
Oxidative Stress and Cancer Heterogeneity Orchestrate NRF2 Roles Relevant for Therapy Response

 , , ,
, , ,  ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. The NRF2 Structure and Regulation
3. Regulation of NRF2 Expression in Cancer
4. Multilevel Models of Communication between NRF2 and TP53
5. The Role of NRF2 in Cancer Stem Cells
6. NRF2 Mediated Polarization of Neutrophils and Macrophages in Cancer
7. NRF2 and Cancer Resistance to Therapies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Meng, Q.; Liu, L.-Z.; Rojanasakul, Y.; Wang, X.-R.; Jiang, B.-H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef] [PubMed]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Sandalio, L.M.; Rodríguez-Serrano, M.; Romero-Puertas, M.C.; del Río, L.A. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell. Biochem. 2013, 69, 231–255. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Naidoo, K.; Birch-Machin, M. Oxidative Stress and Ageing: The Influence of Environmental Pollution, Sunlight and Diet on Skin. Cosmetics 2017, 4, 4. [Google Scholar] [CrossRef]
- Parke, D.V.; Sapota, A. Chemical toxicity and reactive oxygen species. Int. J. Occup. Med. Environ. Health 1996, 9, 331–340. [Google Scholar] [PubMed]
- Murphy, M.P.; Holmgren, A.; Larsson, N.-G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Rybicka, J.M.; Balce, D.R.; Khan, M.F.; Krohn, R.M.; Yates, R.M. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 10496–10501. [Google Scholar] [CrossRef]
- Yang, H.-C.; Cheng, M.-L.; Ho, H.-Y.; Chiu, D.T.-Y. The microbicidal and cytoregulatory roles of NADPH oxidases. Microbes Infect. 2011, 13, 109–120. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- Cinat, D.; Coppes, R.P.; Barazzuol, L. DNA Damage-Induced Inflammatory Microenvironment and Adult Stem Cell Response. Front. Cell Dev. Biol. 2021, 9, 729136. [Google Scholar] [CrossRef] [PubMed]
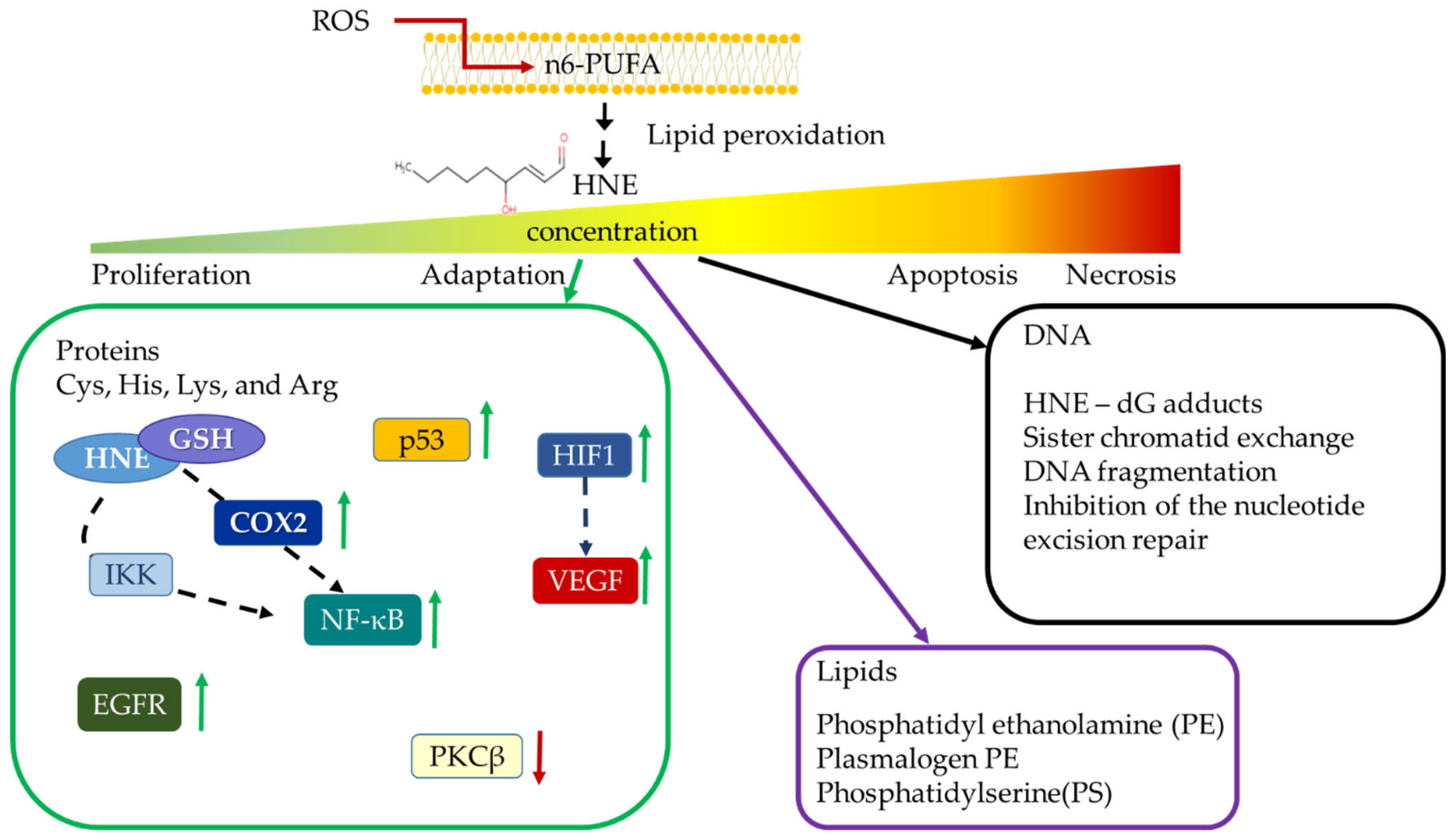
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Zarkovic, N. Overview on major lipid peroxidation bioactive factor 4-hydroxynonenal as pluripotent growth-regulating factor. Free Radic. Res. 2015, 49, 850–860. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef]
- Davies, S.S.; Guo, L. Lipid peroxidation generates biologically active phospholipids including oxidatively N-modified phospholipids. Chem. Phys. Lipids 2014, 181, 1–33. [Google Scholar] [CrossRef]
- Sunjic, S.B.; Cipak, A.; Rabuzin, F.; Wildburger, R.; Zarkovic, N. The influence of 4-hydroxy-2-nonenal on proliferation, differentiation and apoptosis of human osteosarcoma cells. Biofactors 2005, 24, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Borovic, S.; Cipak, A.; Meinitzer, A.; Kejla, Z.; Perovic, D.; Waeg, G.; Zarkovic, N. Differential sensitivity to 4-hydroxynonenal for normal and malignant mesenchymal cells. Redox Rep. 2007, 12, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sharma, A.; Yang, Y.; Awasthi, S.; Singhal, S.S.; Zimniak, P.; Awasthi, Y.C. Functional reconstitution of Ral-binding GTPase activating protein, RLIP76, in proteoliposomes catalyzing ATP-dependent transport of glutathione conjugate of 4-hydroxynonenal. Acta Biochim. Pol. 2002, 49, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; Tang, M.-S. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: A possible mechanism for lipid peroxidation-induced carcinogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 8598–8602. [Google Scholar] [CrossRef]
- Gasparovic, A.C.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. Cancer growth regulation by 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 226–234. [Google Scholar] [CrossRef]
- Abreu, I.A.; Cabelli, D.E. Superoxide dismutases-a review of the metal-associated mechanistic variations. Biochim. Biophys. Acta 2010, 1804, 263–274. [Google Scholar] [CrossRef]
- Chandimali, N.; Jeong, D.K.; Kwon, T. Peroxiredoxin II Regulates Cancer Stem Cells and Stemness-Associated Properties of Cancers. Cancers 2018, 10, 305. [Google Scholar] [CrossRef]
- Rabilloud, T.; Heller, M.; Rigobello, M.P.; Bindoli, A.; Aebersold, R.; Lunardi, J. The mitochondrial antioxidant defence system and its response to oxidative stress. Proteomics 2001, 1, 1105–1110. [Google Scholar] [CrossRef]
- Cipak, A.; Jaganjac, M.; Tehlivets, O.; Kohlwein, S.D.; Zarkovic, N. Adaptation to oxidative stress induced by polyunsaturated fatty acids in yeast. Biochim. Biophys. Acta 2008, 1781, 283–287. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Geir Møller, S.; Hashemzadeh-Chaleshtori, M.; Bidkhori, G.; Jami, M.-S. Molecular Mechanisms behind Free Radical Scavengers Function against Oxidative Stress. Antioxidants 2017, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Moccia, S.; Russo, M.; Spagnuolo, C. Redox regulation by carotenoids: Evidence and conflicts for their application in cancer. Biochem. Pharmacol. 2021, 194, 114838. [Google Scholar] [CrossRef] [PubMed]
- The ATBC Cancer Prevention Study Group. The alpha-tocopherol, beta-carotene lung cancer prevention study: Design, methods, participant characteristics, and compliance. Ann. Epidemiol. 1994, 4, 1–10. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Meyskens, F.L.; Omenn, G.S.; Valanis, B.; Williams, J.H. The Beta-Carotene and Retinol Efficacy Trial: Incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J. Natl. Cancer Inst. 2004, 96, 1743–1750. [Google Scholar] [CrossRef]
- Ebbing, M.; Bønaa, K.H.; Nygård, O.; Arnesen, E.; Ueland, P.M.; Nordrehaug, J.E.; Rasmussen, K.; Njølstad, I.; Refsum, H.; Nilsen, D.W.; et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA 2009, 302, 2119–2126. [Google Scholar] [CrossRef]
- Bast, A.; Haenen, G.R.M.M. The toxicity of antioxidants and their metabolites. Environ. Toxicol. Pharmacol. 2002, 11, 251–258. [Google Scholar] [CrossRef]
- Huang, J.; Hodis, H.N.; Weinstein, S.J.; Mack, W.J.; Sampson, J.N.; Mondul, A.M.; Albanes, D. Serum Metabolomic Response to Low- and High-Dose Vitamin E Supplementation in Two Randomized Controlled Trials. Cancer Epidemiol. Biomarkers Prev. 2020, 29, 1329–1334. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; Ciucis, C.D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Zhao, T.; Zhu, Y.; Morinibu, A.; Kobayashi, M.; Shinomiya, K.; Itasaka, S.; Yoshimura, M.; Guo, G.; Hiraoka, M.; Harada, H. HIF-1-mediated metabolic reprogramming reduces ROS levels and facilitates the metastatic colonization of cancers in lungs. Sci. Rep. 2014, 4, 3793. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.-Y.; Lai, C.-C.; Chang, C.-J.; Huang, W.-C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed]
- Negi, C.K.; Jena, G. Nrf2, a novel molecular target to reduce type 1 diabetes associated secondary complications: The basic considerations. Eur. J. Pharmacol. 2019, 843, 12–26. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Papagiannakopoulos, T.; Motohashi, H. Metabolic features of cancer cells in NRF2 addiction status. Biophys. Rev. 2020, 12, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329.e18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J.; Li, M.; Kong, L.; Yu, J. The expression of p-p62 and nuclear Nrf2 in esophageal squamous cell carcinoma and association with radioresistance. Thorac. Cancer 2020, 11, 130–139. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Parri, M.; Ippolito, L.; Cirri, P.; Ramazzotti, M.; Chiarugi, P. Metabolic cell communication within tumour microenvironment: Models, methods and perspectives. Curr. Opin. Biotechnol. 2020, 63, 210–219. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.-Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985–1003.e7. [Google Scholar] [CrossRef]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef]
- Bruford, E.A.; Braschi, B.; Denny, P.; Jones, T.E.M.; Seal, R.L.; Tweedie, S. Guidelines for human gene nomenclature. Nat. Genet. 2020, 52, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef]
- Kansanen, E.; Kivelä, A.M.; Levonen, A.-L. Regulation of Nrf2-dependent gene expression by 15-deoxy-Delta12,14-prostaglandin J2. Free Radic. Biol. Med. 2009, 47, 1310–1317. [Google Scholar] [CrossRef]
- Ogura, T.; Tong, K.I.; Mio, K.; Maruyama, Y.; Kurokawa, H.; Sato, C.; Yamamoto, M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc. Natl. Acad. Sci. USA 2010, 107, 2842–2847. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Tao, S.; Liu, P.; Luo, G.; Rojo de la Vega, M.; Chen, H.; Wu, T.; Tillotson, J.; Chapman, E.; Zhang, D.D. p97 Negatively Regulates NRF2 by Extracting Ubiquitylated NRF2 from the KEAP1-CUL3 E3 Complex. Mol. Cell. Biol. 2017, 37, e00660-16. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Sova, M.; Saso, L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug Des. Devel. Ther. 2018, 12, 3181–3197. [Google Scholar] [CrossRef]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yu, S.; Liu, T.; Kim, J.-H.; Blank, V.; Li, H.; Kong, A.-N.T. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim. Biophys. Acta 2008, 1783, 1847–1856. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- Shaw, P.; Chattopadhyay, A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef]
- Stefanson, A.L.; Bakovic, M. Dietary regulation of Keap1/Nrf2/ARE pathway: Focus on plant-derived compounds and trace minerals. Nutrients 2014, 6, 3777–3801. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mittal, R. Nrf2: A potential therapeutic target for diabetic neuropathy. Inflammopharmacology 2017, 25, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Alam, J. Heme oxygenase-1: Function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am. J. Respir. Cell Mol. Biol. 1996, 15, 9–19. [Google Scholar] [CrossRef]
- Sun, J.; Brand, M.; Zenke, Y.; Tashiro, S.; Groudine, M.; Igarashi, K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA 2004, 101, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Jaiswal, A.K. Antioxidant-induced phosphorylation of tyrosine 486 leads to rapid nuclear export of Bach1 that allows Nrf2 to bind to the antioxidant response element and activate defensive gene expression. J. Biol. Chem. 2010, 285, 153–162. [Google Scholar] [CrossRef]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e22. [Google Scholar] [CrossRef]
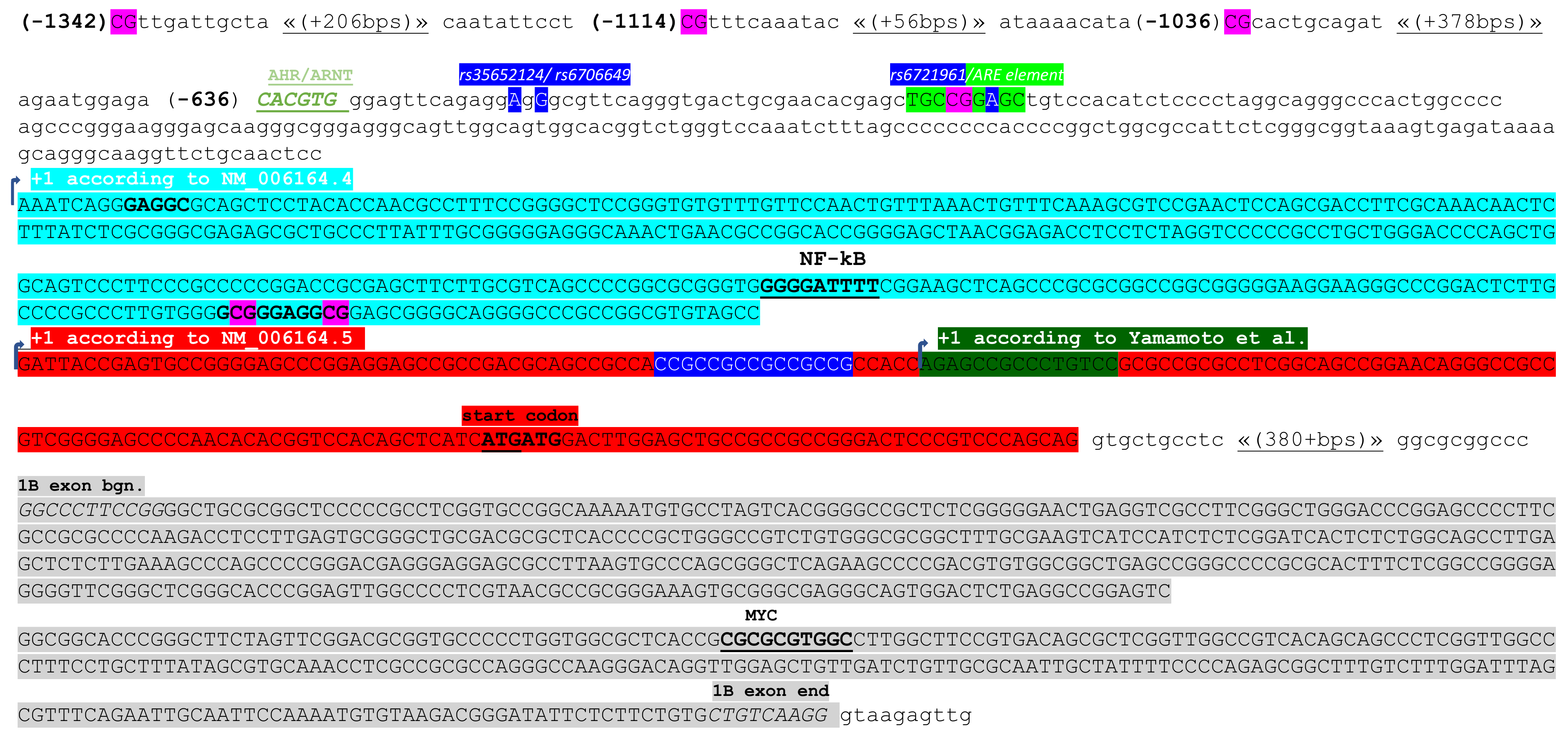
- Yamamoto, T.; Yoh, K.; Kobayashi, A.; Ishii, Y.; Kure, S.; Koyama, A.; Sakamoto, T.; Sekizawa, K.; Motohashi, H.; Yamamoto, M. Identification of polymorphisms in the promoter region of the human NRF2 gene. Biochem. Biophys. Res. Commun. 2004, 321, 72–79. [Google Scholar] [CrossRef]
- Marzec, J.M.; Christie, J.D.; Reddy, S.P.; Jedlicka, A.E.; Vuong, H.; Lanken, P.N.; Aplenc, R.; Yamamoto, T.; Yamamoto, M.; Cho, H.Y.; et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007, 21, 2237–2246. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef]
- Köhle, C.; Bock, K.W. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. Biochem. Pharmacol. 2007, 73, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Milković, L.; Tomljanović, M.; Čipak Gašparović, A.; Novak Kujundžić, R.; Šimunić, D.; Konjevoda, P.; Mojzeš, A.; Đaković, N.; Žarković, N.; Gall Trošelj, K. Nutritional Stress in Head and Neck Cancer Originating Cell Lines: The Sensitivity of the NRF2-NQO1 Axis. Cells 2019, 8, 1001. [Google Scholar] [CrossRef]
- Zgheib, E.; Limonciel, A.; Jiang, X.; Wilmes, A.; Wink, S.; van de Water, B.; Kopp-Schneider, A.; Bois, F.Y.; Jennings, P. Investigation of Nrf2, AhR and ATF4 Activation in Toxicogenomic Databases. Front. Genet. 2018, 9, 429. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Human NAD(P)H:quinone oxidoreductase (NQO1) gene structure and induction by dioxin. Biochemistry 1991, 30, 10647–10653. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Hattori, S.; Kasai, K. 4-hydroxynonenal prevents NO production in vascular smooth muscle cells by inhibiting nuclear factor-kappaB-dependent transcriptional activation of inducible NO synthase. Arter. Thromb. Vasc. Biol. 2001, 21, 1179–1183. [Google Scholar] [CrossRef][Green Version]
- Semlitsch, T.; Tillian, H.M.; Zarkovic, N.; Borovic, S.; Purtscher, M.; Hohenwarter, O.; Schaur, R.J. Differential influence of the lipid peroxidation product 4-hydroxynonenal on the growth of human lymphatic leukaemia cells and human periopherial blood lymphocytes. Anticancer Res. 2002, 22, 1689–1697. [Google Scholar]
- Bae, I.; Fan, S.; Meng, Q.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Xu, J.; Goldberg, I.D.; Jaiswal, A.K.; Rosen, E.M. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004, 64, 7893–7909. [Google Scholar] [CrossRef]
- Kang, H.J.; Hong, Y.B.; Kim, H.J.; Rodriguez, O.C.; Nath, R.G.; Tilli, E.M.; Albanese, C.; Chung, F.L.; Kwon, S.H.; Bae, I. Detoxification: A novel function of BRCA1 in tumor suppression? Toxicol. Sci. 2011, 122, 26–37. [Google Scholar] [CrossRef]
- Brázda, V.; Hároníková, L.; Liao, J.C.; Fridrichová, H.; Jagelská, E.B. Strong preference of BRCA1 protein to topologically constrained non-B DNA structures. BMC Mol. Biol. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kim, H.J.; Kim, S.K.; Barouki, R.; Cho, C.H.; Khanna, K.K.; Rosen, E.M.; Bae, I. BRCA1 modulates xenobiotic stress-inducible gene expression by interacting with ARNT in human breast cancer cells. J. Biol. Chem. 2006, 281, 14654–14662. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Hsiao, J.R.; Jiang, S.S.; Chang, J.Y.; Chu, P.Y.; Liu, K.J.; Fang, H.L.; Lin, L.M.; Chen, H.H.; Huang, Y.W.; et al. c-MYC-directed NRF2 drives malignant progression of head and neck cancer via glucose-6-phosphate dehydrogenase and transketolase activation. Theranostics 2021, 11, 5232–5247. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef]
- Khor, T.O.; Fuentes, F.; Shu, L.; Paredes-Gonzalez, X.; Yang, A.Y.; Liu, Y.; Smiraglia, D.J.; Yegnasubramanian, S.; Nelson, W.G.; Kong, A.N. Epigenetic DNA methylation of antioxidative stress regulator NRF2 in human prostate cancer. Cancer Prev. Res. 2014, 7, 1186–1197. [Google Scholar] [CrossRef]
- Hussain, S.P.; Raja, K.; Amstad, P.A.; Sawyer, M.; Trudel, L.J.; Wogan, G.N.; Hofseth, L.J.; Shields, P.G.; Billiar, T.R.; Trautwein, C.; et al. Increased p53 mutation load in nontumorous human liver of wilson disease and hemochromatosis: Oxyradical overload diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 12770–12775. [Google Scholar] [CrossRef]
- Denissenko, M.F.; Chen, J.X.; Tang, M.S.; Pfeifer, G.P. Cytosine methylation determines hot spots of DNA damage in the human P53 gene. Proc. Natl. Acad. Sci. USA 1997, 94, 3893–3898. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Dong, L.J.; Takahashi, Y.; Chen, J.; Liu, X.; Chen, Q.; Ma, J.X.; Li, X.R. miRNA-451a regulates RPE function through promoting mitochondrial function in proliferative diabetic retinopathy. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E443–E452. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Xiao, M.; Yang, F.; Xiao, J.; Zhang, H.; Su, L.; Zhang, X.; Li, X. MicroRNA-431-5p encapsulated in serum extracellular vesicles as a biomarker for proliferative diabetic retinopathy. Int. J. Biochem. Cell Biol. 2021, 135, 105975. [Google Scholar] [CrossRef] [PubMed]
- Pizzimenti, S.; Ferracin, M.; Sabbioni, S.; Toaldo, C.; Pettazzoni, P.; Dianzani, M.U.; Negrini, M.; Barrera, G. MicroRNA expression changes during human leukemic HL-60 cell differentiation induced by 4-hydroxynonenal, a product of lipid peroxidation. Free Radic Biol Med. 2009, 46, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Inoue, J.; Kawano, T.; Kozaki, K.; Omura, K.; Inazawa, J. The impact of miRNA-based molecular diagnostics and treatment of NRF2-stabilized tumors. Mol. Cancer Res. 2014, 12, 58–68. [Google Scholar] [CrossRef]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991. [Google Scholar] [CrossRef]
- Singh, B.; Ronghe, A.M.; Chatterjee, A.; Bhat, N.K.; Bhat, H.K. MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis 2013, 34, 1165–1172. [Google Scholar] [CrossRef]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef]
- van Jaarsveld, M.T.; Helleman, J.; Boersma, A.W.; van Kuijk, P.F.; van Ijcken, W.F.; Despierre, E.; Vergote, I.; Mathijssen, R.H.; Berns, E.M.; Verweij, J.; et al. miR-141 regulates KEAP1 and modulates cisplatin sensitivity in ovarian cancer cells. Oncogene 2013, 32, 4284–4293. [Google Scholar] [CrossRef] [PubMed]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Junn, E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic. Biol. Med. 2015, 89, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Akdemir, B.; Nakajima, Y.; Inazawa, J.; Inoue, J. miR-432 Induces NRF2 Stabilization by Directly Targeting KEAP1. Mol. Cancer Res. 2017, 15, 1570–1578. [Google Scholar] [CrossRef]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.-C.; Lin, P.-L.; Wang, Y.-C.; He, T.-Y.; Lee, M.-C.; Yeh, S.D.; Chen, C.-Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef]
- Hiemstra, S.; Niemeijer, M.; Koedoot, E.; Wink, S.; Klip, J.E.; Vlasveld, M.; de Zeeuw, E.; van Os, B.; White, A.; van de Water, B. Comprehensive Landscape of Nrf2 and p53 Pathway Activation Dynamics by Oxidative Stress and DNA Damage. Chem. Res. Toxicol. 2017, 30, 923–933. [Google Scholar] [CrossRef]
- You, A.; Nam, C.-W.; Wakabayashi, N.; Yamamoto, M.; Kensler, T.W.; Kwak, M.-K. Transcription factor Nrf2 maintains the basal expression of Mdm2: An implication of the regulation of p53 signaling by Nrf2. Arch. Biochem. Biophys. 2011, 507, 356–364. [Google Scholar] [CrossRef]
- Liu, K.; Jin, B.; Wu, C.; Yang, J.; Zhan, X.; Wang, L.; Shen, X.; Chen, J.; Chen, H.; Mao, Z. NQO1 Stabilizes p53 in Response to Oncogene-Induced Senescence. Int. J. Biol. Sci. 2015, 11, 762–771. [Google Scholar] [CrossRef]
- Kahroba, H.; Shirmohamadi, M.; Hejazi, M.S.; Samadi, N. The Role of Nrf2 signaling in cancer stem cells: From stemness and self-renewal to tumorigenesis and chemoresistance. Life Sci. 2019, 239, 116986. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Fan, Y.; Hu, Y.; Ji, X.; Sun, Q.; Liu, H. Knockdown of nuclear factor erythroid 2-related factor 2 by lentivirus induces differentiation of glioma stem-like cells. Oncol. Rep. 2014, 32, 1170–1178. [Google Scholar] [CrossRef]
- Kamble, D.; Mahajan, M.; Dhat, R.; Sitasawad, S. Keap1-Nrf2 Pathway Regulates ALDH and Contributes to Radioresistance in Breast Cancer Stem Cells. Cells 2021, 10, 83. [Google Scholar] [CrossRef]
- Kim, D.; Choi, B.-H.; Ryoo, I.-G.; Kwak, M.-K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: Inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018, 9, 896. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Morine, Y.; Yamada, S.; Saito, Y.; Ikemoto, T.; Tokuda, K.; Takasu, C.; Miyazaki, K.; Shimada, M. Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells. PLoS ONE 2021, 16, e0256755. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Wang, Y.; Lalli, M.A.; Guzman, E.; Godshalk, S.E.; Zhou, H.; Kosik, K.S. Primary Cilium-Autophagy-Nrf2 (PAN) Axis Activation Commits Human Embryonic Stem Cells to a Neuroectoderm Fate. Cell 2016, 165, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86.e6. [Google Scholar] [CrossRef]
- Fiorillo, M.; Sotgia, F.; Lisanti, M.P. “Energetic” Cancer Stem Cells (e-CSCs): A New Hyper-Metabolic and Proliferative Tumor Cell Phenotype, Driven by Mitochondrial Energy. Front. Oncol. 2018, 8, 677. [Google Scholar] [CrossRef]
- Kipp, A.P.; Deubel, S.; Arnér, E.S.J.; Johansson, K. Time- and cell-resolved dynamics of redox-sensitive Nrf2, HIF and NF-κB activities in 3D spheroids enriched for cancer stem cells. Redox Biol. 2017, 12, 403–409. [Google Scholar] [CrossRef]
- Kim, D.-H.; Jang, J.-H.; Kwon, O.-S.; Cha, H.-J.; Youn, H.-J.; Chun, K.-S.; Surh, Y.-J. Nuclear Factor Erythroid-Derived 2-Like 2-Induced Reductive Stress Favors Self-Renewal of Breast Cancer Stem-Like Cells via the FoxO3a-Bmi-1 Axis. Antioxid. Redox Signal. 2020, 32, 1313–1329. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, Z.; Lu, Y.; Liu, Q.; Xu, X.; Xu, J.; Liu, Y.; Yu, H.; Yu, D.; Sun, B. Loss of Xanthine Oxidoreductase Potentiates Propagation of Hepatocellular Carcinoma Stem Cells. Hepatology 2020, 71, 2033–2049. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bu, S.; Xin, D.; Li, B.; Wang, L.; Lai, D. Autophagy Is Indispensable for the Self-Renewal and Quiescence of Ovarian Cancer Spheroid Cells with Stem Cell-Like Properties. Oxid. Med. Cell. Longev. 2018, 2018, 7010472. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bi, Z.; Fu, Y.; Rice, M.K.A.; Zhang, Q.; Wadgaonkar, P.; Almutairy, B.; Zhang, W.; Lu, Y.; Xu, L.; et al. Characterization of Arsenic-Induced Cancer Stem-Like Cells. Methods Mol. Biol. 2020, 2117, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Chen, B.; Thakur, C.; Lu, Y.; Chen, F. Arsenic-induced sub-lethal stress reprograms human bronchial epithelial cells to CD61¯ cancer stem cells. Oncotarget 2014, 5, 1290–1303. [Google Scholar] [CrossRef] [PubMed]
- Waalkes, M.P.; Liu, J.; Germolec, D.R.; Trempus, C.S.; Cannon, R.E.; Tokar, E.J.; Tennant, R.W.; Ward, J.M.; Diwan, B.A. Arsenic exposure in utero exacerbates skin cancer response in adulthood with contemporaneous distortion of tumor stem cell dynamics. Cancer Res. 2008, 68, 8278–8285. [Google Scholar] [CrossRef]
- Bi, Z.; Zhang, Q.; Fu, Y.; Wadgaonkar, P.; Zhang, W.; Almutairy, B.; Xu, L.; Rice, M.; Qiu, Y.; Thakur, C.; et al. Nrf2 and HIF1α converge to arsenic-induced metabolic reprogramming and the formation of the cancer stem-like cells. Theranostics 2020, 10, 4134–4149. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Taylor, W.R.; Fedorka, S.R.; Gad, I.; Shah, R.; Alqahtani, H.D.; Koranne, R.; Kuganesan, N.; Dlamini, S.; Rogers, T.; Al-Hamashi, A.; et al. Small-Molecule Ferroptotic Agents with Potential to Selectively Target Cancer Stem Cells. Sci. Rep. 2019, 9, 5926. [Google Scholar] [CrossRef]
- Cipak, A.; Mrakovcic, L.; Ciz, M.; Lojek, A.; Mihaylova, B.; Goshev, I.; Jaganjac, M.; Cindric, M.; Sitic, S.; Margaritoni, M.; et al. Growth suppression of human breast carcinoma stem cells by lipid peroxidation product 4-hydroxy-2-nonenal and hydroxyl radical-modified collagen. Acta Biochim. Pol. 2010, 57, 165–171. [Google Scholar] [CrossRef]
- Čipak Gašparović, A.; Milković, L.; Dandachi, N.; Stanzer, S.; Pezdirc, I.; Vrančić, J.; Šitić, S.; Suppan, C.; Balic, M. Chronic Oxidative Stress Promotes Molecular Changes Associated with Epithelial Mesenchymal Transition, NRF2, and Breast Cancer Stem Cell Phenotype. Antioxidants 2019, 8, 633. [Google Scholar] [CrossRef]
- Sunjic, S.B.; Gasparovic, A.C.; Jaganjac, M.; Rechberger, G.; Meinitzer, A.; Grune, T.; Kohlwein, S.D.; Mihaljevic, B.; Zarkovic, N. Sensitivity of Osteosarcoma Cells to Concentration-Dependent Bioactivities of Lipid Peroxidation Product 4-Hydroxynonenal Depend on Their Level of Differentiation. Cells 2021, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.-G.; Choi, B.-H.; Kwak, M.-K. Activation of NRF2 by p62 and proteasome reduction in sphere-forming breast carcinoma cells. Oncotarget 2015, 6, 8167–8184. [Google Scholar] [CrossRef]
- Goto, S.; Kawabata, T.; Li, T.-S. Enhanced Expression of ABCB1 and Nrf2 in CD133-Positive Cancer Stem Cells Associates with Doxorubicin Resistance. Stem Cells Int. 2020, 2020, 8868849. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.S.M.; Parag, R.R.; Rashid, M.I.; Rahman, M.Z.; Chowdhury, A.A.; Sultana, A.; Jerin, C.; Siddiqua, A.; Rahman, L.; Shirin, A.; et al. Widespread expression of Sonic hedgehog (Shh) and Nrf2 in patients treated with cisplatin predicts outcome in resected tumors and are potential therapeutic targets for HPV-negative head and neck cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920911229. [Google Scholar] [CrossRef]
- Suzuki, S.; Yamamoto, M.; Sanomachi, T.; Togashi, K.; Sugai, A.; Seino, S.; Yoshioka, T.; Okada, M.; Kitanaka, C. Dexamethasone Sensitizes Cancer Stem Cells to Gemcitabine and 5-Fluorouracil by Increasing Reactive Oxygen Species Production through NRF2 Reduction. Life 2021, 11, 885. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Oh, J.; Kim, J.-S. Suppression of Nrf2 Activity by Chestnut Leaf Extract Increases Chemosensitivity of Breast Cancer Stem Cells to Paclitaxel. Nutrients 2017, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.-C.; Li, J.; Yu, W.-F.; Zhang, G.-Z.; Wang, H.-M.; Ma, H.-M. Elevated expression of Nrf2 mediates multidrug resistance in CD133(+) head and neck squamous cell carcinoma stem cells. Oncol. Lett. 2016, 12, 4333–4338. [Google Scholar] [CrossRef][Green Version]
- Li, D.; Hong, X.; Zhao, F.; Ci, X.; Zhang, S. Targeting Nrf2 may reverse the drug resistance in ovarian cancer. Cancer Cell Int. 2021, 21, 116. [Google Scholar] [CrossRef]
- Achuthan, S.; Santhoshkumar, T.R.; Prabhakar, J.; Nair, S.A.; Pillai, M.R. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J. Biol. Chem. 2011, 286, 37813–37829. [Google Scholar] [CrossRef]
- Qin, S.; He, X.; Lin, H.; Schulte, B.A.; Zhao, M.; Tew, K.D.; Wang, G.Y. Nrf2 inhibition sensitizes breast cancer stem cells to ionizing radiation via suppressing DNA repair. Free Radic. Biol. Med. 2021, 169, 238–247. [Google Scholar] [CrossRef]
- Godoy, P.R.D.V.; Pour Khavari, A.; Rizzo, M.; Sakamoto-Hojo, E.T.; Haghdoost, S. Targeting NRF2, Regulator of Antioxidant System, to Sensitize Glioblastoma Neurosphere Cells to Radiation-Induced Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 2534643. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, M.; Poljak-Blazi, M.; Zarkovic, K.; Mihaljevic, D.; Schaur, R.J.; Zarkovic, N. Oxidative burst of neutrophils against melanoma B16-F10. Cancer Lett. 2007, 246, 100–108. [Google Scholar] [CrossRef]
- Jaganjac, M.; Poljak-Blazi, M.; Zarkovic, K.; Schaur, R.J.; Zarkovic, N. The involvement of granulocytes in spontaneous regression of Walker 256 carcinoma. Cancer Lett. 2008, 260, 180–186. [Google Scholar] [CrossRef]
- Jaganjac, M.; Poljak-Blazi, M.; Kirac, I.; Borovic, S.; Joerg Schaur, R.; Zarkovic, N. Granulocytes as effective anticancer agent in experimental solid tumor models. Immunobiology 2010, 215, 1015–1020. [Google Scholar] [CrossRef]
- Zivkovic, M.; Poljak-Blazi, M.; Egger, G.; Sunjic, S.B.; Schaur, R.J.; Zarkovic, N. Oxidative burst and anticancer activities of rat neutrophils. BioFactors 2005, 24, 305–312. [Google Scholar] [CrossRef]
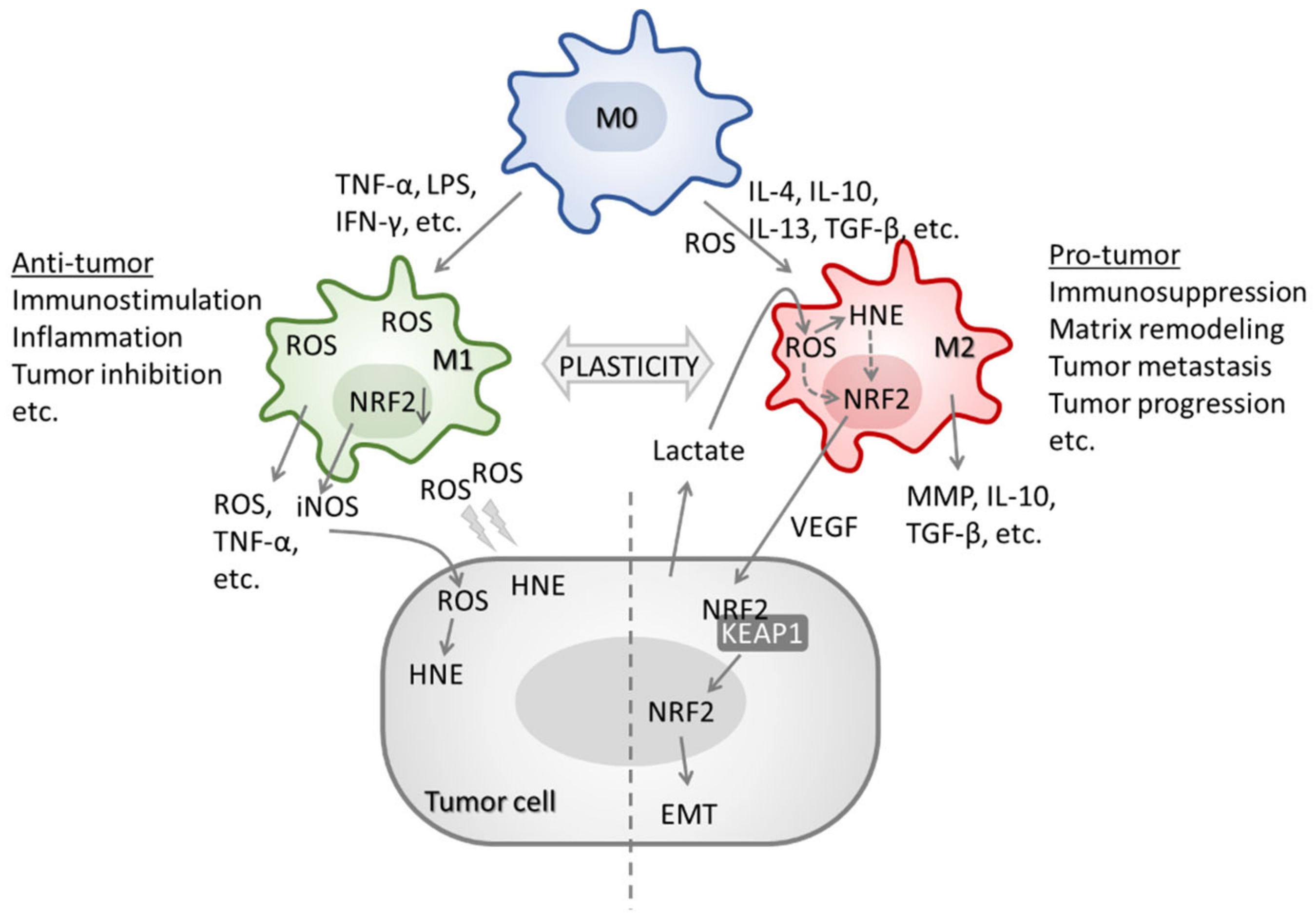
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef]
- Kim, J.; Bae, J.-S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediators Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.-F.; Gereke, M.; von Köckritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Griess, B.; Mir, S.; Datta, K.; Teoh-Fitzgerald, M. Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radic. Biol. Med. 2020, 147, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Ohms, M.; Möller, S.; Laskay, T. An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in vitro. Front. Immunol. 2020, 11, 532. [Google Scholar] [CrossRef]
- Jaganjac, M.; Matijevic Glavan, T.; Zarkovic, N. The Role of Acrolein and NADPH Oxidase in the Granulocyte-Mediated Growth-Inhibition of Tumor Cells. Cells 2019, 8, 292. [Google Scholar] [CrossRef]
- Jaganjac, M.; Cipak, A.; Schaur, R.J.; Zarkovic, N. Pathophysiology of neutrophil-mediated extracellular redox reactions. Front. Biosci. Landmark Ed. 2016, 21, 839–855. [Google Scholar] [CrossRef]
- Al-Menhali, A.S.; Anderson, C.; Gourine, A.V.; Abramov, A.Y.; D’Souza, A.; Jaganjac, M. Proteomic Analysis of Cardiac Adaptation to Exercise by High Resolution Mass Spectrometry. Front. Mol. Biosci. 2021, 8, 723858. [Google Scholar] [CrossRef]
- Al-Menhali, A.S.; Banu, S.; Angelova, P.R.; Barcaru, A.; Horvatovich, P.; Abramov, A.Y.; Jaganjac, M. Lipid peroxidation is involved in calcium dependent upregulation of mitochondrial metabolism in skeletal muscle. Biochim. Biophys. Acta. Gen. Subj. 2020, 1864, 129487. [Google Scholar] [CrossRef]
- Elrayess, M.A.; Almuraikhy, S.; Kafienah, W.; Al-Menhali, A.; Al-Khelaifi, F.; Bashah, M.; Zarkovic, K.; Zarkovic, N.; Waeg, G.; Alsayrafi, M.; et al. 4-hydroxynonenal causes impairment of human subcutaneous adipogenesis and induction of adipocyte insulin resistance. Free Radic. Biol. Med. 2017, 104, 129–137. [Google Scholar] [CrossRef]
- Jaganjac, M.; Milkovic, L.; Gegotek, A.; Cindric, M.; Zarkovic, K.; Skrzydlewska, E.; Zarkovic, N. The relevance of pathophysiological alterations in redox signaling of 4-hydroxynonenal for pharmacological therapies of major stress-associated diseases. Free Radic. Biol. Med. 2020, 157, 128–153. [Google Scholar] [CrossRef]
- Zarkovic, N.; Cipak, A.; Jaganjac, M.; Borovic, S.; Zarkovic, K. Pathophysiological relevance of aldehydic protein modifications. J. Proteomics 2013, 92, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhuang, H.; Lee, P.Y.; Li, M.; Yang, L.; Nigrovic, P.A.; Reeves, W.H. NF-E2-Related Factor 2 Regulates Interferon Receptor Expression and Alters Macrophage Polarization in Lupus. Arthritis Rheumatol. 2020, 72, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Al Haq, A.T.; Tseng, H.-Y.; Chen, L.-M.; Wang, C.-C.; Hsu, H.-L. Targeting prooxidant MnSOD effect inhibits triple-negative breast cancer (TNBC) progression and M2 macrophage functions under the oncogenic stress. Cell Death Dis. 2022, 13, 49. [Google Scholar] [CrossRef]
- El Hadri, K.; Mahmood, D.F.D.; Couchie, D.; Jguirim-Souissi, I.; Genze, F.; Diderot, V.; Syrovets, T.; Lunov, O.; Simmet, T.; Rouis, M. Thioredoxin-1 promotes anti-inflammatory macrophages of the M2 phenotype and antagonizes atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1445–1452. [Google Scholar] [CrossRef]
- Kwon, D.H.; Lee, H.; Park, C.; Hong, S.-H.; Hong, S.H.; Kim, G.-Y.; Cha, H.-J.; Kim, S.; Kim, H.-S.; Hwang, H.-J.; et al. Glutathione Induced Immune-Stimulatory Activity by Promoting M1-Like Macrophages Polarization via Potential ROS Scavenging Capacity. Antioxidants 2019, 8, 413. [Google Scholar] [CrossRef]
- Kim, S.H.; Saeidi, S.; Zhong, X.; Gwak, S.-Y.; Muna, I.A.; Park, S.-A.; Kim, S.H.; Na, H.-K.; Joe, Y.; Chung, H.T.; et al. Breast cancer cell debris diminishes therapeutic efficacy through heme oxygenase-1-mediated inactivation of M1-like tumor-associated macrophages. Neoplasia 2020, 22, 606–616. [Google Scholar] [CrossRef]
- Chiaradia, E.; Tancini, B.; Emiliani, C.; Delo, F.; Pellegrino, R.M.; Tognoloni, A.; Urbanelli, L.; Buratta, S. Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles. Cells 2021, 10, 1763. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, S.; Zhang, X.; Liu, F.; Yang, K.; Du, G.; Rui, X. Dasatinib protects against acute respiratory distress syndrome via Nrf2-regulated M2 macrophages polarization. Drug Dev. Res. 2021, 8, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, L.-P.; Qin, W.-T.; Yang, Q.; Chen, D.-Y.; Li, Q.; Zhou, K.-X.; Huang, P.-Q.; Xu, C.-J.; Li, J.; et al. Identification of a subset of immunosuppressive P2RX1-negative neutrophils in pancreatic cancer liver metastasis. Nat. Commun. 2021, 12, 174. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, J.A. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 2009, 11, e17. [Google Scholar] [CrossRef]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef]
- O’Cathail, S.M.; Wu, C.-H.; Thomas, R.; Hawkins, M.A.; Maughan, T.S.; Lewis, A. NRF2 Mediates Therapeutic Resistance to Chemoradiation in Colorectal Cancer through a Metabolic Switch. Antioxidants 2021, 10, 1380. [Google Scholar] [CrossRef]
- Payandeh, Z.; Tazehkand, A.P.; Mansoori, B.; Khaze, V.; Asadi, M.; Baradaran, B.; Samadi, N. The Impact of Nrf2 Silencing on Nrf2-PD-L1 Axis to Overcome Oxaliplatin Resistance and Migration in Colon Cancer Cells. Avicenna J. Med. Biotechnol. 2021, 13, 122. [Google Scholar] [CrossRef]
- Sadeghi, M.R.; Jeddi, F.; Soozangar, N.; Somi, M.H.; Shirmohamadi, M.; Khaze, V.; Samadi, N. Nrf2/P–glycoprotein axis is associated with clinicopathological characteristics in colorectal cancer. Biomed. Pharmacother. 2018, 104, 458–464. [Google Scholar] [CrossRef]
- Duong, H.-Q.; Yi, Y.W.; Kang, H.J.; Hong, Y.B.; Tang, W.; Wang, A.; Seong, Y.-S.; Bae, I. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2014, 44, 959–969. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Gocho, T.; Aguilar, M.; Wu, M.; Zhuang, Z.-N.; Fu, J.; Yanaga, K.; Huang, P.; Chiao, P.J. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol. Cancer Ther. 2015, 14, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, P.S.; Devegowda, D.; Singh, A.; Thimmulappa, R.K. NRF2, p53, and p16: Predictive biomarkers to stratify human papillomavirus associated head and neck cancer patients for de-escalation of cancer therapy. Crit. Rev. Oncol. Hematol. 2020, 148, 102885. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Bodas, M.; Wakabayashi, N.; Bunz, F.; Biswal, S. Gain of Nrf2 Function in Non-Small-Cell Lung Cancer Cells Confers Radioresistance. Antioxid. Redox Signal. 2010, 13, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Cescon, D.W.; She, D.; Sakashita, S.; Zhu, C.Q.; Pintilie, M.; Shepherd, F.A.; Tsao, M.S. NRF2 pathway activation and adjuvant chemotherapy benefit in lung squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence inKRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef]
- Saigusa, D.; Motoike, I.N.; Saito, S.; Zorzi, M.; Aoki, Y.; Kitamura, H.; Suzuki, M.; Katsuoka, F.; Ishii, H.; Kinoshita, K.; et al. Impacts of NRF2 activation in non-small-cell lung cancer cell lines on extracellular metabolites. Cancer Sci. 2020, 111, 667–678. [Google Scholar] [CrossRef]
- Hamada, S.; Matsumoto, R.; Tanaka, Y.; Taguchi, K.; Yamamoto, M.; Masamune, A. Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition. Int. J. Mol. Sci. 2021, 22, 870. [Google Scholar] [CrossRef]
- Chuang, H.-Y.; Hsu, L.-Y.; Pan, C.-M.; Pikatan, N.W.; Yadav, V.K.; Fong, I.-H.; Chen, C.-H.; Yeh, C.-T.; Chiu, S.-C. The E3 Ubiquitin Ligase NEDD4-1 Mediates Temozolomide-Resistant Glioblastoma through PTEN Attenuation and Redox Imbalance in Nrf2–HO-1 Axis. Int. J. Mol. Sci. 2021, 22, 10247. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| NRF2 Transcript Variants (TVs) | Exons Included | Exon 1A | Exon 1B | Exon 2 | Exon 3 | Exon 4 | Exon 5 |
|---|---|---|---|---|---|---|---|
| 1 NM_006164.5 | 1..196, 30457..3072, 31389..31478, 32145..32336, 32720..34425 | 196 | 267 | 90 | 192 | 1705 | |
| 2 NM_001145412.3 | 597..1325, 30457..30723, 31389..31478, 32145..32336, 32720..34425 | 729 | 267 | 90 | 192 | 1705 | |
| 3 NM_001145413.3 | 597..1325, 30457..30723, 31389..31478, 32166..32336, 32720..34425 | 729 | 267 | 90 | 192 | 1705 | |
| 4 NM_001313900.1 | 597..1199, 30457..30723, 31389..31478, 32145..32336, 32720..34425 | 603 | 267 | 90 | 192 | 1705 | |
| 5 NM_001313901.1 | 597..1291, 30457..30723, 31389..31478, 32145..32336, 32720..34425 | 695 | 267 | 90 | 192 | 1705 | |
| 6 NM_001313902.1 | 1..196, 30457..30723, 32145..32336, 32720..34425 | 196 | 267 | 192 | 1705 | ||
| 7 NM_001313903.1 | 1..196, 30457..30504, 31389..31478, 32145..32336, 32720..34425 | 196 | 267 | 90 | 192 | 1705 | |
| 8 NM_001313904.1 | 597..1325, 30457..30652, 31389..31478, 32145..32336, 32720..34425 | 729 | 267 | 90 | 192 | 1705 |
| Hsa-miR | Locus | Mature miRNA Sequence | Experimental Seed Sequence | In Silico Seed Sequence | Beginning of the Seed, In Silico |
|---|---|---|---|---|---|
| NRF2: NC_000002 REGION: complement (177230303..177265131) | |||||
| 507 | Xq27.3 | UUUUGCACCUUUUGGAGUGAA | not specified | TGCAAAA | 34449 and 34686 (two binding sites) |
| 634 | 17q24.2 | AACCAGCACCCCAACUUUGGAC | not specified | GCTGGTA | 34791 |
| 450a-5p | Xq26.3 | UUUUGCGAUGUGUUCCUAAUAU | not specified | No binding according to in silico analysis | N/A |
| 129–5p | 7q32.1 | CUUUUUGCGGUCUGGGCUUGC | not specified | GCAAAAAA | 34730 and 34769 (two binding sites) |
| 144–3p | 17q11.2 | UACAGUAUAGAUGAUGUACU | AUACUGUA | ATACTGTA | 34613 and 34718 (two binding sites) |
| 153–3p | 2q35 | UUGCAUAGUCACAAAAGUGAUC | CUAUGCAA | CTATGCAA | 34446 |
| 27a-3p | 19p13.12 | UUCACAGUGGCUAAGUUCCGC | ACUGUGA | ACTGTGA | 34410 |
| 142–5p | 17q22 | CAUAAAGUAGAAAGCACUACU | ACUUUAUA | ACTTTATA | 34431 |
| 28–5p | 3q28 | AAGGAGCUCACAGUCUAUUGAG | AGCUCCUA | AGCTCCTA | 34403 |
| KEAP1: NC_000019 REGION: complement (10486125..10503356) | |||||
| 200a-3p | 1p36.33 | UAACACUGUCUGGUAACGAUGU | CAGUGUUA | CAGTGTTA | 16838 |
| 141–3p | 12p13.31 | UAACACUGUCUGGUAAAGAUGG | CAGUGUUA | CAGTGTTA | 16838 |
| 7–5p | 9q21.32 | UGGAAGACUAGUGAUUUUGUUGUU | UGGAAGA | No binding according to in silico analysis | N/A |
| 432–5p | 14q32.2 | UCUUGGAGUAGGUCAUUGGGUGG | UGGAUGG (exon 2) | TCCAAGA (3′UTR) | 16897 |
| Cancer Cell Type | NRF2 Role | Reference |
|---|---|---|
| Breast cancer | Regulation of ALDH and contribution to radioresistance | [133] |
| Ovarian cancer | Regulation of CSC markers, chemoresistance, colony/sphere formation, and tumor growth | [134] |
| Hepatocellular carcinoma | Promotion of cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant cells | [135] |
| Glioma | Induction of stem markers | [132] |
| Breast cancer | Antioxidant response | [137] |
| Breast cancer | Antioxidant response | [138] |
| Colorectal cancer | Proliferation of differentiated spheroids | [139] |
| Breast cancer | Self-renewal of breast cancer stem-like cells | [140] |
| Hepatocellular carcinoma | CSCs enrichment | [141] |
| Breast cancer | Drug resistance acquisition | [152] |
| Head and neck cancer | Chemoresistance | [154] |
| Head and neck cancer | Multidrug resistance | [157] |
| Ovarian cancer | Drug resistance | [158] |
| Breast cancer | Resistance to radiation therapy | [160] |
| Glioblastoma | Resistance to radiation therapy | [161] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gall Trošelj, K.; Tomljanović, M.; Jaganjac, M.; Matijević Glavan, T.; Čipak Gašparović, A.; Milković, L.; Borović Šunjić, S.; Buttari, B.; Profumo, E.; Saha, S.; et al. Oxidative Stress and Cancer Heterogeneity Orchestrate NRF2 Roles Relevant for Therapy Response. Molecules 2022, 27, 1468. https://doi.org/10.3390/molecules27051468
Gall Trošelj K, Tomljanović M, Jaganjac M, Matijević Glavan T, Čipak Gašparović A, Milković L, Borović Šunjić S, Buttari B, Profumo E, Saha S, et al. Oxidative Stress and Cancer Heterogeneity Orchestrate NRF2 Roles Relevant for Therapy Response. Molecules. 2022; 27(5):1468. https://doi.org/10.3390/molecules27051468
Chicago/Turabian StyleGall Trošelj, Koraljka, Marko Tomljanović, Morana Jaganjac, Tanja Matijević Glavan, Ana Čipak Gašparović, Lidija Milković, Suzana Borović Šunjić, Brigitta Buttari, Elisabetta Profumo, Sarmistha Saha, and et al. 2022. "Oxidative Stress and Cancer Heterogeneity Orchestrate NRF2 Roles Relevant for Therapy Response" Molecules 27, no. 5: 1468. https://doi.org/10.3390/molecules27051468
APA StyleGall Trošelj, K., Tomljanović, M., Jaganjac, M., Matijević Glavan, T., Čipak Gašparović, A., Milković, L., Borović Šunjić, S., Buttari, B., Profumo, E., Saha, S., Saso, L., & Žarković, N. (2022). Oxidative Stress and Cancer Heterogeneity Orchestrate NRF2 Roles Relevant for Therapy Response. Molecules, 27(5), 1468. https://doi.org/10.3390/molecules27051468