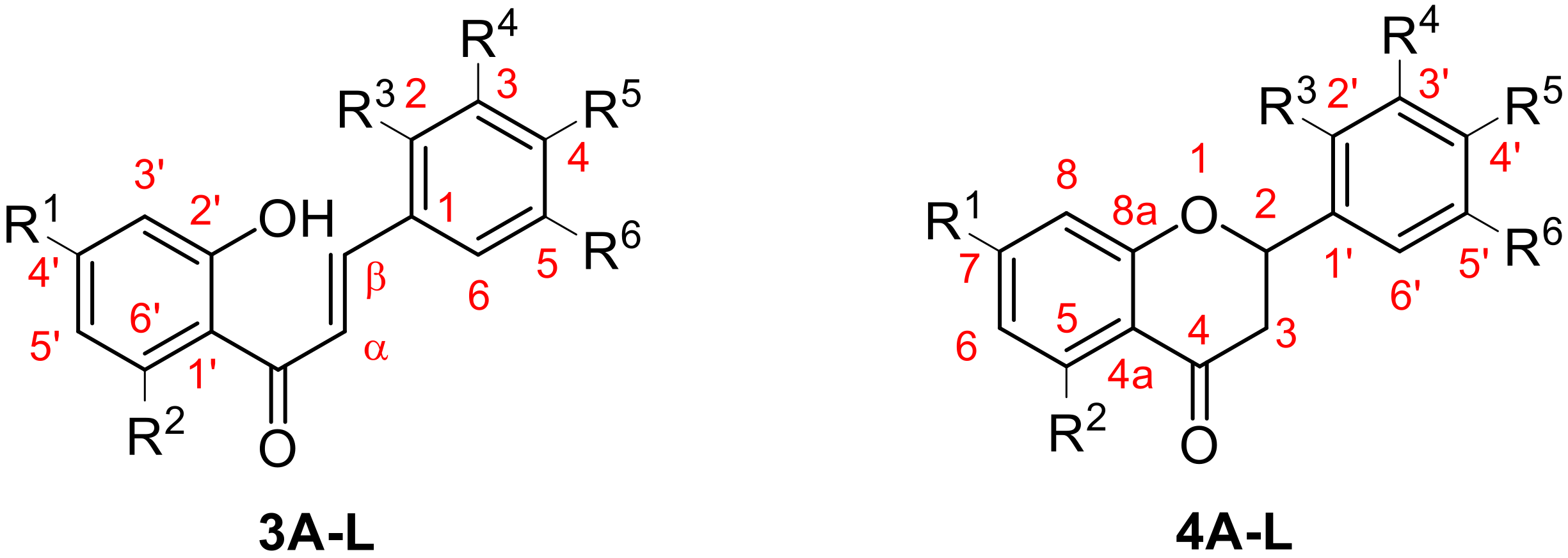
3.1. Syntheses
3.1.1. General Information
3.1.2. Synthetic Methods
- General Procedure for the synthesis of chalcones (3A–L).
- Flavanone 3A synthesis by photochemical activation.
- General Procedure for flavanones synthesis by base activation (4B–L).
3.2. Biological Assays
3.2.1. Cell Culture and Treatments
3.2.2. Determination of Cell Mortality
3.2.3. Quantification of Nitric Oxide (NO)
3.2.4. Predictive Analysis of Drug-Like Absorption
3.2.5. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| NMR. | Nuclear magnetic resonance |
| 1H | Proton nuclear magnetic resonance |
| 13C | Carbon nuclear magnetic resonance |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| LPS | Lipopolysaccharide |
| MeOH | Methanol |
| EtOAc | Ethyl acetate |
| EtOH | Ethanol |
| TLC | Thin Layer chromatography |
| PC | Pinocembrin |
| IC50 | Half maximal inhibitory concentration |
| LDH | Lactate dehydrogenase |
| SD | Standard deviation |
| DMSO | Dimethyl sulfoxide |
| DEX | Dexamethasone |
| DCM | Dichloromethane |
| CC | Column chromatography |
| m.p. | Melting point |
| Hz | Hertz |
| HRMS | High-resolution mass spectrometry |
| ESI | Electrospray ionization |
| M | Mass |
| CAS | Chemical abstracts service |
| Da | Dalton |
| W | Watt |
| C6H12 | Cyclohexane |
| SAR | Structure–activity relationship |
| CI | Confidence Interval |
References
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory Effects of Flavonoids. Food Chem. 2019, 299, 125124. [Google Scholar] [CrossRef] [PubMed]
- Tutunchi, H.; Naeini, F.; Ostadrahimi, A.; Hosseinzadeh-Attar, M.J. Naringenin, a Flavanone with Antiviral and Anti-Inflammatory Effects: A Promising Treatment Strategy against COVID-19. Phytother. Res. 2020, 34, 3137–3147. [Google Scholar] [CrossRef] [PubMed]
- Gautam, R.; Jachak, S.M. Recent Developments in Anti-Inflammatory Natural Products. Med. Res. Rev. 2009, 29, 767–820. [Google Scholar] [CrossRef]
- Ialenti, A.; Moncada, S.; Di Rosa, M. Modulation of Adjuvant Arthritis by Endogenous Nitric Oxide. Br. J. Pharmacol. 1993, 110, 701–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stichtenoth, D.O.; Frolin, J.C. Nitric Oxide and Inflammatory Joint Diseases. Br. J. Rheumatol. 1998, 37, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.H.; Lee, H.J.; Jeong, Y.S.; Ryu, S.Y.; Han, Y.N. Yomogin, an Inhibitor of Nitric Oxide Production in LPS-Activated Macrophages. Arch. Pharmacal Res. 1998, 21, 481–484. [Google Scholar] [CrossRef]
- Kobuchi, H.; Droy-lefaix, M.T.; Christen, Y.; Packer, L. Ginkgo Biloba Extract (Egb 761): Inhibitory Effect on Nitric Oxide Production in the Macrophage Cell Line RAW 264.7. Biochem. Pharm. 1997, 53, 897–903. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Schwedhelm, C.; Hoffmann, G.; Knüppel, S.; Preterre, A.; Iqbal, K.; Bechthold, A. Food Groups and Risk of Colorectal Cancer. Int. J. Cancer 2018, 142, 1748–1758. [Google Scholar] [CrossRef] [Green Version]
- Van Breda, S.G.; De Kok, T.M.C.M. Smart Combinations of Bioactive Compounds in Fruits and Vegetables May Guide New Strategies for Personalized Prevention of Chronic Diseases. Mol. Nutr. Food Res. 2018, 62, 1700597. [Google Scholar] [CrossRef]
- Pietta, P.G. Flavonoids as Antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Kanadaswami, C.; Lee, L.T.; Lee, P.H.; Hwang, J.J.; Ke, F.C.; Huang, Y.T.; Lee, M.T. The Antitumor Activities of Flavonoids. Vivo 2005, 19, 895–910. [Google Scholar]
- Lolli, G.; Cozza, G.; Mazzorana, M.; Tibaldi, E.; Cesaro, L.; Donella-Deana, A.; Meggio, F. Inhibition of Protein Kinase CK2 by Flavonoids and Tyrphostins. A Structural Insight. Biochemistry 2012, 51, 6097–6107. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Liu, Y.; Luo, X.; Yang, Z. Advances in Biosynthesis, Pharmacology, and Pharmacokinetics of Pinocembrin, a Promising Natural Small-Molecule Drug. Molecules 2019, 24, 2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soromou, L.W.; Chu, X.; Jiang, L.; Wei, M.; Huo, M.; Chen, N.; Guan, S.; Yang, X.; Chen, C.; Feng, H.; et al. In Vitro and in Vivo Protection Provided by Pinocembrin against Lipopolysaccharide-Induced Inflammatory Responses. Int. Immunopharmacol. 2012, 14, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Li, X.; Cui, Y.; Xiao, T.; Liu, R.; Wang, M.; Wei, Y. Pinocembrin Relieves Lipopolysaccharide and Bleomycin Induced Lung Inflammation via Inhibiting TLR4-NF-ΚB-NLRP3 Inflammasome Signaling Pathway. Int. Immunopharmacol. 2021, 90, 107230. [Google Scholar] [CrossRef]
- Gabaston, J.; Richard, T.; Cluzet, S.; Pinto, A.P.; Dufour, M.; Corio-Costet, M.; Mérillon, J. Pinus Pinaster Knot: A Source of Polyphenols against Plasmopara Viticola. J. Agric. Food Chem. 2017, 65, 8884–8891. [Google Scholar] [CrossRef]
- French, D.; Schifano, P.; Cortés-Concepcion, J.; Hargrove-Leak, S. Li-Al Layered Double Hydroxides as Catalysts for the Synthesis of Flavanone. Catal. Commun. 2010, 12, 92–94. [Google Scholar] [CrossRef]
- Shi, L.; Feng, X.E.; Cui, J.R.; Fang, L.H.; Du, G.H.; Li, Q.S. Synthesis and Biological Activity of Flavanone Derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 5466–5468. [Google Scholar] [CrossRef]
- Brennan, C.M.; Hunt, I.; Jarvis, T.C.; Johnson, D.; McDonnell, P.D. Stereoelectronic effects in ring closure reactions: The 2′-hydroxychalcone—flavanone equilibrium, and related systems. Can. J. Chem. 1990, 1, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Keane, D.D.; Marathe, K.G.; O’Sullivan, W.I.; Philbin, E.M.; Simons, R.M.; Teague, P.C. Configuration and Conformation of 3-Arylideneflavanones. J. Org. Chem. 1970, 35, 2286–2290. [Google Scholar] [CrossRef]
- Hoshino, Y.; Takeno, N. Thermal Isomerization Equilibrium between 2′-Hydroxychalcones and Flavanones. Bull. Chem. Soc. Jpn. 1986, 59, 2903–2904. [Google Scholar] [CrossRef]
- Pandey, G.; Krishna, A.; Kumaraswamy, G. Photosensitized (Set) Conversion of 2′-Hydroxychalcones to Flavonoids a Probable Biogenetic Pathway. Tetrahedron Lett. 1987, 28, 4615–4616. [Google Scholar] [CrossRef]
- Geresh, S.; Levy, O.; Markovits, Y.; Shani, A. On the Mechanism of Intramolecular Photocycloaddition of Substituted O-Allylphenols to Cyclic Ethers. Tetrahedron 1975, 31, 2803–2807. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Hoffmann, N. Studies in Organic and Physical Photochemistry-an Interdisciplinary Approach. Org. Biomol. Chem. 2016, 14, 7392–7442. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, R.; Hiroyuki, K. Photochemical Cyclization of 2′-Hydroxychalcones. J. Chem. Soc. Perkin Trans. 2 1985, 6, 743–748. [Google Scholar] [CrossRef]
- Tundis, R.L.; Frattaruolo, G.C.; Armentano, B.; Badolato, M.; Loizzo, M.; Aiello, F.; Cappello, A. An ancient remedial repurposing: Synthesis of new pinocembrin fatty acid acyl derivatives as potential antimicrobial/anti-inflammatory agents. Nat. Prod. Res. 2018, 33, 1–7. [Google Scholar] [CrossRef]
- Forest, V.; Figarol, A.; Boudard, D.; Cottier, M.; Grosseau, P.; Pourchez, J. Adsorption of lactate dehydrogenase enzyme on carbon nanotubes: How to get accurate results about the cytotoxicity of these nanomaterials. Langmuir 2015, 31, 3635–3643. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H. In-Vitro Evaluation for Antioxidant and Anti-Inflammatory Property of Flavanone Derivatives. Food Biosci. 2015, 11, 1–7. [Google Scholar] [CrossRef]
- Kim, H.K.; Cheon, B.S.; Kim, Y.H.; Kim, S.Y.; Kim, H.P. Effects of Naturally Occurring Flavonoids on Nitric Oxide Production in the Macrophage Cell Line RAW 264.7 and Their Structure-Activity Relationships. Biochem. Pharm. 1999, 58, 759–765. [Google Scholar] [CrossRef]
- Shin, S.Y.; Woo, Y.; Hyun, J.; Yong, Y.; Koh, D.; Lee, Y.H.; Lim, Y. Relationship between the Structures of Flavonoids and Their NF-ΚB-Dependent Transcriptional Activities. Bioorganic Med. Chem. Lett. 2011, 21, 6036–6041. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Boeck, P.; Bandeira Falcão, C.A.; Leal, P.C.; Yunes, R.A.; Filho, V.C.; Torres-Santos, E.C.; Rossi-Bergmann, B. Synthesis of Chalcone Analogues with Increased Antileishmanial Activity. Bioorganic Med. Chem. 2006, 14, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Thieury, C.; Lebouvier, N.; Le Guével, R.; Barguil, Y.; Herbette, G.; Antheaume, C.; Hnawia, E.; Asakawa, Y.; Nour, M.; Guillaudeux, T. Mechanisms of Action and Structure-Activity Relationships of Cytotoxic Flavokawain Derivatives. Bioorganic Med. Chem. 2017, 25, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Detsi, A.; Majdalani, M.; Kontogiorgis, C.A.; Hadjipavlou-Litina, D.; Kefalas, P. Natural and synthetic 2’-hydroxy-chalcones and aurones: Synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorganic Med. Chem. 2009, 17, 8073–8085. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.X.; Chen, X.; Hughes, R.A.; Williams, S.J.; Woodman, O.L. Understanding the Cardioprotective Effects of Flavonols: Discovery of Relaxant Flavonols without Antioxidant Activity. J. Med. Chem. 2008, 51, 1874–1884. [Google Scholar] [CrossRef]
- Krüger, K.; Lüdke, V.; Pettinger, J.; Ashton, L.; Bonnet, L.; Motti, C.A.; Lex, J.; Oelgemöller, M. Photochemical synthesis of cyclic peptide models from phthalimido acetamides and phthaloyl dipeptide esters. Tetrahedron Lett. 2018, 59, 1427–1430. [Google Scholar] [CrossRef]
- Kavala, V.; Lin, C.; Kuo, C.W.; Fang, H.; Yao, C.F. Iodine Catalyzed One-Pot Synthesis of Flavanone and Tetrahydropyrimidine Derivatives via Mannich Type Reaction. Tetrahedron 2012, 68, 1321–1329. [Google Scholar] [CrossRef]
- Mazzio, E.A.; Li, N.; Bauer, D.; Mendonca, P.; Taka, E.; Darb, M.; Thomas, L.; Williams, H.; Soliman, K.F.A. Natural product HTP screening for antibacterial (E.coli 0157:H7) and anti-inflammatory agents in (LPS from E. coli O111:B4) activated macrophages and microglial cells; focus on sepsis. BMC Complementary Altern. Med. 2016, 16, 467. [Google Scholar] [CrossRef] [Green Version]
- Haslam, G.; Wyatt, D.; Kitos, P.A. Estimating the Number of Viable Animal Cells in Multi-Well Cultures Based on Their Lactate Dehydrogenase Activities. Cytotechnology 2000, 32, 63–75. [Google Scholar] [CrossRef]
- Riss, T.; Niles, A.; Moravec, R.; Karassina, N.; Vidugiriene, J. Cytotoxicity Assays: In Vitro methods to measure dead cells. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Kumar-Roiné, S.; Matsui, M.; Reybier, K.; Darius, H.T.; Chinain, M.; Pauillac, S.; Laurent, D. Ability of Certain Plant Extracts Traditionally Used to Treat Ciguatera Fish Poisoning to Inhibit Nitric Oxide Production in RAW 264.7 Macrophages. J. Ethnopharmacol. 2009, 123, 369–377. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | R’ | Chalcones 3 | Flavanones 4 |
|---|---|---|---|---|
| Yield (%) | Yield (%) | |||
| A | 5, 7-(OCH3)2 | H | 27 | 7 * |
| B | 5, 7-(OCH3)2 | 3′-OCH3 | 82 | 74 |
| C | 5, 7-(OCH3)2 | 4′-OCH3 | 92 | 66 |
| D | 5, 7-(OCH3)2 | 4′-Br | 52 | 69 |
| E | 5, 7-(OCH3)2 | 4′-Cl | 23 | 66 |
| F | 5, 7-(OCH3)2 | 2′-COOH | 36 | 72 |
| G | H | H | 62 | 59 |
| H | H | 4′-Cl | 86 | 37 |
| I | H | 4′-OCH3 | 56 | 37 |
| J | H | 2′-COOH | 32 | 81 |
| K | H | 2′-OCH3, 5′-Br | 84 | 56 |
| L | 5-OCH3 | H | 30 | 9 |
| Compound | Inhibitory Response (%) a | p Value b |
|---|---|---|
| Pinocembrin (PC) | −21.81 ± 37.42 | 0.1732 ns |
| Dexamethasone | 100.4 ± 16.14 | <0.0001 **** |
| 4A | 58.99 ± 45.19 | 0.0009 *** |
| 4B | 40.45 ± 49.71 | 0.0272 * |
| 4C | 19.14 ± 33.83 | 0.1470ns |
| 4D | 61.10 ± 25.50 | <0.0001 **** |
| 4E | 26.85 ± 24.43 | 0.0318 * |
| 4F | 78.65 ± 24.73 | <0.0001 **** |
| 4G | 75.65 ± 46.88 | <0.0001 **** |
| 4H | 3.128 ± 34.64 | 0.5605 ns |
| 4I | −0.87 ± 37.54 | 0.8116 ns |
| 4J | 72.56 ± 22.70 | <0.0001 **** |
| 4K | 73.29 ± 86.10 | 0.0105 * |
| 4L | 64.97 ± 42.37 | 0.0003 *** |
| Compound | IC50 (µg/mL) a | 95% CI b |
|---|---|---|
| Pinocembrin (PC) c | 203.60 c | 101.30−569.31 c |
| Dexamethasone | 0.005 | 0.003−0.008 |
| 4D | 1.030 | 0.675−1.382 |
| 4F | 0.906 | 0.550−1.765 |
| 4G | 0.603 | 0.366−1.003 |
| 4J | 1.830 | 1.467−2.677 |
| Compound | IC50 (µg/mL) | MW (Da) | Log p | Hydrogen Bond Acceptors | Hydrogen Bond Donors | Polar Surface Area (Å2) |
|---|---|---|---|---|---|---|
| 4D | 1.030 | 363.21 | 4.32 ± 0.41 | 4 | 0 | 44.77 |
| 4F | 0.906 | 328.32 | 3.23 ± 0.38 | 6 | 1 | 82.07 |
| 4G | 0.603 | 224.26 | 3.62 ± 0.26 | 2 | 0 | 26.30 |
| 4J | 1.830 | 268.27 | 3.29 ± 0.27 | 4 | 1 | 63.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinyeue, C.; Matsui, M.; Oelgemöller, M.; Bregier, F.; Chaleix, V.; Sol, V.; Lebouvier, N. Synthesis and Investigation of Flavanone Derivatives as Potential New Anti-Inflammatory Agents. Molecules 2022, 27, 1781. https://doi.org/10.3390/molecules27061781
Sinyeue C, Matsui M, Oelgemöller M, Bregier F, Chaleix V, Sol V, Lebouvier N. Synthesis and Investigation of Flavanone Derivatives as Potential New Anti-Inflammatory Agents. Molecules. 2022; 27(6):1781. https://doi.org/10.3390/molecules27061781
Chicago/Turabian StyleSinyeue, Cynthia, Mariko Matsui, Michael Oelgemöller, Frédérique Bregier, Vincent Chaleix, Vincent Sol, and Nicolas Lebouvier. 2022. "Synthesis and Investigation of Flavanone Derivatives as Potential New Anti-Inflammatory Agents" Molecules 27, no. 6: 1781. https://doi.org/10.3390/molecules27061781
APA StyleSinyeue, C., Matsui, M., Oelgemöller, M., Bregier, F., Chaleix, V., Sol, V., & Lebouvier, N. (2022). Synthesis and Investigation of Flavanone Derivatives as Potential New Anti-Inflammatory Agents. Molecules, 27(6), 1781. https://doi.org/10.3390/molecules27061781