
Virtual Screening of Natural Chemical Databases to Search for Potential ACE2 Inhibitors

Abstract
1. Introduction
2. Results
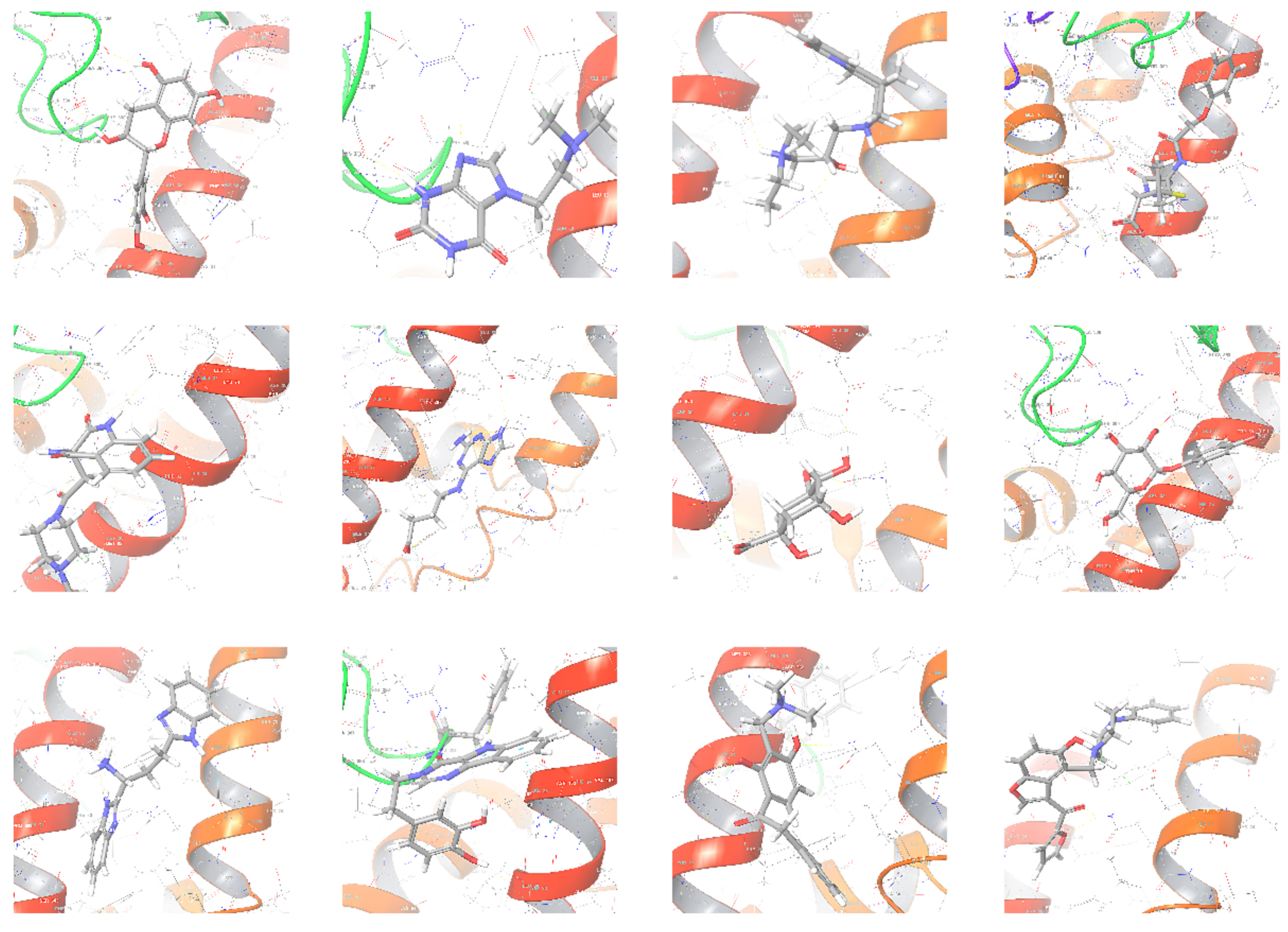
2.1. Molecular Docking Screening
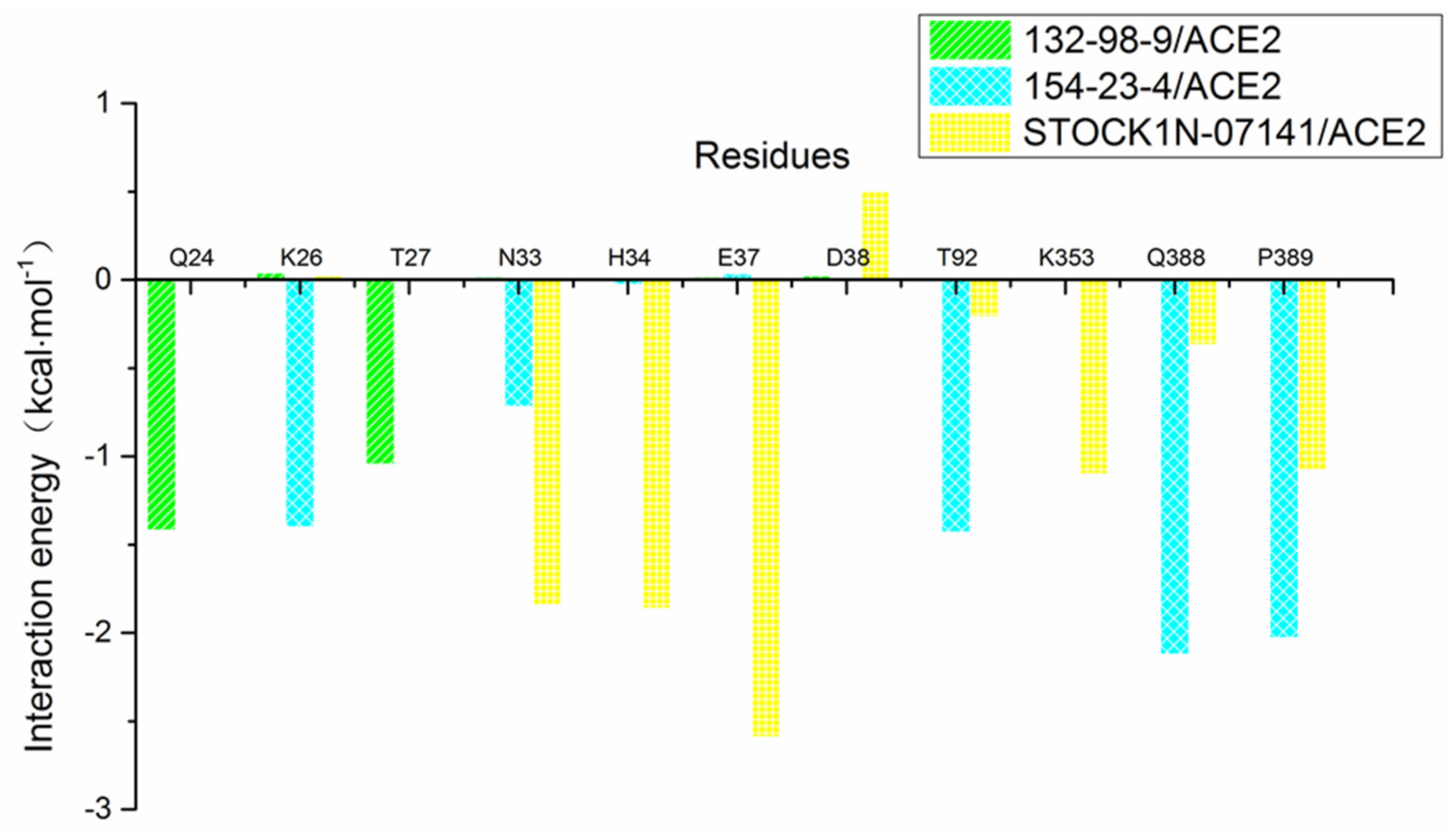
2.2. MM-GBSA
2.3. ADME Analysis
2.4. Cluster Analysis
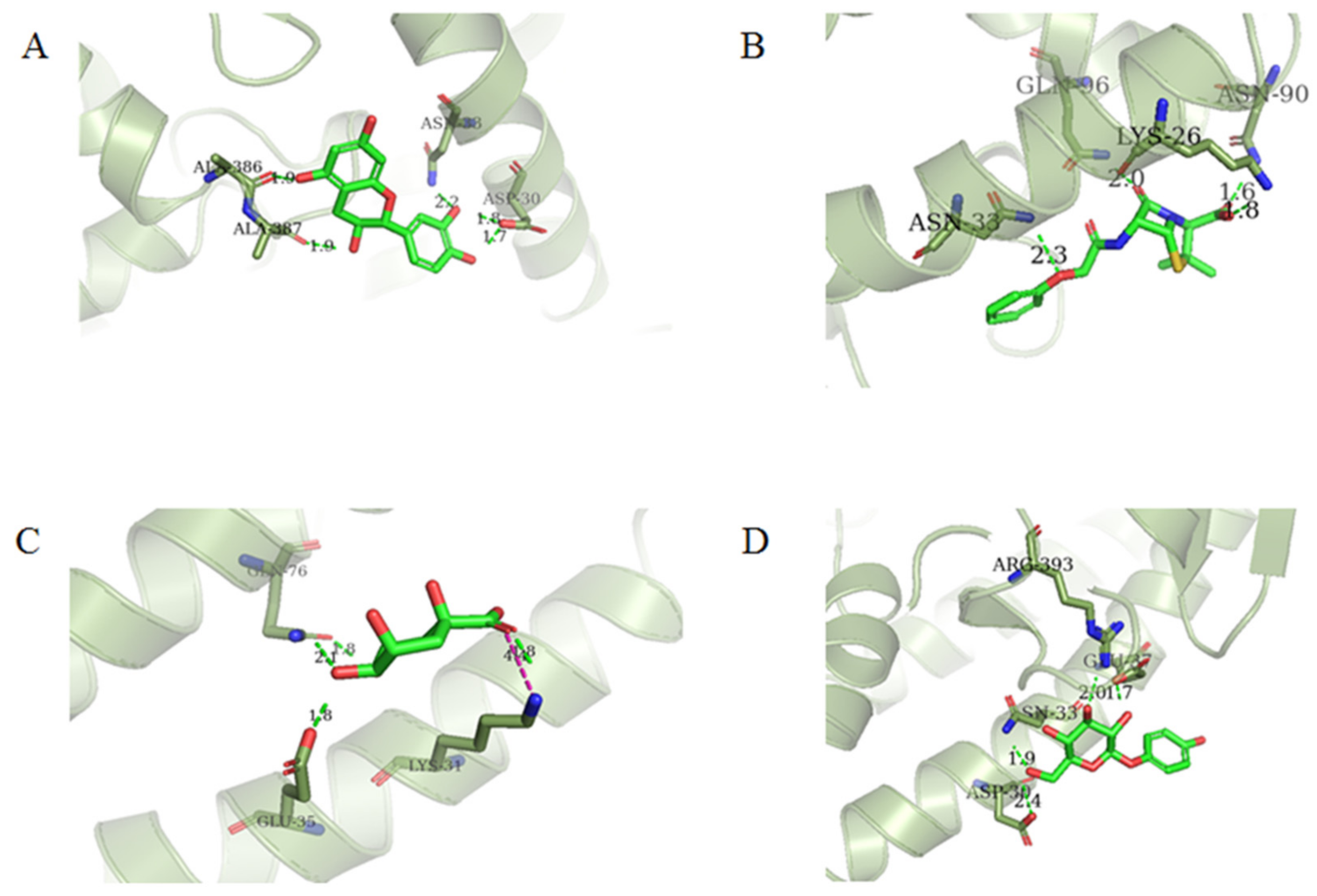
2.5. Virtual Screening Results
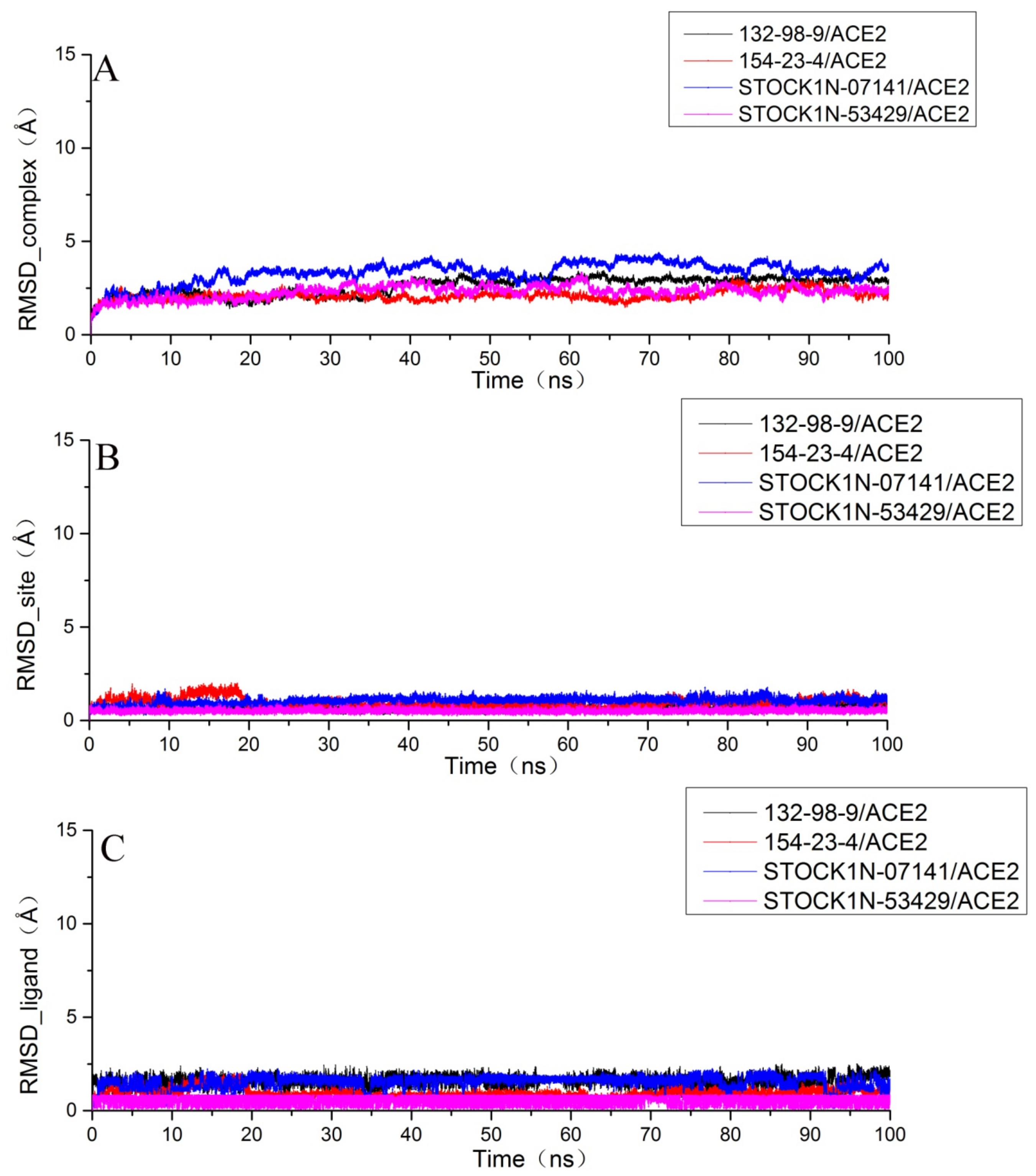
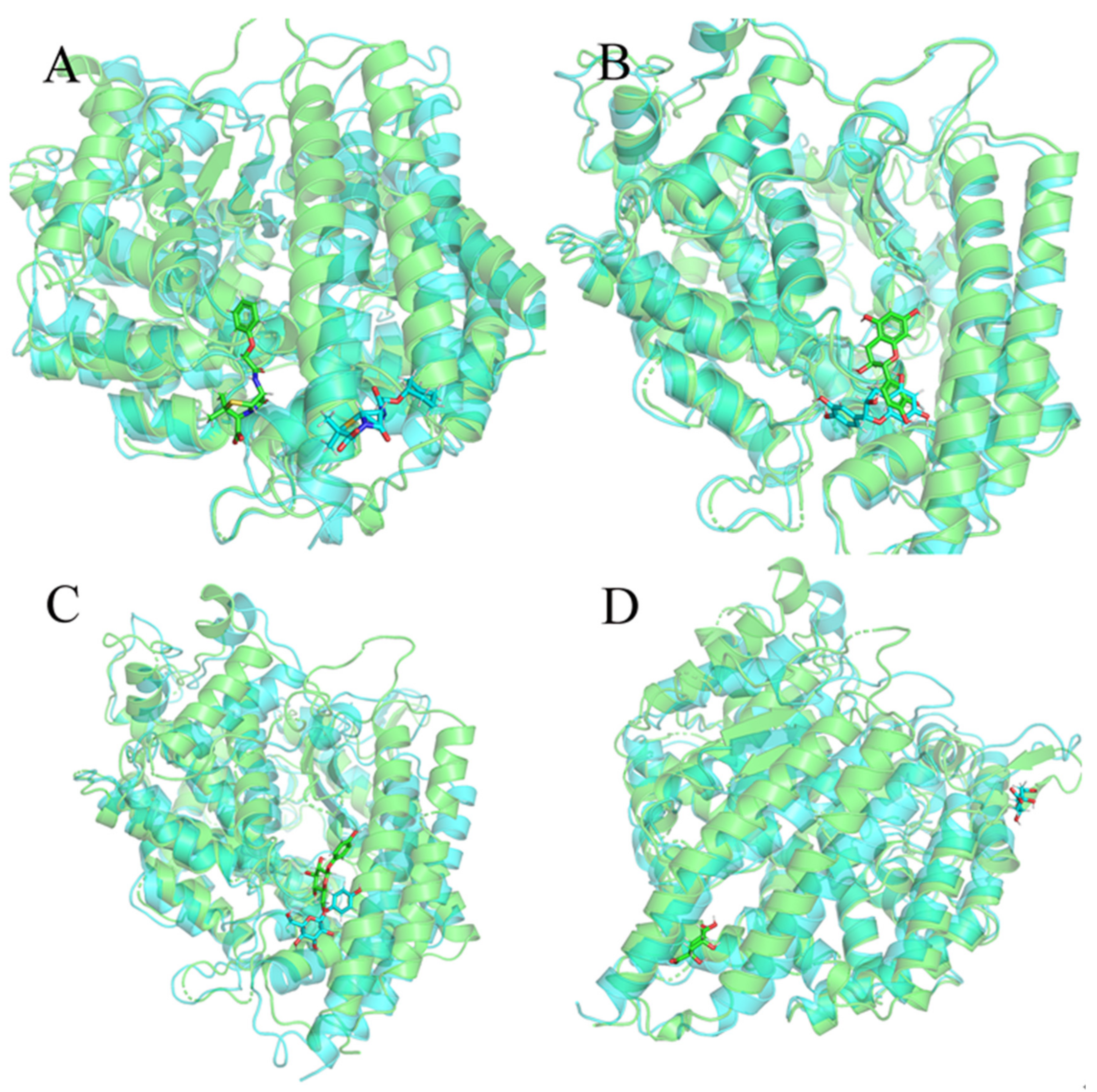
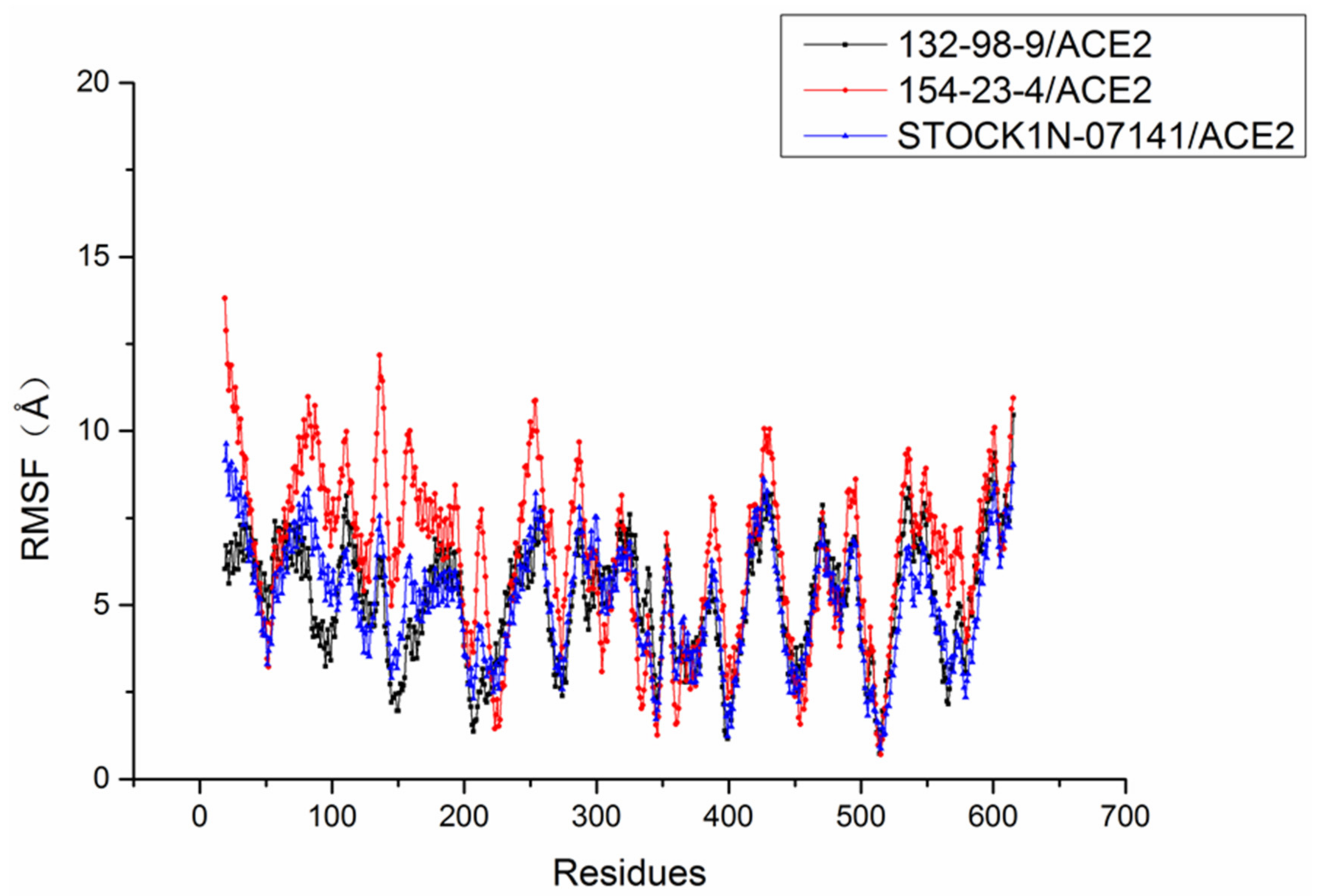
2.6. Molecular Dynamics Simulation Results
3. Materials and Methods
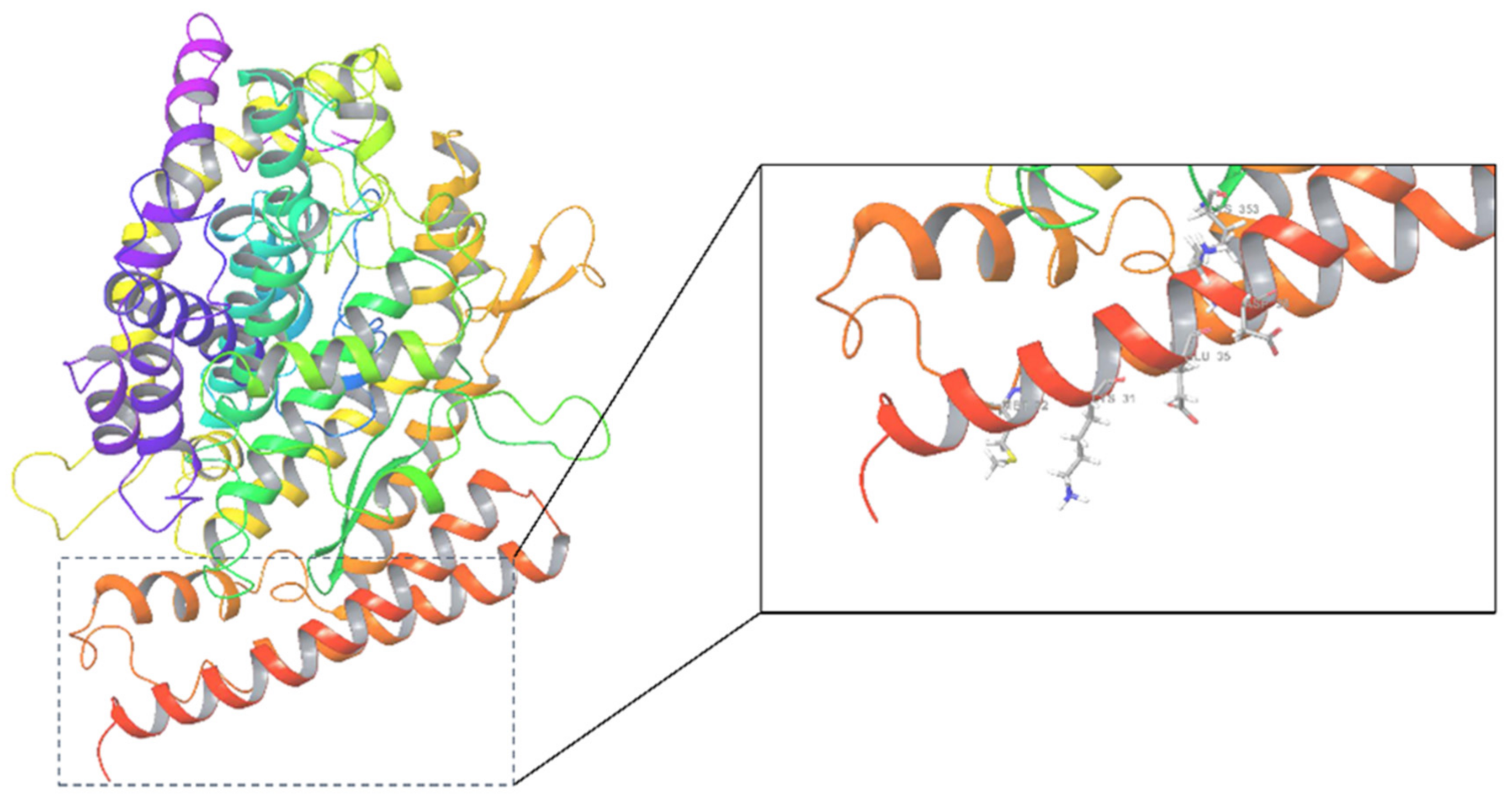
3.1. Protein Preparation
3.2. Ligand Preparation
3.3. Molecular Docking
3.4. ADME Analysis
3.5. Cluster Analysis
3.6. Molecular Dynamics Simulation
3.7. Binding Free Energy Calculations
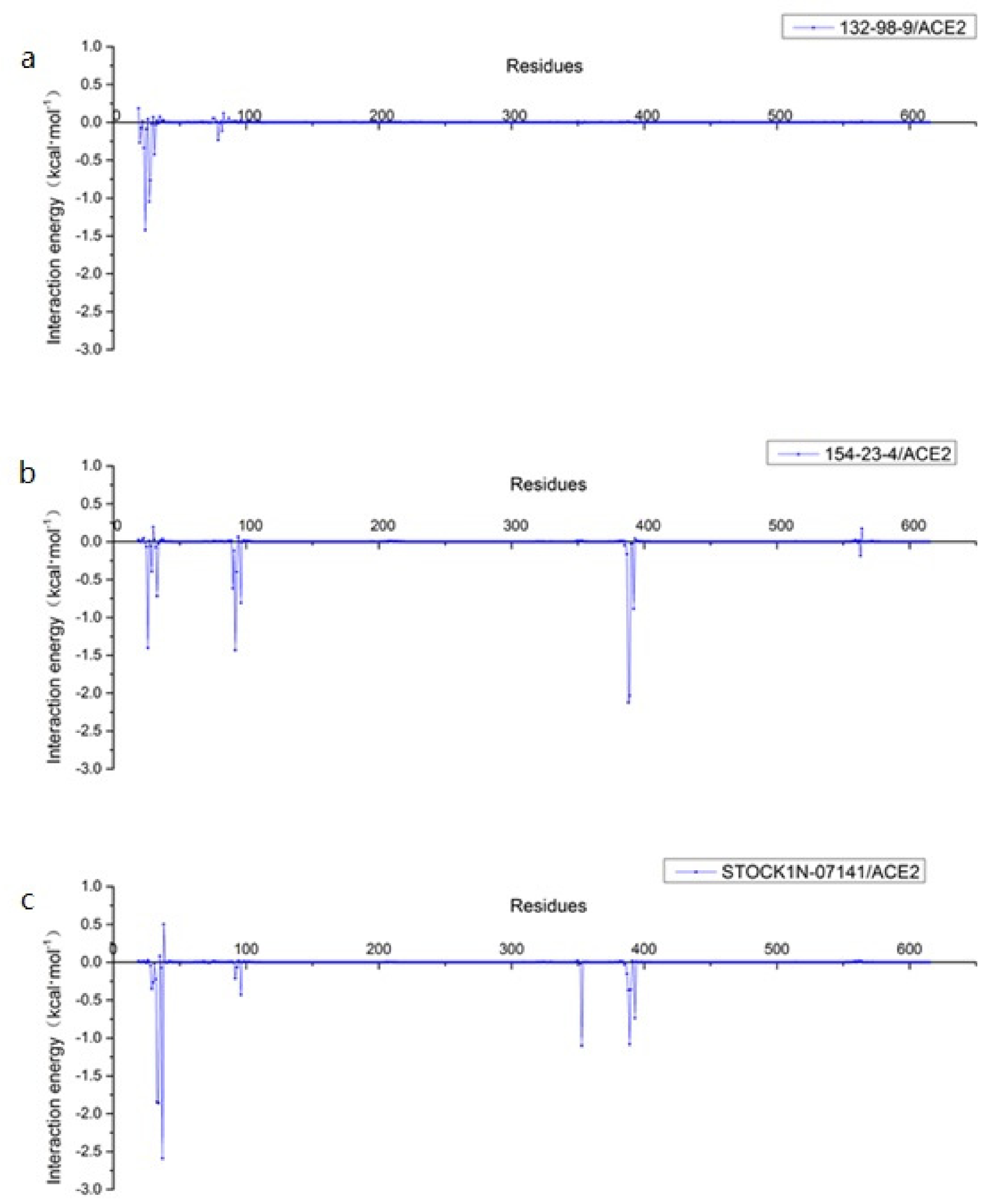
3.8. Per-Residue Free Energy Decomposition Analysis
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, C.; Acuña, M.J.; Torrejón, J.; Rebolledo, D.; Cabrera, D.; Santos, R.A.; Brandan, E. ACE2 is augmented in dystrophic skeletal muscle and plays a role in decreasing associated fibrosis. PLoS ONE 2014, 9, e93449. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Huang, X.; Tan, J.; Wu, P.; He, L. The expression of angiotensin-converting enzyme 2 in ovarian cancer and its correlation with prognosis. Chin. J. Front. Med. Sci. 2021, 3, 74–78. [Google Scholar]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- El Zowalaty, M.E.; Jarhult, J.D. From SARS to COVID-19: A previously unknown SARS- related coronavirus (SARS-CoV-2) of pandemic potential infecting humans—Call for a One Health approach. One Health 2020, 9, 100124. [Google Scholar] [CrossRef]
- Chen, T.M.; Rui, J.; Wang, Q.P.; Zhao, Z.Y.; Cui, J.A.; Yin, L. A mathematical model for simulating the phase-based transmissibility of a novel coronavirus. Infect. Dis. Poverty 2020, 9, 24. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Peng, X.; Xu, X.; Li, Y.; Cheng, L.; Zhou, X.; Ren, B. Transmission routes of 2019-nCoV and controls in dental practice. Int. J. Oral Sci. 2020, 12, 9. [Google Scholar] [CrossRef]
- She, J.; Jiang, J.; Ye, L.; Hu, L.; Bai, C.; Song, Y. 2019 novel coronavirus of pneumonia in Wuhan, China: Emerging attack and management strategies. Clin. Transl. Med. 2020, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Cai, L.; Cheng, Z.S.; Cheng, H.; Deng, T.; Fan, Y.P.; Fang, C.; Huang, D.; Huang, L.-Q.; Huang, Q.; et al. A rapid advice guideline for the diagnosis and treatment of 2019 novel coronavirus (2019-nCoV) infected pneumonia (standard version). Mil. Med. Res. 2020, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Malik, Y.S.; Sircar, S.; Bhat, S.; Sharun, K.; Dhama, K.; Dadar, M.; Tiwari, R.; Chaicumpa, W. Emerging novel coronavirus (2019-nCoV)-current scenario, evolutionary perspective based on genome analysis and recent developments. Vet. Q. 2020, 40, 68–76. [Google Scholar] [CrossRef]
- Jiang, S.; Du, L.; Shi, Z. An emerging coronavirus causing pneumonia outbreak in Wuhan, China: Calling for developing therapeutic and prophylactic strategies. Emerg. Microbes Infect. 2020, 9, 275–277. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Gomez-Puertas, P.; Cavanagh, D.; Gorbalenya, A.E.; Enjuanes, L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch. Virol. 2003, 148, 2207–2235. [Google Scholar] [CrossRef]
- Neuman, B.W.; Adair, B.D.; Yoshioka, C.; Quispe, J.D.; Orca, G.; Kuhn, P.; Milligan, R.A.; Yeager, M.; Buchmeier, M.J. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J. Virol. 2006, 80, 7918–7928. [Google Scholar] [CrossRef]
- Barcena, M.; Oostergetel, G.T.; Bartelink, W.; Faas, F.G.A.; Verkleij, A.; Rottier, P.J.M.; Koster, A.J.; Bosch, B.J. Cryo-electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc. Natl. Acad. Sci. USA 2009, 106, 582–587. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.-H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef]
- Lau, S.K.; Feng, Y.; Chen, H.; Luk, H.K.; Yang, W.H.; Li, K.S.; Zhang, Y.-Z.; Huang, Y.; Song, Z.-Z.; Chow, W.-N.; et al. Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Berardi, M.; Li, W.; Farzan, M.; Dormitzer, P.R.; Harrison, S.C. Conformational states of the severe acute respiratory syndrome coronavirus spike protein ectodomain. J. Virol. 2006, 80, 6794–6800. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/media/155071/download (accessed on 2 January 2022).
- Muralidharan, A.R.; Selvaraj, C.; Singh, S.K.; Sheu, J.R.; Thomas, P.A.; Geraldine, P. Structure-Based Virtual Screening and Biological Evaluation of a Calpain Inhibitor for Prevention of Selenite-Induced Cataractogenesis in an in Vitro System. J. Chem. Inf. Model. 2015, 55, 1686–1697. [Google Scholar] [CrossRef]
- Sugappriya, M.; Sudarsanam, D.; Bhaskaran, R.; Joseph, J.; Suresh, A. Druggability and Binding Site Interaction Studies of Potential Metabolites Isolated from Marine Sponge Aurora globostellata against Human Epidermal Growth Factor Receptor-2. Bioinformation 2017, 13, 261–268. [Google Scholar] [CrossRef][Green Version]
- Suenaga, A.; Okimoto, N.; Hirano, Y.; Fukui, K. An efficient computational method for calculating ligand binding affinities. PLoS ONE 2012, 7, e42846. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–245. [Google Scholar] [CrossRef]
- Wang, K.; Wan, M.; Wang, R.S.; Weng, Z. Opportunities for Web-based Drug Repositioning: Searching for Potential Antihypertensive Agents with Hypotension Adverse Events. J. Med. Internet Res. 2016, 18, e76. [Google Scholar] [CrossRef]
- Islam, S.; Shajib, M.S.; Rashid, R.B.; Khan, M.F.; Al-Mansur, M.A.; Datta, B.K.; Rashid, M.A. Antinociceptive activities of Artocarpus lacucha Buch-ham (Moraceae) and its isolated phenolic compound, catechin, in mice. BMC Complement. Altern. Med. 2019, 19, 214. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.H.; Huang, J.; Lin, Z.; Brown, A.C. Catechin-mediated restructuring of a bacterial toxin inhibits activity. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.W.; Tan, X.; Sun, J.Y.; Gu, C.M.; Liu, C.; Guo, X. Catechin attenuates TNF-alpha induced inflammatory response via AMPK-SIRT1 pathway in 3T3-L1 adipocytes. PLoS ONE 2019, 14, e0217090. [Google Scholar] [CrossRef]
- Lee, S.M.; Ko, I.G.; Kim, S.E.; Kim, D.H.; Kang, B.N. Protective effect of catechin on apoptosis of the lens epithelium in rats with N-methyl-N-nitrosourea-induced cataracts. Korean J. Ophthalmol. 2010, 24, 101–107. [Google Scholar] [CrossRef][Green Version]
- You, H.L.; Huang, C.C.; Chen, C.J.; Chang, C.C.; Liao, P.L.; Huang, S.T. Anti-pandemic influenza A (H1N1) virus potential of catechin and gallic acid. J. Chin. Med. Assoc. 2018, 81, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, M.; Guo, P.; Li, H.; Hu, Z.; Liu, X.; Zhang, Q. Isolation, Screening, and Characterization of Antibiotic-Degrading Bacteria for Penicillin V Potassium (PVK) from Soil on a Pig Farm. Int. J. Environ. Res. Public Health 2019, 16, 2166. [Google Scholar] [CrossRef] [PubMed]
- Barco, A.; Benetti, S.; Risi, C.D.; Marchetti, P.; Pollini, G.P.; Zanirato, V. D-(-)-Quinic acid: A chiron store for natural product synthesis. Tetrahedron Asymmetry 1997, 8, 3515–3545. [Google Scholar] [CrossRef]
- Indahl, S.R.; Scheline, R.R. Quinic acid aromatization in the rat. Urinary hippuric acid and catechol excretion following the singular or repeated administration of quinic acid. Xenobiotica 1973, 3, 549–556. [Google Scholar] [CrossRef]
- Liu, H.; Garrett, T.J.; Su, Z.; Khoo, C.; Zhao, S.; Gu, L. Modifications of the urinary metabolome in young women after cranberry juice consumption were revealed using the UHPLC-Q-orbitrap-HRMS-based metabolomics approach. Food Funct. 2020, 11, 2466–2476. [Google Scholar] [CrossRef]
- Pero, R.W.; Lund, H.; Leanderson, T. Antioxidant metabolism induced by quinic acid. Increased urinary excretion of tryptophan and nicotinamide. Phytother. Res. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Zhao, B.; Zhao-Wilson, X. Quinic acid could be a potential rejuvenating natural compound by improving survival of Caenorhabditis elegans under deleterious conditions. Rejuvenation Res. 2012, 15, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.H.; Liang, Q.; Zhang, Y.J.; Zhao, P. Naturally Occurring Arbutin Derivatives and Their Bioactivities. Chem. Biodivers. 2015, 12, 54–81. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fu, X.; Jiang, L.; Wang, L.; Bai, S.; Jiao, Y.; Xing, S.; Li, W.; Ma, J. Arbutin increases Caenorhabditis elegans longevity and stress resistance. Peer J. 2017, 5, e4170. [Google Scholar] [CrossRef]
- Bang, S.H.; Han, S.J.; Kim, D.H. Hydrolysis of arbutin to hydroquinone by human skin bacteria and its effect on antioxidant activity. J. Cosmet. Dermatol. 2008, 7, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, K.W. Anti-inflammatory effects of arbutin in lipopolysaccharide-stimulated BV2 microglial cells. Inflamm. Res. 2012, 61, 817–825. [Google Scholar] [CrossRef]
- Migas, P.; Krauze-Baranowska, M. The significance of arbutin and its derivatives in therapy and cosmetics. Phytochem. Lett. 2015, 13, 35–40. [Google Scholar] [CrossRef]
- Debnath, S.; Debnath, T.; Bhaumik, S.; Majumdar, S.; Kalle, A.M.; Aparna, V. Discovery of novel potential selective HDAC8 inhibitors by combine ligand-based, structure-based virtual screening and in-vitro biological evaluation. Sci. Rep. 2019, 9, 17174. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Gudipati, S.; Muttineni, R.; Mankad, A.U.; Pandya, H.A.; Jasrai, Y.T. Molecular docking based screening of Noggin inhibitors. Bioinformation 2018, 14, 15–20. [Google Scholar] [CrossRef]
- Chacko, S.; Samanta, S. A novel approach towards design, synthesis and evaluation of some Schiff base analogues of 2-aminopyridine and 2-aminobezothiazole against hepatocellular carcinoma. Biomed. Pharmacother. 2017, 89, 162–176. [Google Scholar] [CrossRef]
- Subramaniyan, V.; Mathiyalagan, S.; Praveenkumar, A.; Srinivasan, P.; Palani, M.; Ravichandran, V.; Nallasamy, P. Molecular docking and ADME properties of bioactive molecules against human acid-beta-glucosidase enzyme, cause of Gaucher’s disease. Silico Pharmacol. 2018, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- David, T.I.; Adelakun, N.S.; Omotuyi, O.I.; Metibemu, D.S.; Ekun, O.E. Molecular docking analysis of phyto-constituents from Cannabis sativa with pfDHFR. Bioinformation 2018, 14, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Betz, R.; Botello-Smith, W.; Cerutti, D.; Cheatham IIIT, E.; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.; et al. AMBER 2014; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Cruz, J.N.; Costa, J.F.S.; Khayat, A.S.; Kuca, K.; Barros, C.A.L.; Neto, A.M.J.C. Molecular dynamics simulation and binding free energy studies of novel leads belonging to the benzofuran class inhibitors of Mycobacterium tuberculosis Polyketide Synthase 13. J. Biomol. Struct. Dyn. 2019, 37, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, Y.; Wan, S.; Zhang, J.J. Investigation on the binding mechanism of loratinib with the c-ros oncogene 1 (ROS1) receptor tyrosine kinase via molecular dynamics simulation and binding free energy calculations. J. Biomol. Struct. Dyn. 2018, 36, 3106–3113. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Qiu, M.; Guo, L.; Yao, X. Computational study of the binding mechanism between farnesoid X receptor and antagonist Nbenzyl-N-(3-(tertbutyl)-4-hydroxyphenyl)-2,6-dichloro-4-(dimethylamino) benzamide. J. Biomol. Struct. Dyn. 2019, 37, 1628–1640. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Van der Spoel, D.; van Maaren, P.J. The Origin of Layer Structure Artifacts in Simulations of Liquid Water. J. Chem. Theory Comput. 2006, 1, 1–11. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Shen, M.; Tian, S.; Xu, L.; Pan, P.; Guan, Y.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 2014, 16, 22035–22045. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| categories | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| numbers | 26 | 3 | 6 | 1 | 3 | 10 | 112 | 12 | 1 | 2 | 121 | 1 |
| Molecule | Structures | a mol MW | b QPlogPo/w | c Donor HB | d Accept HB | e PSA | f %Human Oral Absorption |
|---|---|---|---|---|---|---|---|
| 154-23-4 |  | 290.272 | 0.459 | 5 | 5.450 | 114.862 | 60.524 |
| STOCK1N-25862 |  | 223.234 | −0.792 | 2.000 | 7.000 | 105.840 | 52.040 |
| STOCK1N-20317 |  | 319.442 | 0.732 | 1.000 | 8.700 | 59.123 | 66.608 |
| 132-98-9 |  | 350.389 | 2.076 | 1.250 | 7.000 | 116.295 | 62.569 |
| STOCK1N-81825 |  | 316.359 | −0.373 | 1.000 | 9.000 | 109.685 | 52.306 |
| STOCK1N-79835 |  | 236.233 | −0.301 | 5.000 | 6.500 | 139.745 | 40.010 |
| STOCK1N−53429 |  | 192.168 | −1.256 | 5.000 | 7.850 | 127.861 | 38.810 |
| STOCK1N-07141 |  | 272.254 | −0.976 | 5.000 | 10.000 | 120.231 | 56.911 |
| STOCK1N-05528 |  | 291.355 | 2.229 | 4.000 | 4.000 | 79.958 | 80.263 |
| STOCK1N-20017 |  | 321.335 | 2.216 | 3.000 | 5.500 | 96.236 | 84.644 |
| STOCK1N-74592 |  | 285.342 | 3.083 | 1.000 | 4.500 | 69.235 | 86.381 |
| STOCK1N-88912 |  | 402.449 | 3.630 | 1.000 | 6.750 | 65.991 | 100.000 |
| Cluster No. | Compound ID | Docking Interaction | Interacting Residues | Glide Score | Docking Score | ΔG (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | 154-23-4 |  | Asp30, Asn33, Ala386, Ala387 | −5.418 | −5.148 | −37.592 |
| 2 | STOCK1N-25862 |  | Glu37, Ala387, Arg393 | −4.677 | −4.584 | −22.063 |
| 3 | STOCK1N-20317 |  | Glu35, Glu75, Gln76 | −5.156 | −4.905 | −42.895 |
| 4 | 132-98-9 |  | Lys26, Asn33, Asn90, Gln96 | −4.335 | −4.335 | −18.899 |
| 5 | STOCK1N-81825 |  | Asp30, Asn33, Glu37, Phe390, | −4.839 | −4.813 | −33.580 |
| 6 | STOCK1N-79835 |  | Gln24, Glu35, Gln76, Tyr83 | −4.959 | −4.947 | −22.213 |
| 7 | STOCK1N−53429 |  | Lys31, Glu35, Gln76, | −5.923 | −5.923 | −19.312 |
| 8 | STOCK1N-07141 |  | Asp30, Asn33, Glu37, Arg393 | −4.898 | −4.898 | −27.518 |
| 9 | STOCK1N-05528 |  | Glu35, Glu75 | −5.002 | −4.855 | −41.649 |
| 10 | STOCK1N-20017 |  | Asp30, His34, Asn33, Glu37, Arg393 | −4.543 | −4.542 | −37.204 |
| 11 | STOCK1N-74592 |  | Lys31, Glu35, Gln76 | −6.193 | −5.881 | −33.179 |
| 12 | STOCK1N-88912 |  | Gln75, Glu76 | −4.134 | −4.094 | −32.160 |
| Contribution (kcal/mol) | Complexes | ||
|---|---|---|---|
| 132-98-9/ACE2 | 154-23-4/ACE2 | STOCK1N-07141/ACE2 | |
| ∆Eele | 173.35(23.03) | −38.55(11.61) | −48.92(20.00) |
| ∆EvdW | −15.22(7.94) | −17.99(3.39) | −14.62(4.28) |
| ∆GGB | −166.61(21.36) | 49.47(10.29) | 54.00(15.97) |
| ∆GSA | −2.12(1.09) | −3.29(0.39) | −2.92(0.49) |
| ∆Egas | 158.12(22.65) | −56.54(11.58) | −63.54(19.00) |
| ∆Esolv | −168.73(21.46) | 46.18(10.12) | 51.08(15.66) |
| ∆Gbind | −10.61(6.97) | −10.36(3.61) | −12.46(5.52) |
| Complex | Acceptor | DonorH | Donor | Frac |
|---|---|---|---|---|
| 132-98-9/ACE2 | MOL@O1 | TYR_83@HH | TYR_83@OH | 0.1460 |
| 154-23-4/ACE2 | MOL@O2 | ASN_90@HD21 | ASN_90@ND2 | 0.3216 |
| GLN_388@OE1 | MOL@H13 | MOL@O5 | 0.3006 | |
| STOCK1N-07141/ACE2 | GLU_37@OE22 | MOL@H6 | MOL@O3 | 0.5430 |
| GLU_37@OE2 | MOL@H7 | MOL@O4 | 0.5300 | |
| ALA_387@O | MOL@H15 | MOL@O6 | 0.5072 | |
| GLU_37@OE1 | MOL@H7 | MOL@O4 | 0.4849 | |
| GLU_37@OE1 | MOL@H6 | MOL@O3 | 0.4359 | |
| MOL@O4 | HIP_34@HD1 | HIP_34@ND1 | 0.3574 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, H. Virtual Screening of Natural Chemical Databases to Search for Potential ACE2 Inhibitors. Molecules 2022, 27, 1740. https://doi.org/10.3390/molecules27051740
Yao H. Virtual Screening of Natural Chemical Databases to Search for Potential ACE2 Inhibitors. Molecules. 2022; 27(5):1740. https://doi.org/10.3390/molecules27051740
Chicago/Turabian StyleYao, Huiping. 2022. "Virtual Screening of Natural Chemical Databases to Search for Potential ACE2 Inhibitors" Molecules 27, no. 5: 1740. https://doi.org/10.3390/molecules27051740
APA StyleYao, H. (2022). Virtual Screening of Natural Chemical Databases to Search for Potential ACE2 Inhibitors. Molecules, 27(5), 1740. https://doi.org/10.3390/molecules27051740