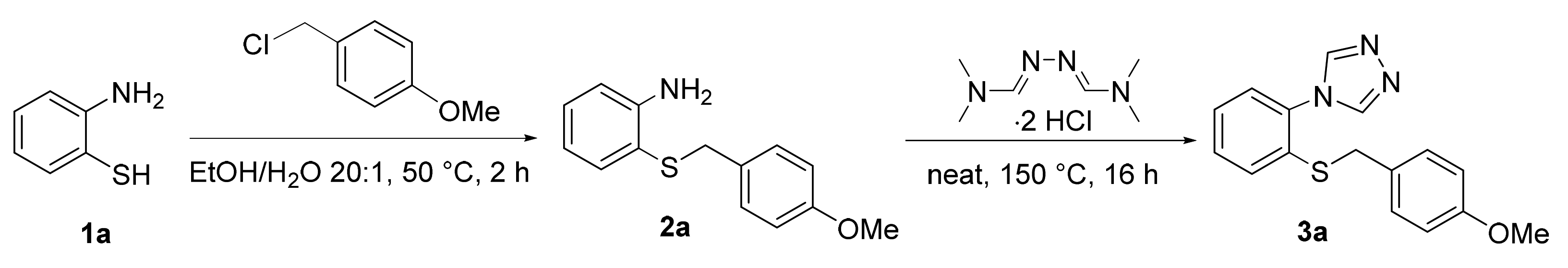
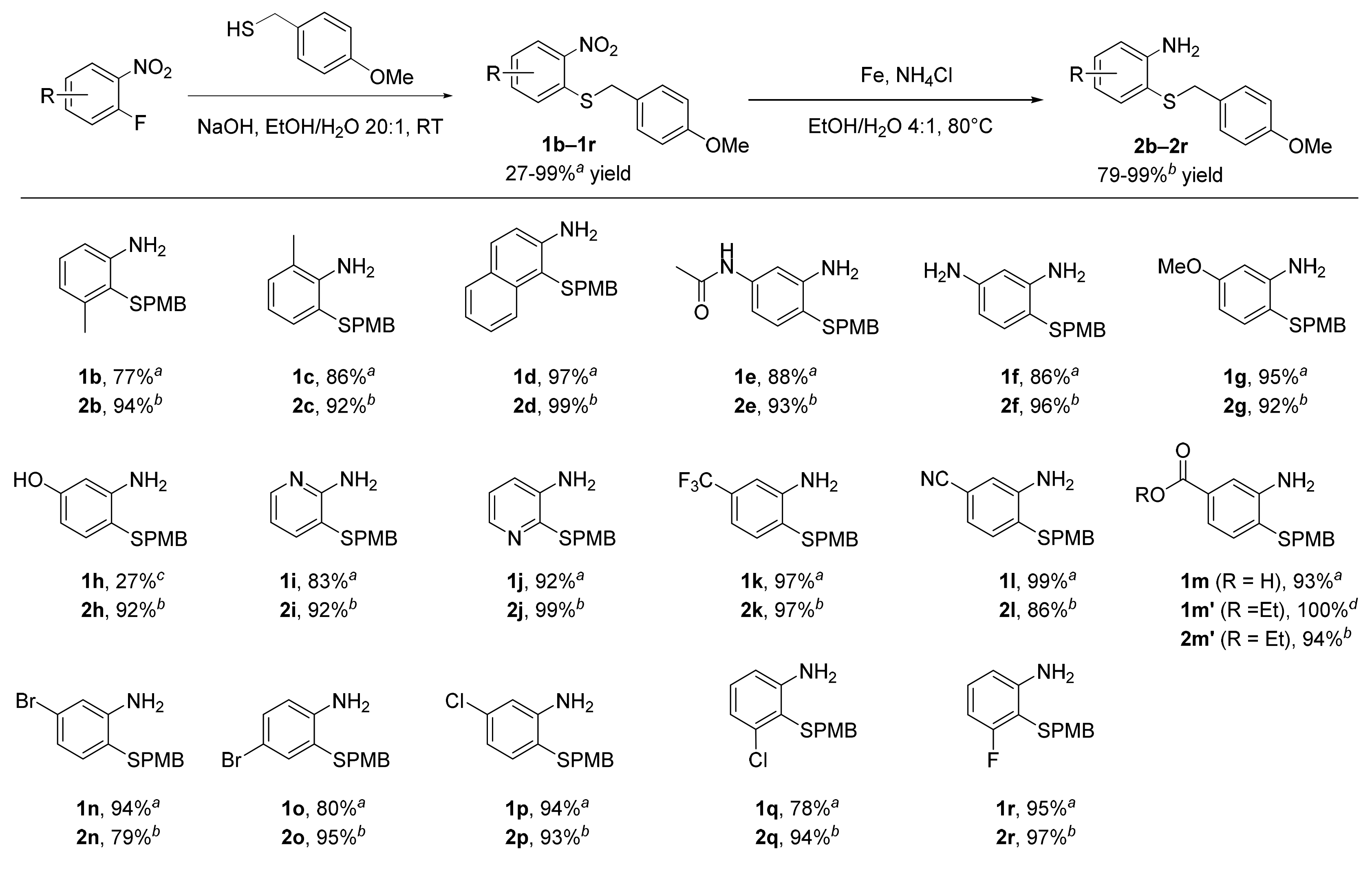
3.4. General Procedure 1 for the Synthesis of (4-Methoxybenzyl)(2-Nitrophenyl)sulfanes (1b–1r)
The following description is for a 30 mmol scale reaction. The solvent quantities and flask size were adjusted accordingly for smaller-scale reactions.
A 500 mL round-bottomed flask equipped with a stir bar was loaded with the 1-fluoro-2-nitrobenzene derivative (1 equiv.) and 200 mL of ethanol and placed under an atmosphere of argon. (4-methoxyphenyl)methanethiol (1 equiv.) was added with a syringe, followed by a dropwise addition of NaOH (1 equiv.) dissolved in 10 mL of H2O. The reaction mixture was stirred at room temperature until TLC indicated the completion of the reaction (typically within 2 h). After removing the solvent under reduced pressure, the residue was diluted with 150 mL of H2O and extracted twice with dichloromethane. The organic phases were combined, dried over MgSO4, filtered, and concentrated. The resulting crude product was purified by recrystallization or column chromatography as described below.
3.4.1. (4-Methoxybenzyl)(2-Methyl-6-Nitrophenyl)sulfane (1b)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from diethyl ether/hexanes (1:1) afforded the product as an off-white powder in 77% (6.701 g, 23.18 mmol) yield; m.p. 63–64 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.62 (dd, J = 7.4 Hz, J = 1.0 Hz, 1H), 7.55 (dd, J = 7.6 Hz, J = 0.8 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.02 (dt, J = 8.6 Hz, J = 2.9 Hz, 2H), 6.80 (dt, J = 8.4 Hz, J = 3.0 Hz, 1H), 3.95 (s, 2H), 3.71 (s, 3H), and 2.39 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.6, 156.2, 145.6, 133.0, 130.0, 130.0, 128.8, 124.2, 120.5, 113.8, 55.0, and 20.6.
3.4.2. (4-Methoxybenzyl)(3-Methyl-2-Nitrophenyl)sulfane (1c)
The title compound was prepared according to general procedure 1 on a 5 mmol scale. Recrystallization from diethyl ether afforded the product as a bright yellow powder in 86% (1.238 g, 4.28 mmol) yield; m.p. 90–92 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.52 (d, J = 7.5 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 7.22 (dt, J = 8.6 Hz, J = 2.8 Hz, 2H), 6.85 (dt, J = 8.7 Hz, J = 2.9 Hz, 2H), 4.23 (s, 2H), 3.71 (s, 3H), and 3.32 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.5, 151.0, 130.7, 130.1 (2 signals), 129.4, 128.8, 128.5, 128.0, 113.9, 55.0, 37.0, and 17.0.
3.4.3. (4-Methoxybenzyl)(2-Nitronaphthalen-1-yl)sulfane (1d)
The title compound was prepared according to general procedure 1 on a 5.21 mmol scale. Recrystallization from acetone/hexanes (1:4) afforded the product as a yellow powder in 97% (1.636 g, 5.03 mmol) yield; m.p. 68–70 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.57 (d, J = 8.2 Hz, 1H), 8.21 (d, J = 8.8 Hz, 1H), 8.13 (dd, J = 7.8 Hz, J = 1.4 Hz, 1H), 7.85 (d, J = 8.8 Hz, 1H), 7.81–7.73 (m, 2H), 6.92 (dt, J = 8.6 Hz, J = 2.6 Hz, 2H), 6.72 (dt, J = 8.6 Hz, J = 2.6 Hz, 2H), 4.07 (s, 2H), and 3.67 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.6, 153.7, 133.8, 133.6, 131.5, 129.9, 129.0, 128.5, 128.5, 127.3, 123.8, 119.4, 113.8, 55.0, and 40.5.
3.4.4. N-(4-((4-Ethoxybenzyl)thio)-3-Nitrophenyl)acetamide (1e)
The title compound was prepared according to general procedure 1 from
N-(4-fluoro-3-nitrophenyl)acetamide (see
Section 3.3.1) on a 30 mmol scale. Extraction was carried out with ethyl acetate (instead of dichloromethane) and afforded the pure product as a bright yellow powder in 88% (6.150 g, 19.04 mmol) yield; m.p. 140–141 °C.
1H NMR (400 MHz, DMSO-
d6, 298 K): δ = 10.35 (s, 1H), 8.55 (d,
J = 2.2 Hz, 1H), 7.80 (dd,
J = 8.8 Hz,
J = 2.3 Hz, 1H), 7.67 (d,
J = 8.8 Hz, 1H), 7.33 (d,
J = 8.6 Hz, 2H), 6.89 (d,
J = 8.6 Hz, 2H), 4.44 (s, 2H), 3.73 (s, 3H), and 2.07 (s, 3H);
13C{
1H} NMR (100 MHz, DMSO-
d6, 298 K): δ = 168.9, 158.6, 145.4, 136.8, 130.4, 129.6, 128.5, 127.4, 124.4, 114.9, 114.0, 55.0, 35.7, and 24.0.
3.4.5. (2,4-. Dinitrophenyl)(4-Methoxybenzyl)sulfane (1f)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from diethyl ether afforded the product as a light brown powder in 86% (8.718 g, 25.84 mmol) yield; m.p. 113–114 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.86 (d, J = 2.6 Hz, 2H), 8.44 (dt, J = 9.0 Hz, J = 2.6 Hz, 1H), 7.96 (d, J = 9.1 Hz, 1H), 7.40 (dt, J = 8.7 Hz, J = 2.9 Hz, 2H), 6.92 (dt, J = 8.7 Hz, J = 3.0 Hz, 1H), 4.44 (s, 2H), and 3.74 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.9, 145.7, 144.0, 143.6, 130.6, 128.4, 127.4, 126.1, 121.2, 114.2, 55.1, and 35.8.
3.4.6. (4-. Methoxy-2-Nitrophenyl)(4-Methoxybenzyl)sulfane (1g)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from acetone/hexanes (1:4) afforded the product as a bright orange powder in 95% (8.718 g, 28.55 mmol) yield; m.p. 116–118 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.65 (d, J = 2.8 Hz, 1H), 7.62 (d, J = 9.0 Hz, 1H), 7.33–7.28 (m, 3 H), 6.87 (dt, J = 8.6 Hz, J = 2.8 Hz, 2H), 4.22 (s, 2 H), 3.83 (s, 3H), and 3.72 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.6, 157.0, 147.1, 130.3, 130.0, 127.6, 126.1, 121.2, 113.9, 109.5, 56.0, 55.0, and 36.2.
3.4.7. 4-((4-Methoxybenzyl)thio)-3-Nitrophenol (1h)
The title compound was prepared according to general procedure 1 from tert-butyl(4-fluoro-3-nitrophenoxy)dimethylsilane (see 3.3.2) on a 26.5 mmol scale. Purification by column chromatography (silica gel, 1. diethyl ether/hexanes (1:3), and 2. diethyl ether/hexanes (1:2), Rf = 0.21) afforded the product as a bright yellow powder in 27% (8.718 g, 7.06 mmol) yield; m.p. 115–116 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 10.35 (s, 1H), 7.53 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 2.7 Hz, 1H), 7.28 (d, J = 8.6 Hz, 2H), 7.13 (dd, J = 8.8 Hz, J = 2.7 Hz, 1H), 8.87 (d, J = 8.6 Hz, 2H), 4.18 (s, 2H), and 3.73 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.5, 155.5, 147.3, 130.4, 130.3, 127.8, 123.8, 122.0, 113.9, 111.2, 55.0, and 36.4.
3.4.8. 3-((4-Methoxybenzyl)thio)-2-Nitropyridine (1i)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from acetone/hexanes (1:4) afforded the product as a bright powder in 83% (6.889 g, 24.75 mmol) yield; m.p. 140–142 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.39 (d, J = 3.6 Hz, 1H), 8.30 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 4.4 Hz, 1H), 7.34 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 4.34 (s, 2H), and 3.72 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.7, 153.5, 144.4, 138.8, 131.3, 130.4, 128.7, 126.9, 114.1, 55.1, and 35.4.
3.4.9. 2-((4-Methoxybenzyl)thio)-3-Nitropyridine (1j)
The title compound was prepared according to general procedure 1 on a 27.9 mmol scale. Trituration with hexanes afforded the product as a yellow powder in 92% (7.629 g, 27.61 mmol) yield; m.p. 88–90 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.86 (dd, J = 4.6 Hz, J = 1.5 Hz, 2H), 8.60 (dd, J = 8.3 Hz, J = 1.5 Hz, 1H), 7.46 (dd, J = 8.3 Hz, J = 4.6 Hz, 1H), 7.35 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 4.42 (s, 2H), and 3.72 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.5, 156.0, 153.8, 141.4, 134.4, 130.5, 128.4, 120.0, 113.9, 55.0, and 33.9.
3.4.10. (4-. Methoxybenzyl)(2-Nitro-4-(Trifluoromethyl)phenyl)sulfane (1k)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Trituration with hexanes afforded the product as a bright yellow powder in 97% (9.967 g, 29.03 mmol) yield; m.p. 111–112 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.45 (d, J = 1.1 Hz, 1H), 8.04 (dd, J = 8.6 Hz, J = 1.7 Hz, 1H), 7.94 (d, J = 8.6 Hz, 1H), 7.38 (d, J = 8.6 Hz, 2H), 6.91 (d, J = 8.6 Hz, 2H), 4.39 (s, 2H), and 3.74 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 159.3, 145.2, 142.9, 131.0, 130.4 (q, JC–F = 3.3 Hz), 129.2, 126.9, 125.7 (q, JC–F = 33.6 Hz), 123.6 (q, JC–F = 270.4 Hz), 123.4 (q, JC–F = 4.0 Hz), 114.6, 55.5, and 36.1; 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = −61.3.
3.4.11. 4-((4-Methoxybenzyl)thio)-3-Nitrobenzonitrile (1l)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from diethyl ether afforded the product as a bright yellow powder in 99% (8.934 g, 29.74 mmol) yield; m.p. 202–204 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.70 (d, J = 1.8 Hz, 2H), 8.12 (dt, J = 8.5 Hz, J = 1.8 Hz, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.38 (dt, J = 8.6 Hz, J = 2.9 Hz, 2H), 6.92 (dt, J = 8.7 Hz, J = 2.9 Hz, 2H), 4.40 (s, 2H), and 3.74 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.8, 144.7, 143.4, 136.2, 130.6, 130.0, 128.4, 126.3, 117.1, 114.2, 107.4, 55.1, and 35.6.
3.4.12. 4-((4-Methoxybenzyl)thio)-3-Nitrobenzoic acid (1m)
The title compound was prepared according to procedure 1 on an 85 mmol scale. Recrystallization from acetone/hexanes (1:4) afforded the product as a bright yellow powder in 93% (25.374 g, 79.46 mmol) yield., m.p. 269–271 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 13.55 (s, 1H), 8.61 (d, J = 1.9 Hz, 1H), 8.15 (dd, J = 8.5 Hz, J = 1.9 Hz, 1H), 7.86 (d, J = 8.6 Hz, 1H), 7.39 (d, J = 8.6 Hz, 2H), 6.92 (d, J = 8.7 Hz, 2H), 4.37 (s, 2H), and 3.74 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 165.4, 158.8, 144.6, 142.5, 133.7, 130.6, 127.8, 127.5, 126.6, 126.4, 114.1, 55.1, and 35.6.
3.4.13. Ethyl 4-((4-Methoxybenzyl)thio)-3-Nitrobenzoate (1m′)
A 250 mL round-bottomed flask was charged with compound 1m (23 mmol, 1 equiv.), oxalyl chloride (3 equiv.), and dichloromethane (50 mL). After adding DMF (3 drops), the reaction mixture was stirred for 1 h at 0 °C in an ice bath. EtOH (1.3 equiv.), triethylamine (1.3 equiv), and dichloromethane (50 mL) were added at 0 °C, the ice bath was removed, and the reaction flask was fitted with a reflux condenser. The reaction mixture was stirred at 40 °C for 2 h, then cooled to room temperature. After washing the dichloromethane solution with water, the organic phase was dried over MgSO4, filtered, and evaporated. Recrystallization of the crude product from diethyl ether/hexanes (1:2) afforded the product as a bright yellow solid in 99.99% (7.989 g, 22.998 mmol) yield; m.p. 149–150 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.69 (d, J = 1.9 Hz, 1H), 8.16 (dd, J = 8.5 Hz, J = 1.9 Hz, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.39 (d, J = 8.6 Hz, 2H), 6.92 (d, J = 8.7 Hz, 2H), 4.39–4.33 (m, 4H), 3.74 (s, 3H), and 1.34 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 164.1, 158.9, 144.7, 143.2, 133.6, 130.8, 128.0, 126.6, 126.5, 126.4, 114.3, 61.7, 55.2, 35.8, and 14.2.
3.4.14. (4-. Bromo-2-Nitrophenyl)(4-Methoxybenzyl)sulfane (1n)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from diethyl ether/hexanes (1:2) afforded the product as a bright yellow powder in 94% (9.968 g, 28.14mmol) yield; m.p. 121–124 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.33 (d, J = 2.2 Hz, 1H), 7.89 (dd, J = 8.7 Hz, J = 2.2 Hz, 1H), 7.67 (d, J = 8.8 Hz, 1H), 7.34 (dt, J = 8.7 Hz, J = 2.8 Hz, 2H), 6.90 (dt, J = 8.7 Hz, J = 3.0 Hz, 2H), 4.31 (s, 2H), and 3.73 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.7, 145.9, 136.6, 136.0, 130.4, 129.6, 128.0, 126.8, 117.0, 114.1, 55.1, and 35.6.
3.4.15. (5-. Bromo-2-nitrophenyl)(4-methoxybenzyl)sulfane (1o)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from diethyl ether/hexanes (1:5) afforded the product as a bright yellow powder in 80% (8.501 g, 24.00 mmol) yield; m.p. 139–140 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.10 (d, J = 8.8 Hz, 1H), 7.85 (d, J = 2.0 Hz, 1H), 7.57 (dd, J = 8.8 Hz, J = 2.0 Hz, 1H), 7.35 (dt, J = 8.7 Hz, J = 2.8 Hz, 2H), 7.91 (dt, J = 8.7 Hz, J = 3.0 Hz, 2H), 4.36 (s, 2H), and 3.74 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.7, 144.1, 139.2, 130.5, 129.6, 128.7, 128.1, 127.5, 126.7, 114.1, 55.1, and 35.6.
3.4.16. (4-. Chloro-2-Nitrophenyl)(4-Methoxybenzyl)sulfane (1p)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from diethyl ether/hexanes (1:2) afforded the product as an orange powder in 94% (8.258 g, 28.23 mmol) yield; m.p. 113–114 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.23 (d, J = 2.2 Hz, 1H), 7.78 (dd, J = 8.7 Hz, J = 2.2 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.35 (dt, J = 8.6 Hz, J = 3.0 Hz, 2H), 6.90 (dt, J = 8.7 Hz, J = 3.0 Hz, 2H), 4.31 (s, 2H), and 3.73 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.7, 145.8, 135.6, 133.8, 130.4, 129.4, 126.9, 125.3, 114.1, 55.1, and 35.6.
3.4.17. (2-. Chloro-6-Nitrophenyl)(4-Methoxybenzyl)sulfane (1q)
The title compound was prepared according to general procedure 1 on a 30 mmol scale. Recrystallization from diethyl ether/hexanes (1:1) afforded the product as a bright yellow powder in 78% (7.290 g, 23.53 mmol) yield; m.p. 86–88 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.84 (dd, J = 8.1 Hz, J = 1.2 Hz, 1H), 7.76 (dd, J = 8.0 Hz, J = 1.1 Hz, 1H), 7.58 (t, J = 8.1 Hz, 1H), 7.02 (dt, J = 8.6 Hz, J = 2.7 Hz, 2H), 6.78 (dt, J = 8.6 Hz, J = 2.6 Hz, 2H), 4.09 (s, 2H), and 3.69 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.6, 156.1, 140.5, 132.9, 131.4, 130.0, 128.1, 125.0, 121.9, 113.8, 55.0, and 38.6.
3.4.18. (2-. Fluoro-6-Nitrophenyl)(4-Methoxybenzyl)sulfane (1r)
The title compound was prepared according to general procedure 1 on a 15 mmol scale. Recrystallization from acetone/hexanes (1:4) afforded the product as a bright yellow powder in 95% (4.167 g, 14.20 mmol) yield; m.p. 105–107 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.74–7.71 (m, 1H), 7.63–7.56 (m, 2H), 7.08 (dt, J = 8.6 Hz, J = 2.0 Hz, 2H), 6.78 (dt, J = 8.7 Hz, J = 2.0 Hz, 2H), 4.14 (s, 2H), and 3.69 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 162.0 (d, JC–F = 246.7 Hz), 158.6, 153.5 (d, JC–F = 3.0 Hz), 130.7 (d, JC–F = 9.4 Hz), 130.0, 128.4, 120.0 (d, JC–F = 5.9 Hz), 119.9 (d, JC–F = 14.8 Hz), 116.6 (d, JC–F = 23.2 Hz), 113.8, 55.0, and 38.0 (d, JC–F = 7.0 Hz); 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = −102.14
3.5. General Procedure 2 for the Synthesis of Substituted Anilines (2b–2r)
The following description is for a 20–25 mmol scale reaction. The solvent quantities and flask size were adjusted accordingly for smaller-scale reactions.
A 250 mL round-bottomed flask equipped with a stir bar was loaded with the (4-methoxybenzyl)(2-nitrophenyl)sulfane derivative (1b–1r, 1 equiv.), iron power (5.0 equiv.), NH4Cl (5.0 equiv.), and 150 mL of EtOH/H2O (4:1). The reaction flask was placed under inert atmosphere (argon), fitted with a reflux condenser, and stirred at 80 °C until TLC indicated a complete reduction (typically between 1 and 4 h). After cooling to room temperature, the reaction mixture was filtered through celite and concentrated under reduced pressure. The residue was then basified with 1M NaOH and extracted twice with dichloromethane, dried over MgSO4, and evaporated. The crystalline solid was washed with hexanes or diethyl ether as described below.
3.5.1. 2-((4-Methoxybenzyl)thio)-3-Methylaniline (2b)
The title compound was prepared according to general procedure 2 on a 20.0 mmol scale. The crystalline light brown solid was washed with hexanes, which gave the desired product in 94% (4.897 g, 18.88 mmol) yield; m.p. 74–76 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.06 (d, J = 8.3 Hz, 2H), 6.92 (t, J = 7.6 Hz, 1H), 7.76 (d, J = 8.3 Hz, 2H), 6.60 (d, J = 7.9 Hz, 1H), 6.41 (d, J = 7.2 Hz, 1H), 5.39 (s, 2H), 3.74 (s, 2H), 3.70 (s, 3H), and 2.13 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.2, 150.5, 143.0, 130.0, 129.9, 129.2, 117.7, 114.8, 113.5, 112.0, 55.0, 36.4, and 21.4. HRMS (ESI) m/z calculated for [M + H]+ = [C15H18NOS]+ 260.1104; observed, 260.1105.
3.5.2. 2-((4-Methoxybenzyl)thio)-6-Methylaniline (2c)
The title compound was prepared according to general procedure 2 on a 2.00 mmol scale. The crystalline white solid was washed with hexanes, which gave the desired product in 92% (0.474 g, 1.83 mmol) yield; m.p. 92–93 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.14 (d, J = 8.1 Hz, 2H), 7.03 (d, J = 7.4 Hz, 1H), 6.94 (d, J = 7.0 Hz, 1H), 8.82 (d, J = 7.0 Hz, 2H), 6.44 (t, J = 7.4 Hz, 1H), 5.01 (s, 2H), 3.89 (s, 2H), 3.71 (s, 3H), and 2.11 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.2, 146.9, 132.5, 130.4, 130.0, 129.9, 121.7, 116.2, 116.1, 113.6, 55.0, 37.5, and 18.2. HRMS (ESI) m/z calculated for [M + H]+ = [C15H18NOS]+ 260.1104 observed, 260.1104.
3.5.3. 1-((4-Methoxybenzyl)thio)naphthalen-2-Amine (2d)
The title compound was prepared according to general procedure 2 on a 4.45 mmol scale. The yellow oil was washed with hexanes, which gave the desired product as a light brown solid in 99% (1.300 g, 4.41 mmol) yield; m.p. 58–60 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.20 (d, J = 8.5 Hz, 2H), 7.66 (t, J = 8.4 Hz, 2H), 7.42–7.38 (m, 1H), 7.17–7.12 (m, 3H), 7.09 (d, J = 8.8 Hz, 1H), 6.78 (dt, J = 8.7 Hz, J = 2.1 Hz, 2H), 5.83 (s, 2H), 3.79 (s, 2H), and 3.69 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.2, 149.5, 136.3, 130.3, 130.0 129.9, 128.3, 127.1, 126.9, 123.3, 121.1, 117.4, 113.6, 104.9, 55.0, and 37.3. HRMS (ESI) m/z calculated for [M + H]+ = [C18H18NOS]+ 296.1104; observed, 296.1104.
3.5.4. N-(3-Amino-4-((4-Methoxybenzyl)thio)phenyl)acetamide (2e)
The title compound was prepared according to general procedure 2 on a 17.05 mmol scale. The crystalline beige powder solid was washed with hexanes, which gave the desired product in 93% (4.807 g, 15.91 mmol) yield; m.p. 111–112 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.71 (s, 1H), 7.11 (t, J = 8.3 Hz, 3H), 6.97 (d, J = 8.2 Hz, 1H), 6.80 (d, J = 8.3 Hz, 2H), 6.62 (d, J = 7.8 Hz, 1H), 5.33 (s, 2H), 3.81 (s, 2H), 3.70 (s, 3H), and 2.01 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 168.1, 158.1, 149.9, 140.6, 135.9, 130.2, 130.0, 113.6, 109.7, 107.7, 104.3, 55.0, 37.7, and 24.1. HRMS (ESI) m/z calculated for [M + H]+ = [C16H19N2O2S]+ 303.1162; observed, 303.1162.
3.5.5. 4-((4-Methoxybenzyl)thio)benzene-1,3-Diamine (2f)
The title compound was prepared according to general procedure 2 on a 20.0 mmol scale. The crystalline beige solid was washed with hexanes, which gave the desired product in 96% (4.979 g, 19.12 mmol) yield; m.p. 91–92 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.08 (d, J = 8.6 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 6.72 (d, J = 8.2 Hz, 1H), 5.93 (d, J = 7.9 Hz, 1H), 5.73 (dd, J = 8.2 Hz, J = 2.3 Hz, 1H), 5.00 (s, 2H), 4.97 (s, 2H), 3.71 (s, 3H), and 3.67 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.0, 150.6, 150.5, 137.4, 130.6, 130.0, 113.5, 104.3, 102.0, 99.0, 55.0, and 38.8. HRMS (ESI) m/z calculated for [M + H]+ = [C14H17N2OS]+ 261.1056; observed, 261.1057.
3.5.6. 5-Methoxy-2-((4-Methoxybenzyl)thio)aniline (2g)
The title compound was prepared according to general procedure 2 on a 27.9 mmol scale. The crystalline dark beige solid was washed with hexanes, which gave the desired product in 92% (7.082 g, 25.72 mmol) yield; m.p. 113–114 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.10 (dt, J = 8.6 Hz, J = 2.0 Hz, 1H), 6.96 (d, J = 8.4 Hz, 2H), 7.76 (dt, J = 8.6 Hz, J = 2.0 Hz, 2H), 6.30 (d, J = 2.7 Hz, 1H), 6.05 (dd, J = 8.5 Hz, J = 2.7 Hz, 1H), 5.34 (s, 2H), 3.77 (s, 2H), 3.71 (s, 3H), and 3.65 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 160.9, 158.1, 151.0, 137.2, 130.2, 130.0, 113.6, 107.1, 102.9, 98.9, 55.0, 54.7, and 38.0. HRMS (ESI) m/z calculated for [M + H]+ = [C15H18NO2S]+ 276.1053; observed, 276.1052.
3.5.7. 3-Amino-4-((4-Methoxybenzyl)thio)phenol (2h)
The title compound was prepared according to general procedure 2 on a 2.75 mmol scale. The crystalline light brown powder was washed with diethyl ether/hexanes (1:6), which gave the desired product in 92% (546.4 mg, 2.52 mmol) yield; m.p. 116–118 °C. 1H NMR (400 MHz, CD3CN, 298 K): δ = 7.07 (dt, J = 8.7 Hz, J = 2.0 Hz, 2H), 6.96 (d, J = 8.3 Hz, 1H), 6.86 (s, 2H), 6.79 (dt, J = 8.7 Hz, J = 2.1 Hz, 2H), 6.20 (d, J = 2.6 Hz, 1H), 6.03 (dd, J = 8.3 Hz, J = 2.6 Hz, 1H), 4.62 (s, 2H), 3.76 (s, 2H), and 3.74 (s, 3H); 13C{1H} NMR (100 MHz, CD3CN, 298 K): δ = 159.8, 159.6, 151.9, 139.0, 131.6, 131.0, 114.5, 108.2, 106.2, 101.7, 55.8, and 39.6. HRMS (ESI) m/z calculated for [M + H]+ = [C15H15NO2S]+ 262.0896; observed, 262.0898.
3.5.8. 3-((4-Methoxybenzyl)thio)pyridin-2-Amine (2i)
The title compound was prepared according to general procedure 2 on a 20.0 mmol scale. The crystalline beige solid was washed with hexanes, which gave the desired product in 92% (4.553 g, 18.48 mmol) yield; m.p. 76–78 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.87 (dd, J = 4.8 Hz, J = 1.7 Hz, 1H), 7.66 (dd, J = 7.4 Hz, J = 1.6 Hz, 1H), 7.15 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 6.46 (dd, J = 7.4 Hz, J = 4.9 Hz, 1H), 6.03 (s, 2H), 3.97 (s, 2H), and 3.70 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 159.1, 158.3, 147.4, 141.6, 130.1, 129.4, 113.7, 112.8, 111.9, 55.0, and 36.4. HRMS (ESI) m/z calculated for [M + H]+ = [C13H15N2OS]+ 247.0900; observed, 247.0899.
3.5.9. 2-((4-Methoxybenzyl)thio)pyridin-3-Amine (2j)
The title compound was prepared according to general procedure 2 on a 20.0 mmol scale. The crystalline brown powder was washed with hexanes, which gave the desired product in 99% (4.898 g, 19.88 mmol) yield. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.79 (dd, J = 7.6 Hz, J = 1.4 Hz, 1H), 7.29 (d, J = 7.9 Hz, 1H), 6.90 (t, J = 7.9 Hz, 2H), 6.84 (d, J = 7.6 Hz, 2H), 5.01 (s, 2H), 4.34 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.2, 141.6, 141.5, 137.2, 130.2, 130.0, 120.5, 119.1, 113.7, 55.0, and 32.7. HRMS (ESI) m/z calculated for [M + H]+ = [C13H15N2OS]+ 247.0900; observed, 247.0899.
3.5.10. 2-((4-Methoxybenzyl)thio)-5-(Trifluoromethyl)aniline (2k)
The title compound was prepared according to general procedure 2 on a 27.0 mmol scale. The crystalline off-white solid was washed with hexanes, which gave the desired product in 97% (8.167 g, 26.07 mmol) yield; m.p. 79–80 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.28 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 7.02 (s, 2H), 6.83 (d, J = 8.4 Hz, 2H), 6.75 (d, J = 7.8 Hz, 1H), 5.66 (s, 2H), 4.04 (s, 3H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.4, 148.6, 133.1, 130.1, 129.2, 128.7 (q, JC–F = 31.0 Hz), 124.4 (q, JC–F = 270.4 Hz), 121.5, 113.7, 112.0 (q, JC–F = 15.4 Hz), 109.8 (q, JC–F = 3.9 Hz), 55.0, and 36.2; 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = −61.45. HRMS (ESI) m/z calculated for [M + H]+ = [C15H15F3NOS]+ 314.0821; observed, 314.0823.
3.5.11. 3-Amino-4-((4-Methoxybenzyl)thio)benzonitrile (2l)
The title compound was prepared according to general procedure 2 on a 10.0 mmol scale. The crystalline yellow solid was washed with hexanes, which gave the desired product in 86% (2.315 g, 8.56 mmol) yield; m.p. 189–190 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.28 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 8.6 Hz, 2H), 6.98 (d, J = 1.6 Hz, 1H), 6.84 (d, J = 8.7 Hz, 2H), 6.74 (dd, J = 8.0 Hz, J = 1.4 Hz, 1H), 5.64 (s, 2H), 4.04 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.8, 144.7, 143.4, 136.2, 130.6, 130.0, 128.4, 126.3, 117.1, 114.2, 107.4, 55.1, and 35.6. HRMS (ESI) m/z calculated for [M + H]+ = [C15H15N2OS]+ 271.0900; observed, 271.0900.
3.5.12. Ethyl 3-Amino-4-((4-Methoxybenzyl)thio)benzoate (2m′)
The title compound was prepared according to general procedure 2 on a 23.0 mmol scale. The crystalline yellow solid was washed with hexanes, which gave the desired product in 94% (6.894 g, 21.72 mmol) yield; m.p. 89–90 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.33 (s, 1H), 7.23–7.19 (m, 3H), 7.07 (d, J = 7.9 Hz, 1H), 6.83 (d, J = 8.3, 2H), 5.44 (s, 2H), 6.88 (q, J = 7.0 Hz, 2H), 4.05 (s, 2H), 3.71 (s, 3H), and 1.29 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 165.9, 158.3, 147.8, 131.8, 130.1, 129.2, 129.2, 123.0, 116.8, 114.3, 113.7, 60.4, 55.0, 55.0, 36.0, and 14.2. HRMS (ESI) m/z calculated for [M + H]+ = [C17H20NO2S]+ 318.1158; observed, 318.1160.
3.5.13. 5-Bromo-2-((4-Methoxybenzyl)thio)aniline (2n)
The title compound was prepared according to general procedure 2 on a 2.86 mmol scale. The crystalline light brown solid was washed with hexanes, which gave the desired product in 79% (728.1 mg, 2.25 mmol) yield; m.p. 100–102 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.13 (d, J = 8.5 Hz, 2H), 6.98 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.82 (d, J = 8.6 Hz, 2H), 6.58 (dd, J = 8.2 Hz, J = 2.1 Hz, 1H), 5.56 (s, 2H), 3.89 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.2, 150.6, 136.3, 130.0, 129.6, 122.2, 118.5, 116.1, 115.2, 113.6, 55.0, and 36.9. HRMS (ESI) m/z calculated for [M + H]+ = [C14H15BrNOS]+ 324.0052; observed, 324.0052.
3.5.14. 4-Bromo-2-((4-Methoxybenzyl)thio)aniline (2o)
The title compound was prepared according to general procedure 2 on a 30 mmol scale. The crystalline beige solid was washed with hexanes, which gave the desired product in 95% (9.287g, 28.64 mmol) yield; m.p. 86–87 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.18–7.11 (m, 4H), 6.83 (dt, J = 8.6 Hz, J = 1.9 Hz, 1H), 6.66 (d, J = 8.6 Hz, 1H), 5.44 (s, 2H), 3.95 (s, 2H), and 3.72 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.3, 148.3, 135.6, 131.4, 130,1, 130.0, 118.2, 115.9, 113.6, 105.9, 55.0, and 36.9. HRMS (ESI) m/z calculated for [M + H]+ = [C14H15BrNOS]+ 324.0052; observed, 324.0050.
3.5.15. 5-Chloro-2-((4-Methoxybenzyl)thio)aniline (2p)
The title compound was prepared according to general procedure 2 on a 2.72 mmol scale. The crystalline pale brown powder was washed with hexanes, which gave the desired product in 93% (7.142 g, 2.55 mmol) yield; m.p. 80–82 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.13 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 8.2 Hz, 1H), 6.81 (d, J = 2.0 Hz, 1H), 6.75 (d, J = 8.6 Hz, 2H), 6.45 (dd, J = 8.2 Hz, J = 2.1 Hz, 1H), 5.58 (s, 2H), 3.89 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.2, 150.5, 136.2, 133.6, 130.0, 129.6, 115.6, 114.7, 113.6, 113.2, 55.0, and 37.0. HRMS (ESI) m/z calculated for [M + H]+ = [C14H15ClNOS]+ 280.0557; observed, 280.0558.
3.5.16. 3-Chloro-2-((4-Methoxybenzyl)thio)aniline (2q)
The title compound was prepared according to general procedure 2 on a 20.0 mmol scale. The crystalline off-white powder was washed with hexanes, which gave the desired product in 94% (5.282 g, 18.88 mmol) yield; m.p. 71–73 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.14 (d, J = 8.5 Hz, 2H), 7.00 (t, J = 8.0 Hz, 1H), 6.80 (d, J = 8.5 Hz, 2H), 6.67–6.63 (m, 2H), 5.66 (s, 2H), 3.86 (s, 2H), and 3.70 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.3, 152.4, 139.8, 130.5, 130.0, 129.4, 116.5, 113.6, 113.4, 112.8, 55.0, and 37.0. HRMS (ESI) m/z calculated for [M + H]+ = [C14H15ClNOS]+ 280.0557; observed, 280.0559.
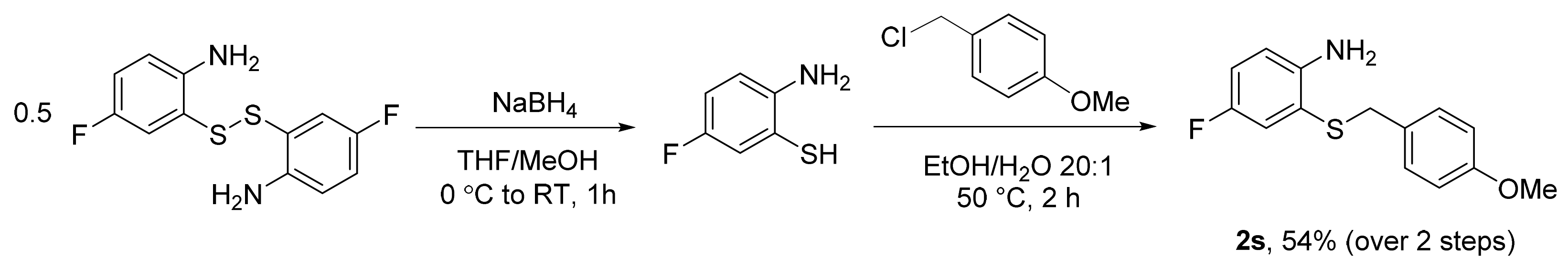
3.5.17. 3-Fluoro-2-((4-Methoxybenzyl)thio)aniline (2r)
The title compound was prepared according to general procedure 2 on a 12 mmol scale. The crystalline off-white powder was washed with hexanes, which gave the desired product in 97% (2.064 g, 11.64 mmol) yield; m.p. 71–72 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 7.12 (d, J = 8.6 Hz, 2H), 7.02 (q, J = 6.7 Hz, 1H), 6.79 (d, J = 8.6 Hz, 2H), 6.52 (d, J = 8.2 Hz, 1H), 6.32–6.28 (m, 1H), 5.62 (s, 2H), 3.83 (s, 2H), and 3.70 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 163.8 (d, JC–F = 239.0 Hz), 158.2, 152.1 (d, JC–F = 3.8 Hz), 130.5 (d, JC–F = 11.0 Hz), 129.9, 129.7, 113.6, 109.8 (d, JC–F = 2.5 Hz), 102.4 (d, JC–F = 21.4 Hz), 101.9 (d, JC–F = 24.0 Hz), 55.0, and 36.9; 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = −108.81. HRMS (ESI) m/z calculated for [M + H]+ = [C14H15FNOS]+ 264.0853; observed, 264.0854.
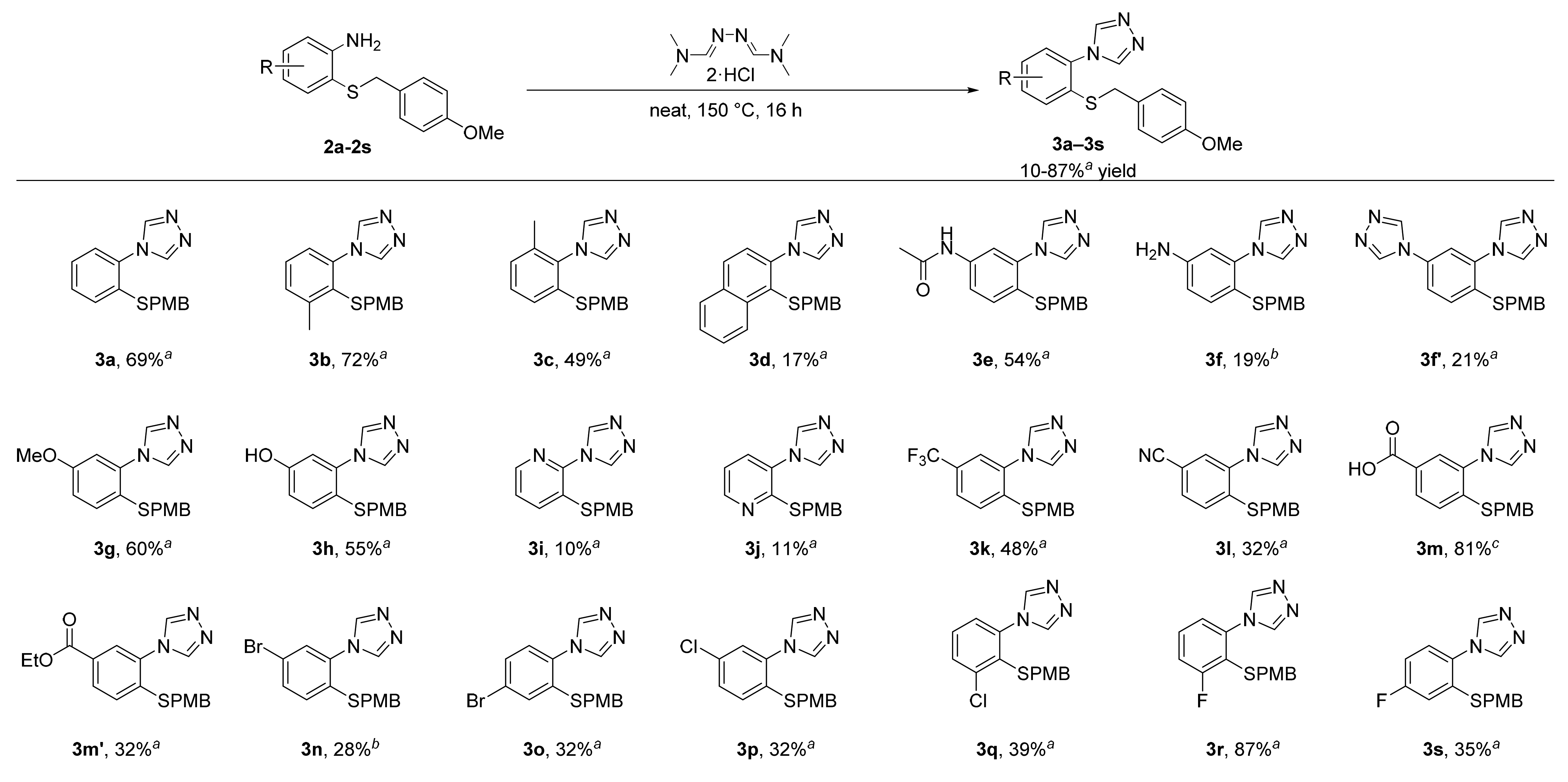
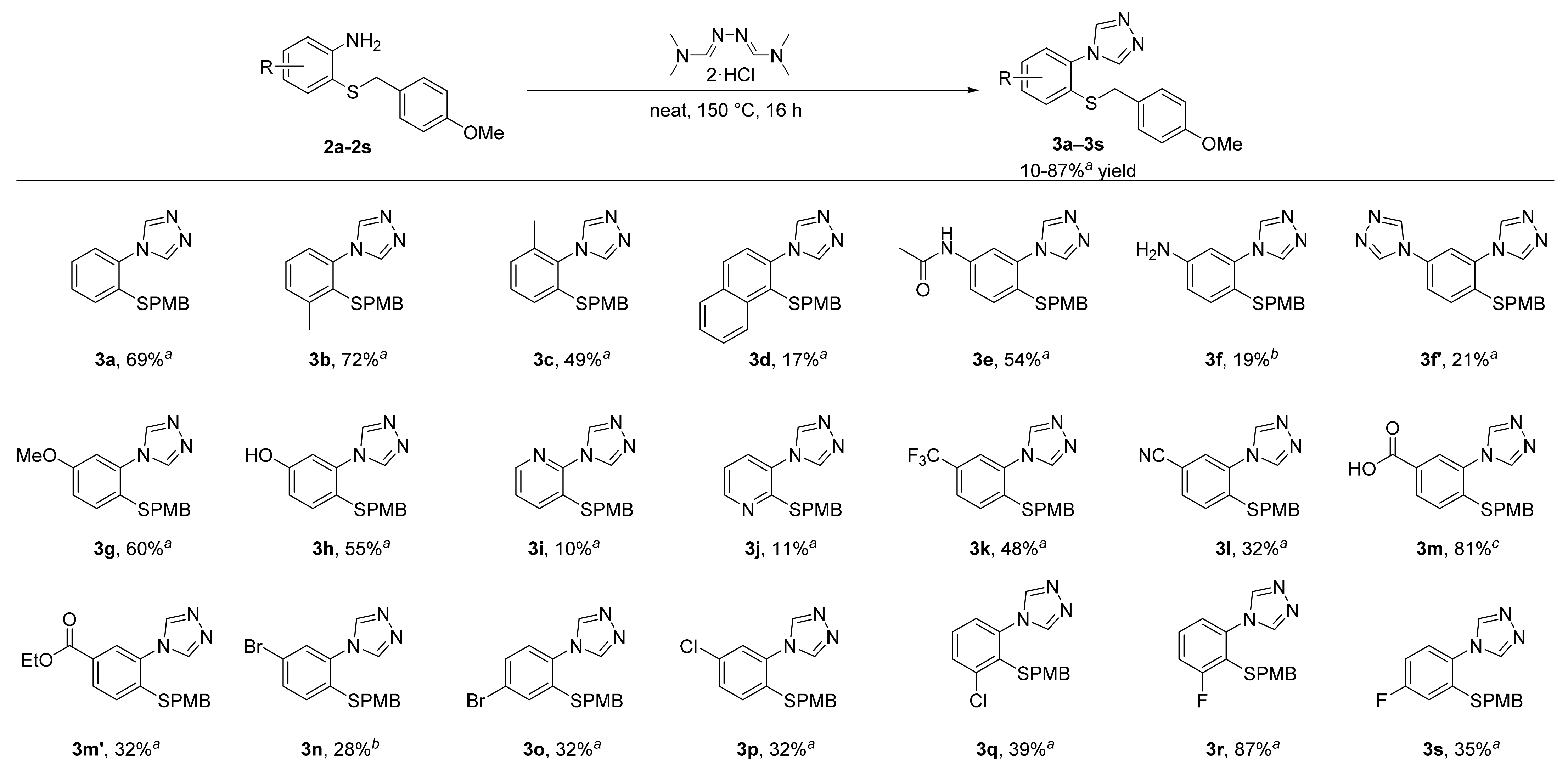
3.7. General Procedure 3 for the Synthesis of Substituted Triazoles (3b–3s)
A 50 mL round-bottomed flask equipped with a stir bar was loaded the 2-((4-methoxybenzyl)thio)aniline derivative (2b–2s, 1 equiv.) and N,N-dimethylformamide azine dihydrochloride (1.1 equiv). After mixing the two compounds thoroughly, the reaction flask was placed under an inert atmosphere (argon) and stirred in an oil bath at 150 °C. The two solid compounds were observed to melt within an hour and turn dark red. After 16 h, the reaction flask was cooled to room temperature. The resulting dark solid was basified with 1 M NaOH and extracted with dichloromethane, dried over MgSO4, filtered, and concentrated. The crude product was purified by column chromatography or recrystallization as described below.
3.7.1. 4-(2-((4-Methoxybenzyl)thio)-3-Methylphenyl)-4H-1,2,4-triazole (3b)
The title compound was prepared according to general procedure 3 on a 3.63 g (14.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.56) gave the product as an off-white powder in 72% (3.15 g, 10.1 mmol) yield; m.p. 116–118 °C. 1H NMR (400 MHz, CDCl3, 298 K): δ = 7.92 (s, 2H), 7.43 (d, J = 7.6 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.9 Hz, 1H), 6.68 (s, 4H), 3.74 (s, 1H), 3.53 (s, 1H), and 2.61 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3, 298 K): δ = 158.7, 145.5, 143.1, 137.8, 131.6, 129.6, 129.5 (2 signals), 128.7, 124.1, 113.7, 55.1, 38.7, and 21.4. HRMS (ESI) m/z: [M + H]+ calculated for C17H17N3OS, 312.1165; observed, 312.1165.
3.7.2. 4-(2-((4-Methoxybenzyl)thio)-6-Methylphenyl)-4H-1,2,4-triazole (3c)
The title compound was prepared according to general procedure 3 on a 4.67 g (18.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.41) gave the product as an off-white powder in 49% (2.76 g, 7.55 mmol) yield; m.p. 191–193 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.59 (s, 2H), 7.46–7.40 (m, 2H), 7.27 (d, J = 6.7 Hz, 1H), 7.21 (d, J = 8.5 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 4.12 (s, 1H), 3.71 (s, 1H), and 1.96 (s, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.5, 143.2, 135.9, 135.7, 131.3, 130.0 (2 signals), 128.0 (2 signals), 126.2, 113.9, 55.1, 35.7, and 17.2. HRMS (ESI) m/z: [M + H]+ calculated for C17H17N3OS, 312.1165; observed, 312.1165.
3.7.3. 4-(1-((4-Methoxybenzyl)thio)naphthalen-2-yl)-4H-1,2,4-triazole (3d)
The title compound was prepared according to general procedure 3 on a 975 mg (3.30 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.67) gave the product as a pale yellow powder in 17% (195 mg, 0.56 mmol) yield; m.p. 129–130 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.65 (d, J = 8.4 Hz, 1H), 8.52 (s, 2H), 8.18 (d, J = 8.6 Hz, 1H), 8.13 (d, J = 7.9 Hz, 1H), 7.81–7.77 (m, 1 H), 7.73–7.69 (m, 1 H), 7.57 (d, J = 8.6 Hz, 1H), 6.70 (s, 4H), 3.81 (s, 3H), and 3.68 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.4, 143.6, 136.3, 134.2, 133.3, 131.0, 129.7, 129.0, 128.7, 128.5, 127.7, 127.4, 126.7, 124.4, 113.7, and 55.1. HRMS (ESI) m/z: [M + H]+ calculated C20H17N3OS, 348.1165; observed, 348.1163.
3.7.4. N-(4-((4-Methoxybenzyl)thio)-3-(4H-1,2,4-Triazol-4-Yl)phenyl)acetamide (3e)
The title compound was prepared according to general procedure 2 on a 3.26 g (10.8 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.19) gave the product as a pale brown powder in 54% (2.06 g, 5.80 mmol) yield; m.p. 176–178 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 10.26 (s, 1H), 8.58 (s, 1H), 7.69 (d, J = 1.8 Hz, 1H), 7.62 (dd, J = 8.6 Hz, J = 2.0 Hz, 1H), 7.56 (d, J = 8.6 Hz, 1H), 7.06 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 3.97 (s, 2H), 3.71 (s, 3H), and 2.06 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 168.9, 158.5, 143.4, 139.1, 134.3, 133.1, 130.0, 128.6, 125.6, 120.1, 117.1, 113.9, 55.1, 37.8, and 24.1. HRMS (ESI) m/z: [M + H]+ calculated C18H18N4O2S, 355.1223; observed, 355.1225.
3.7.5. 4-((4-Methoxybenzyl)thio)-3-(4H-1,2,4-Triazol-4-Yl)aniline (3f)
The title compound was obtained as a by-product of the synthesis of compound 3f′, which was prepared according to general procedure 3 on a 3.64 mg (14.0 mmol) scale; for this reaction, 2.2 equiv. of N,N-dimethylformamide azine dihydrochloride was used. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.26) gave the product as a pale brown powder in 19% (818 mg, 2.24 mmol) yield; m.p. 147–149 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.01 (s, 2H), 7.24 (d, J = 8.2 Hz, 1H), 7.18 (dt, J = 8.6 Hz, J = 2.0 Hz, 2H), 6.92 (d, J = 2.4 Hz, 1H), 6.83 (dt, J = 8.6 Hz, J = 2.0 Hz, 2H), 6.74 (dd, J = 8.2 Hz, J = 2.4 Hz, 2H), 7.96–7.90 (m, 3 H), 7.86–7.80 (m, 3 H), 7.70 (tt, J = 7.4 Hz, J = 1.1 Hz, 1H), 5.46 (s, 2H), 3.96 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.7, 150.7, 141.8, 136.3, 135.0, 130.5, 130.0, 116.5, 114.3, 114.2, 109.3, 106.6, 55.5, and 37.5. HRMS (ESI) m/z: [M + H]+ calculated C16H16N4OS, 313.1118; observed, 313.1120.
3.7.6. 4,4′-(4-((4-. Methoxybenzyl)thio)-1,3-Phenylene)bis(4H-1,2,4-Triazole) (3f′)
The title compound was prepared according to general procedure 3 on a 3.64 g (14.0 mmol) scale; for this reaction, 2.2 equiv. of N,N-dimethylformamide azine dihydrochloride was used. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 10:1; Rf = 0.49) gave the product as a pale yellow powder in 21% (1.08 mg, 2.95 mmol) yield; m.p. 250–253 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.21 (s, 2H), 8.74 (s, 2H), 7.99 (s, 1H), 7.91 (d, J = 7.0 Hz, 1H), 7.81 (d, J = 7.6 Hz, 1H), 7.20 (d, J = 6.2 Hz, 2H), 6.85 (d, J = 6.4 Hz, 2H), 4.20 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.6, 143.3, 141.1, 133.7, 132.8, 132.4, 131.4, 130.0, 127.9, 121.8, 119.7, 113.9, 55.1, and 36.4. HRMS (ESI) m/z: [M + H]+ calculated C18H16N6OS, 365.1179; observed, 365.1180.
3.7.7. 4-(5-Methoxy-2-((4-Methoxybenzyl)thio)phenyl)-4H-1,2,4-triazole (3g)
The title compound was prepared according to general procedure 3 on a 4.13 g (15.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.44) gave the product as a white powder in 60% (2.92 g, 8.93 mmol) yield; m.p. 128–130 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.56 (s, 2H), 7.57 (dd, J = 9.2 Hz, J = 2.1 Hz, 1H), 7.11–7.08 (m, 2H), 7.00 (d, J = 8.6 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 3.89 (s, 2H), 3.80 (s, 3H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 159.3, 158.4, 143.3, 136.0, 135.1, 129.9, 128.9, 122.1, 116.0, 113.8, 112.9, 55.8, 55.1, and 36.7 HRMS (ESI) m/z: [M + H]+ calculated C17H18N3O2S, 328.1114; observed 328.1114.
3.7.8. 4-((4-Methoxybenzyl)thio)-3-(4H-1,2,4-Triazol-4-Yl)phenol (3h)
The title compound was prepared according to general procedure 3 on a 900 mg (3.44 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.22) gave the product as a white powder in 55% (570 mg, 1.91 mmol) yield; m.p. 212–214 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 10.25 (s, 1H), 8.52 (s, 2H), 7.45 (d, J = 8.6 Hz, 1H), 6.97 (d, J = 8.6 Hz, 2H), 6.91 (dd, J = 8.6 Hz, J = 2.6 Hz, 1H), 6.80–6.78 (m, 3H) 3.81 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.3, 158.0, 143.3, 136.4, 136.1, 129.9, 129.0, 119.7, 117.1, 114.1, 113.7, and 55.1. HRMS (ESI) m/z: [M + H]+ calculated C16H15N3O2S, 314.0958; observed, 314.0959.
3.7.9. 3-((4-Methoxybenzyl)thio)-2-(4H-1,2,4-Triazol-4-Yl)pyridine (3i)
The title compound was prepared according to general procedure 3 on a 3.45 g (14.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.44) gave the product as an orange powder in 10% (421 mg, 1.41 mmol) yield; m.p. 154–156 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.71 (s, 2H), 8.46 (d, J = 6.3 Hz, 1H), 7.77 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 8.28 Hz, 2H), 6.99–6.92 (m, 3H), 5.83 (s, 2 H), and 3.74 (s, 3 H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 175.6, 159.0, 143.3, 143.2, 136.1, 133.0, 129.8, 127.5, 114.0, 111.8, 58.2, and 55.1. HRMS (ESI) m/z: [M + H]+ calculated C15H14N4OS, 299.0961; observed, 299.0961.
3.7.10. 2-((4-Methoxybenzyl)thio)-3-(4H-1,2,4-Triazol-4-Yl)pyridine (3j)
The title compound was prepared according to general procedure 3 on a 3.70 g (15.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.48) gave the product as a white powder in 11% yield (504 mg, 1.70 mmol); m.p. 219–220 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.85 (s, 2H), 8.42 (dd, J = 4.7 Hz, J = 1.4 Hz, 1H), 8.16 (dd, J = 8.0 Hz, J = 1.4 Hz, 1H), 7.56 (dd, J = 8.0 Hz, J = 4.7 Hz, 1H), 7.18 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 4.20 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.6, 146.4, 144.9, 142.4, 140.2, 130.1, 128.5, 127.6, 125.0, 113.9, 55.1, and 36.4. HRMS (ESI) m/z: [M + H]+ calculated C15H14N4OS, 299.0961; observed, 299.0960.
3.7.11. 4-(2-((4-Methoxybenzyl)thio)-5-(Trifluoromethyl)phenyl)-4H-1,2,4-triazole (3k)
The title compound was prepared according to general procedure 3 on an 8.15 g (26.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.62) gave the product as an off-white powder in 48% (4.54 g, 12.41 mmol) yield; m.p. 126–127 °C. 1H NMR (400 MHz, CD3CN, 298 K): δ = 8.32 (s, 2H), 7.76 (dd, J = 8.5 Hz, J = 1.6 Hz, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.68 (s, 1H), 7.19 (dt, J = 8.7 Hz, J = 2.0 Hz, 2H), 6.83 (dt, J = 8.7 Hz, J = 2.1 Hz, 2H), 4.16 (s, 2H), and 3.74 (s, 3H); 13C{1H} NMR (100 MHz, CD3CN, 298 K): δ = 160.2, 143.9, 141.2, 133.8, 131.0, 130.7, 128.7 (q, JC–F = 33.2 Hz), 128.4, 127.6 (q, JC–F = 3.7 Hz), 125.3 (q, JC–F = 3.8 Hz), 124.6 (q, JC–F = 269.7 Hz), 115.0, 55.9, and 37.4; 19F{1H} NMR (376 MHz, CD3CN, 298 K, referenced to C6H5F): δ = −61.45. HRMS (ESI) m/z: [M + H]+ calculated C17H14F3N3OS, 366.0883; observed, 366.0882.
3.7.12. 4-((4-Methoxybenzyl)thio)-3-(4H-1,2,4-Triazol-4-yl)benzonitrile (3l)
The title compound was prepared according to general procedure 3 on a 1.94 g (7.16 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.53) gave the product as a pale yellow powder in 32% (750 mg, 2.33 mmol) yield; m.p. 209–210 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.75 (s, 2H), 8.05 (d, J = 1.7 Hz, 2H), 7.97 (dd, J = 8.3 Hz, J = 1.7 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.28 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 4.32 (s, 2H), and 3.72 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.7, 143.2, 142.4, 133.3, 131.7, 130.8, 130.2, 127.9, 126.9, 117.7, 114.0, 108.0, 55.1, and 34.9. HRMS (ESI) m/z: [M + H]+ calculated C17H14N4OS, 323.0961; observed, 323.0962.
3.7.13. 4-((4-Methoxybenzyl)thio)-3-(4H-1,2,4-Triazol-4-Yl)benzoic Acid (3m)
A 50 mL round-bottomed flask was charged with compound 3m′ (1.50 mmol, 1 equiv.), KOH (2 equiv.), and MeOH, and then heated to 35 °C for 1 h. After removal of the solvent under reduced pressure, the crude product was purified by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 10:1, Rf = 0.19), which gave the product as a white powder in 87% (413 mg, 1.21 mmol) yield; m.p. > 360 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 13.29 (s, 1H), 8.74 (s, 2H), 8.02 (d, J = 8.0 Hz, 1H), 7.85 (s, 1H), 7.74 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.1 Hz, J, 2H), 6.86 (d, J = 8.4 Hz, 2H), 4.27 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 166.0, 158.6, 143.3, 140.7, 131.5, 130.4, 130.2, 128.6, 127.8, 127.7, 127.3, 114.0, 7, 55.1, and 35.2. HRMS (ESI) m/z: [M + H]+ calculated C17H15N3O3S, 342.0907; observed, 342.0908.
3.7.14. Ethyl 4-((4-Methoxybenzyl)thio)-3-(4H-1,2,4-Triazol-4-Yl)benzoate (3m′)
The title compound was prepared according to general procedure 3 on a 6.89 g (15.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.64) gave the product as a pale yellow powder in 32% (1.76 g, 4.76 mmol) yield; m.p. 126–127 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.74 (s, 2H), 8.03 (dd, J = 8.4 Hz, J = 1.6 Hz, 1H), 7.89 (d, J = 1.5 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.28 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 4.34–4.28 (m, 4H), 3.71 (s, 3H), and 1.31 (t, J = 7.1 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 164.5, 158.6, 143.2, 141.5, 131.5, 130.2, 127.7, 127.6, 127.3, 127.1, 114.0, 61.2, 55.1, 35.1, and 14.1. HRMS (ESI) m/z: [M + H]+ calculated C19H19N3O3S, 370.1220; observed, 370.1222.
3.7.15. 4-(5-Bromo-2-((4-Methoxybenzyl)thio)phenyl)-4H-1,2,4-triazole (3n)
The title compound was prepared according to general procedure 3 on a 1.30 g (4.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.62) gave the product as a pale brown powder in 28% (421 mg, 1.15 mmol) yield; m.p. 163–165 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.67 (s, 2H), 7.77 (s, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.17 (d, J = 7.3 Hz, 1H), 6.84 (d, J = 7.2 Hz, 1H), 4.13 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.5, 143.2, 133.9, 133.3, 132.7, 131.5, 130.0 (2 signals), 127.8, 119.0, 113.9, 55.1, and 36.3. HRMS (ESI) m/z: [M + H]+ calculated C16H14BrN3OS, 376.0114; observed, 376.0113.
3.7.16. 4-(4-Bromo-2-((4-Methoxybenzyl)thio)phenyl)-4H-1,2,4-triazole (3o)
The title compound was prepared according to general procedure 3 on a 7.78 g (24.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.60) gave the product as a pale brown powder in 32% (2.86 g, 7.83 mmol) yield; m.p. 173–175 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.67 (s, 2H), 7.80 (d, J = 2.1 Hz, 1H), 7.58 (dd, J = 8.6 Hz, J = 2.1 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.20 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 4.22 (s, 2H), and 3.72 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.6, 143.3, 136.6, 131.6, 131.4, 130.2, 129.5, 129.1, 127.5, 123.0, 114.0, 55.1, and 35.9. HRMS (ESI) m/z: [M + H]+ calculated C16H14BrN3OS, 376.0114; observed, 376.0112.
3.7.17. 4-(5-Chloro-2-((4-Methoxybenzyl)thio)phenyl)-4H-1,2,4-triazole (3p)
The title compound was prepared according to general procedure 3 on a 3.36 g (12.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.60) gave the product as a pale yellow powder in 32% (1.29 g, 3.88 mmol) yield; m.p. 158–159 °C. 1H NMR (400 MHz, CDCl3, 298 K): δ = 8.08 (s, 2H), 7.48 (d, J = 8.5 Hz, 1H), 7.39 (dd, J = 8.5 Hz, J = 2.1 Hz, 1H), 7.19 (d, J = 2.1 Hz, 1H), 6.94 (d, J = 8.6 Hz, 1H), 6.73 (d, J = 8.6 Hz, 1H), 3.85 (s, 1H), and 3.73 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3, 298 K): δ = 159.1, 142.7, 134.9, 133.8, 133.6, 131.7, 130.0 129.8, 127.6, 126.8, 114.1, 55.3, and 39.0. HRMS (ESI) m/z: [M + H]+ calculated C16H14ClN3OS, 332.0619; observed, 332.0618.
3.7.18. 4-(3-Chloro-2-((4-Methoxybenzyl)thio)phenyl)-4H-1,2,4-triazole (3q)
The title compound was prepared according to general procedure 3 on a 4.20 mg (15.0 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.43) gave the product as an off-white powder in 39% (1.93 mg, 5.82 mmol) yield; m.p. 121–123 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.41 (s, 2H), 7.79 (dd, J = 8.1 Hz, J = 1.3 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.46 (dd, J = 7.9 Hz, J = 1.3 Hz, 1H), 3.87 (s, 2H), and 3.71 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.5, 143.4, 140.4, 139.1, 131.0 (2 signals), 129.8, 129.8, 128.6, 126.3, 113.8, 55.1, and 38.0. HRMS (ESI) m/z: [M + H]+ calculated C16H14ClN3OS, 332.0619; observed, 332.0617.
3.7.19. 4-(3-Fluoro-2-((4-Methoxybenzyl)thio)phenyl)-4H-1,2,4-triazole (3r)
The title compound was prepared according to general procedure 3 on a 2.11 g (8.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.41) gave the product as a pale brown powder in 87% (2.20 g, 6.984 mmol) yield; m.p. 218–219 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.46 (s, 2H), 7.62–7.56 (m, 1H), 7.50 (td, J = 9.7 Hz, J = 1.0 Hz, 1H), 7.35 (d, J = 7.9 Hz, 1H), 6.91 (d, J = 8.6 Hz, 2H), 6.77 (d, J = 8.6 Hz, 2H), 3.88 (s, 2H), and 3.70 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 163.0 (d, JC–F = 244.5 Hz), 158.5, 143.3, 137.9 (d, JC–F = 3.0 Hz), 131.2 (d, JC–F = 9.9 Hz), 129.8, 128.8, 123.2 (d, JC–F = 3.2 Hz), 118.7 (d, JC–F = 21.0 Hz), 116.9 (d, JC–F = 23.8 Hz), 131.8, 55.1, and 38.0. 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = −102.99. HRMS (ESI) m/z: [M + H]+ calculated C16H14FN3OS, 316.0915; observed, 316.0913.
3.7.20. 4-(4-Fluoro-2-((4-Methoxybenzyl)thio)phenyl)-4H-1,2,4-triazole (3s)
The title compound was prepared according to general procedure 3 on a 2.788 g (10.58 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1; Rf = 0.38) gave the product as a pale brown powder in 35% (1.148 g, 3.64 mmol) yield; m.p. 127–128 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 8.65 (s, 2H), 7.54–7.49 (m, 2H), 7.25–7.20 (m, 3H), 4.23 (s, 2H), and 3.71 (s, 2H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 162.3 (d, JC–F = 247.1 Hz), 158.6, 143.5, 137.3 (d, JC–F = 9.1 Hz), 130.1, 129.4 (d, JC–F = 9.6 Hz), 128.4 (d, JC–F = 2.8 Hz), 127.5, 115.5 (d, JC–F = 25.4 Hz), 114.0, 113.3 (d, JC–F = 23.0 Hz), 131.8, 55.0, and 36.7. 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = -110.77. HRMS (ESI) m/z: [M + H]+ calculated C16H14FN3OS, 316.0915; observed, 316.0913.
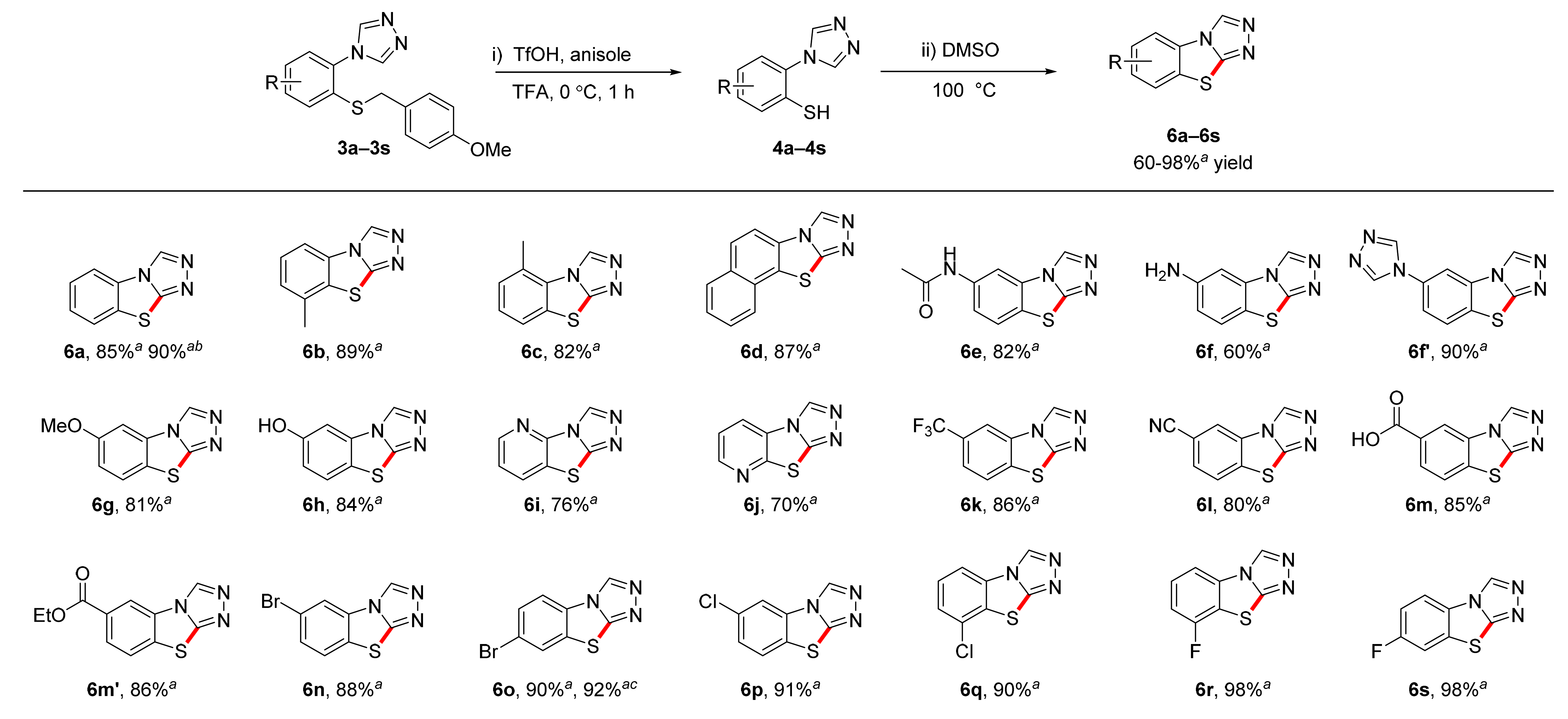
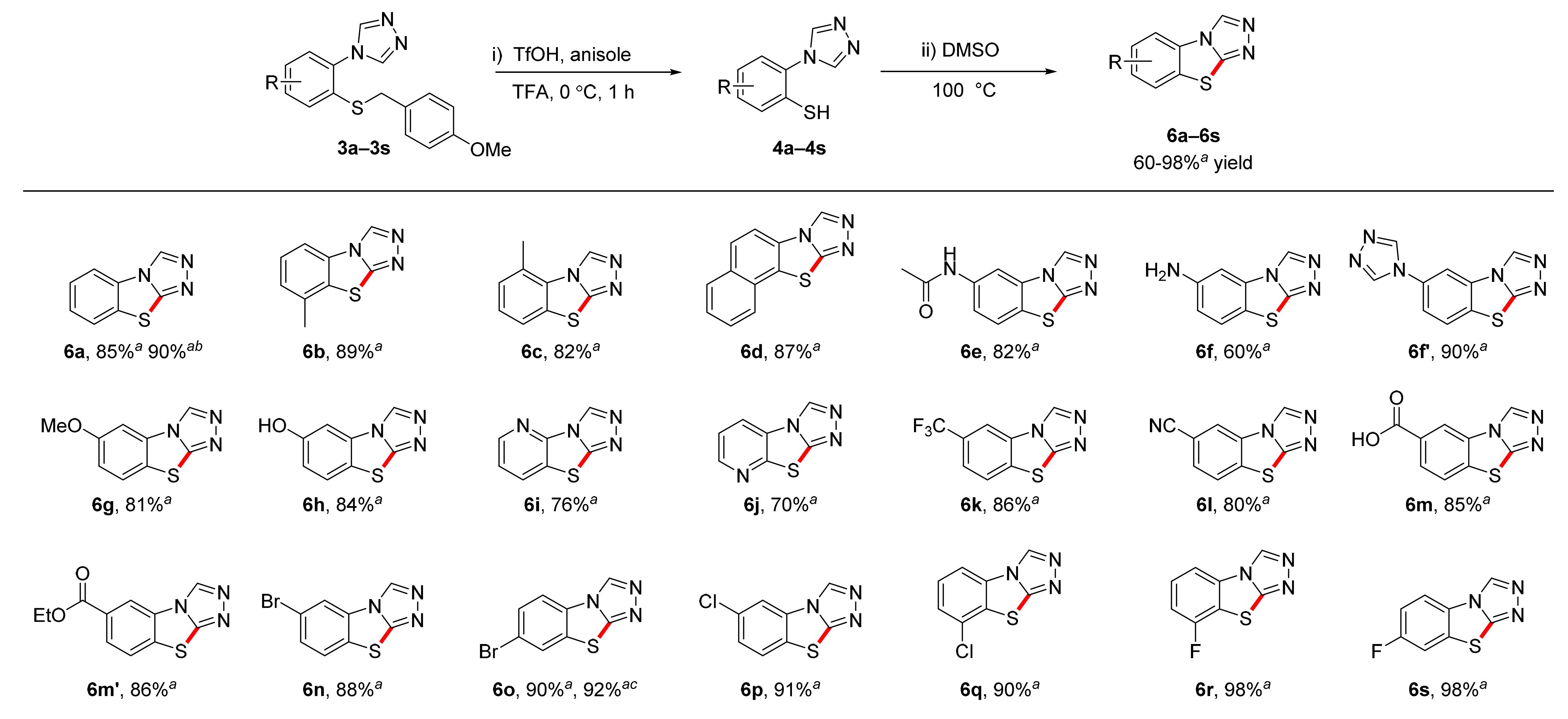
3.9. General Procedure 4 for the Synthesis of benzo[4,5]thiazolo[2,3-c][1,2,4]triazoles (6a–6s)
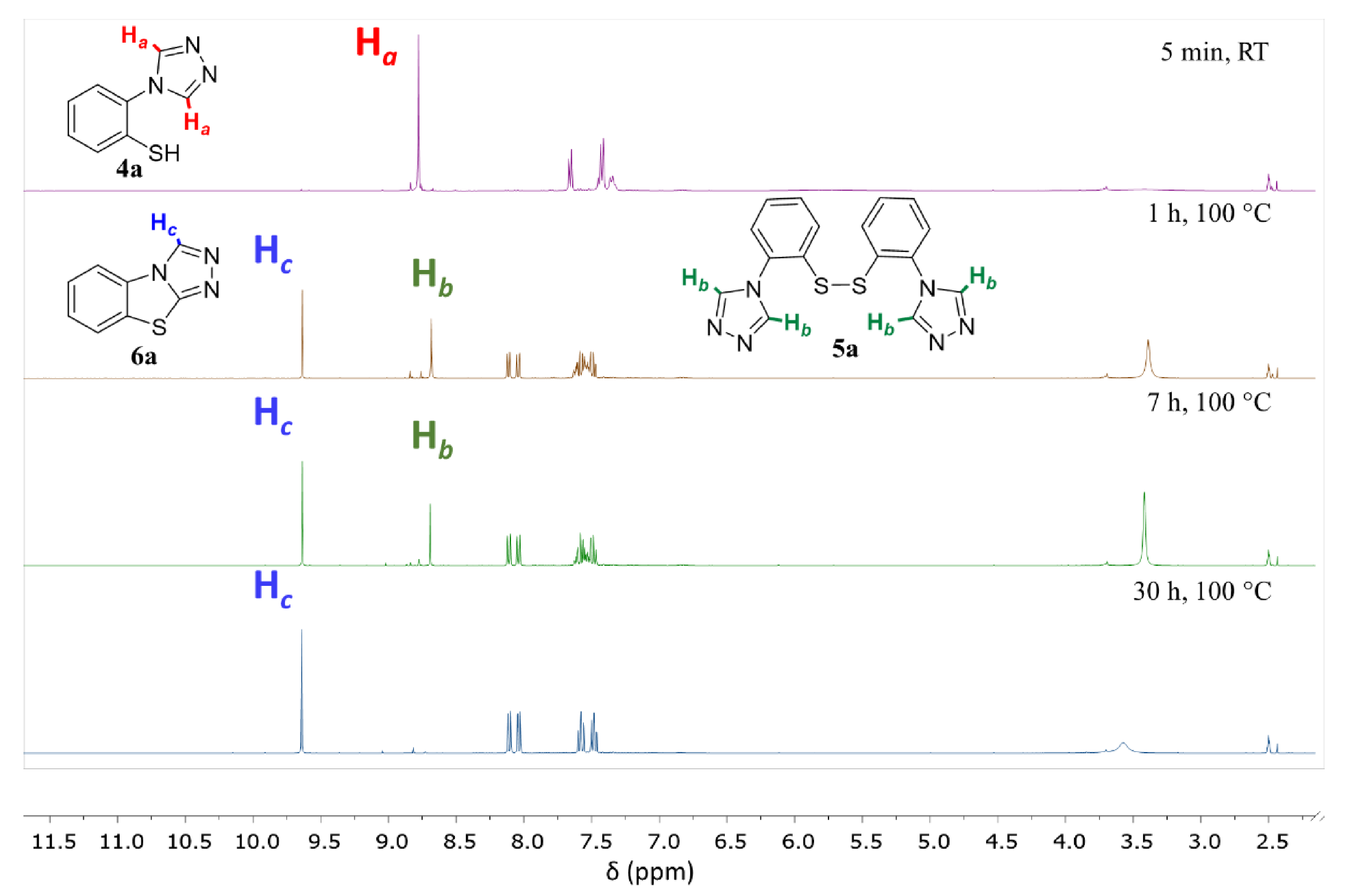
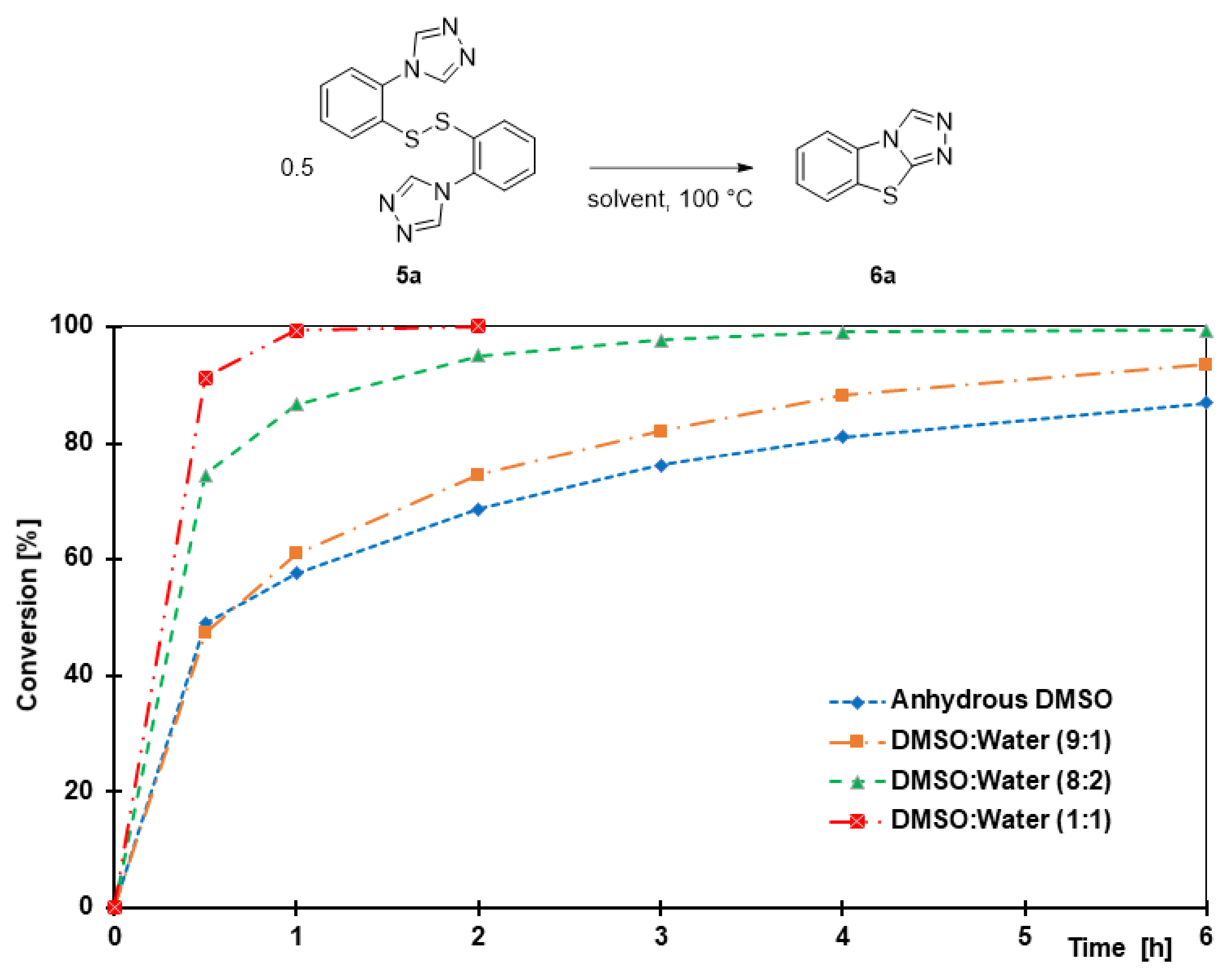
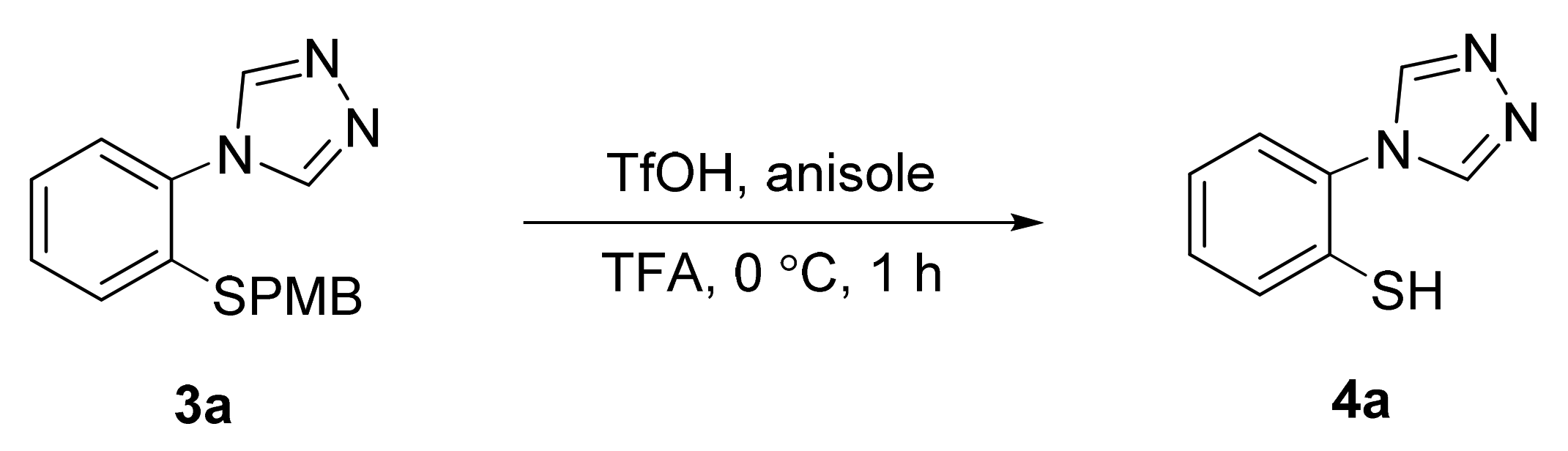
A 25 mL round-bottomed flask equipped with a stir bar was loaded with the triazole (3a–3s, 1 equiv.) and trifluoroacetic acid (50 equiv.). After flushing with argon, anisole (5 equiv.) and trifluoromethanesulfonic acid (5 equiv.) were added to this solution, and the reaction mixture was stirred at 0 °C for 1 h. The solution was then concentrated under high vacuum. The resulting dark red oil was diluted with 20 mL of water and extracted twice with dichloromethane (50 mL). Following concentration of the combined organic phases, the crude thiol was dissolved in 2 mL of DMSO and heated to 100 C. After completion of the cyclization reaction, the reaction mixture was diluted with 20 mL of sat. NaHCO3 and extracted twice with dichloromethane (50 mL). The combined organic phases were dried over MgSO4, filtered, and concentrated. The resulting crude product was purified by column chromatography or recrystallization as described below.
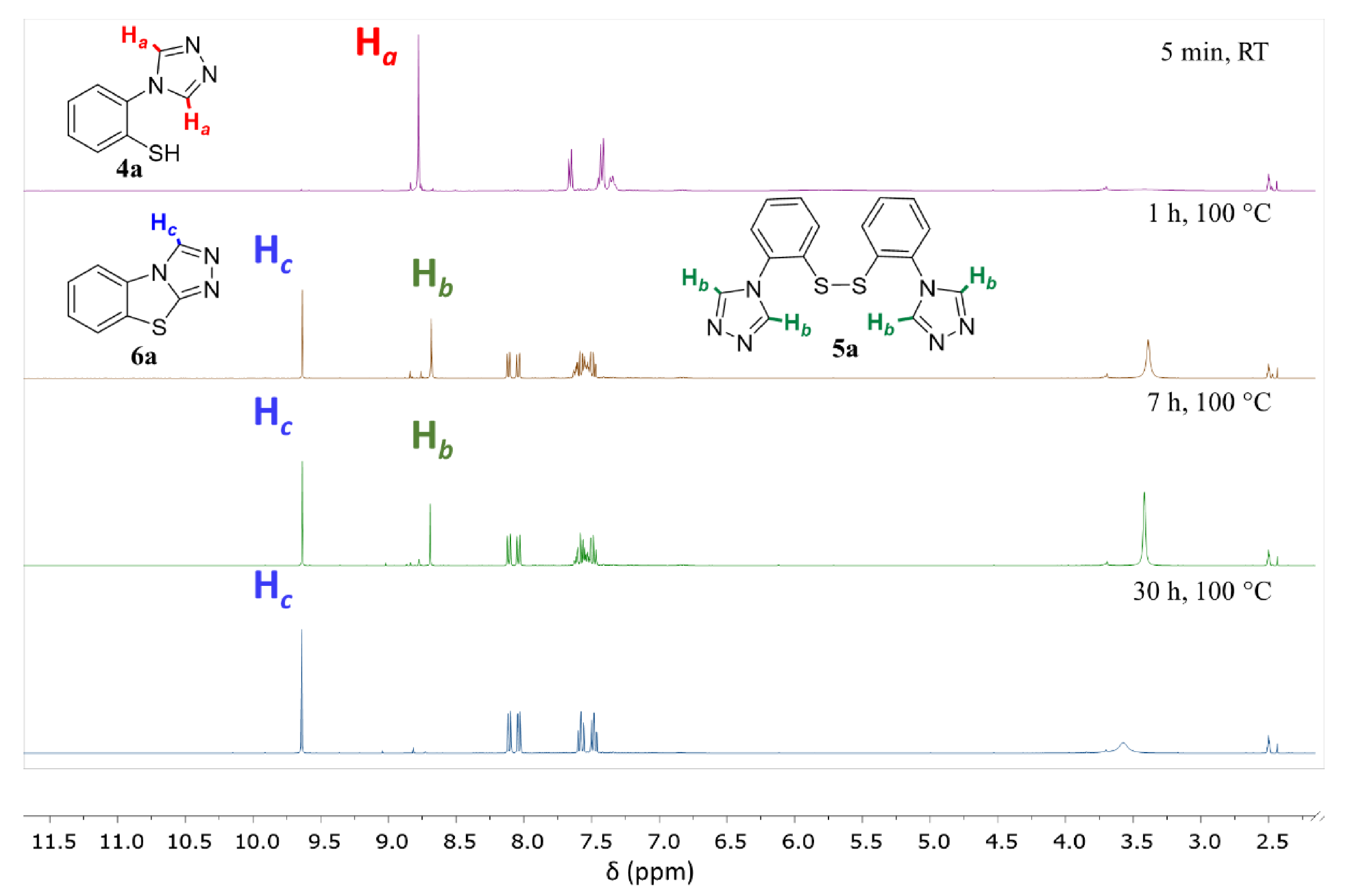
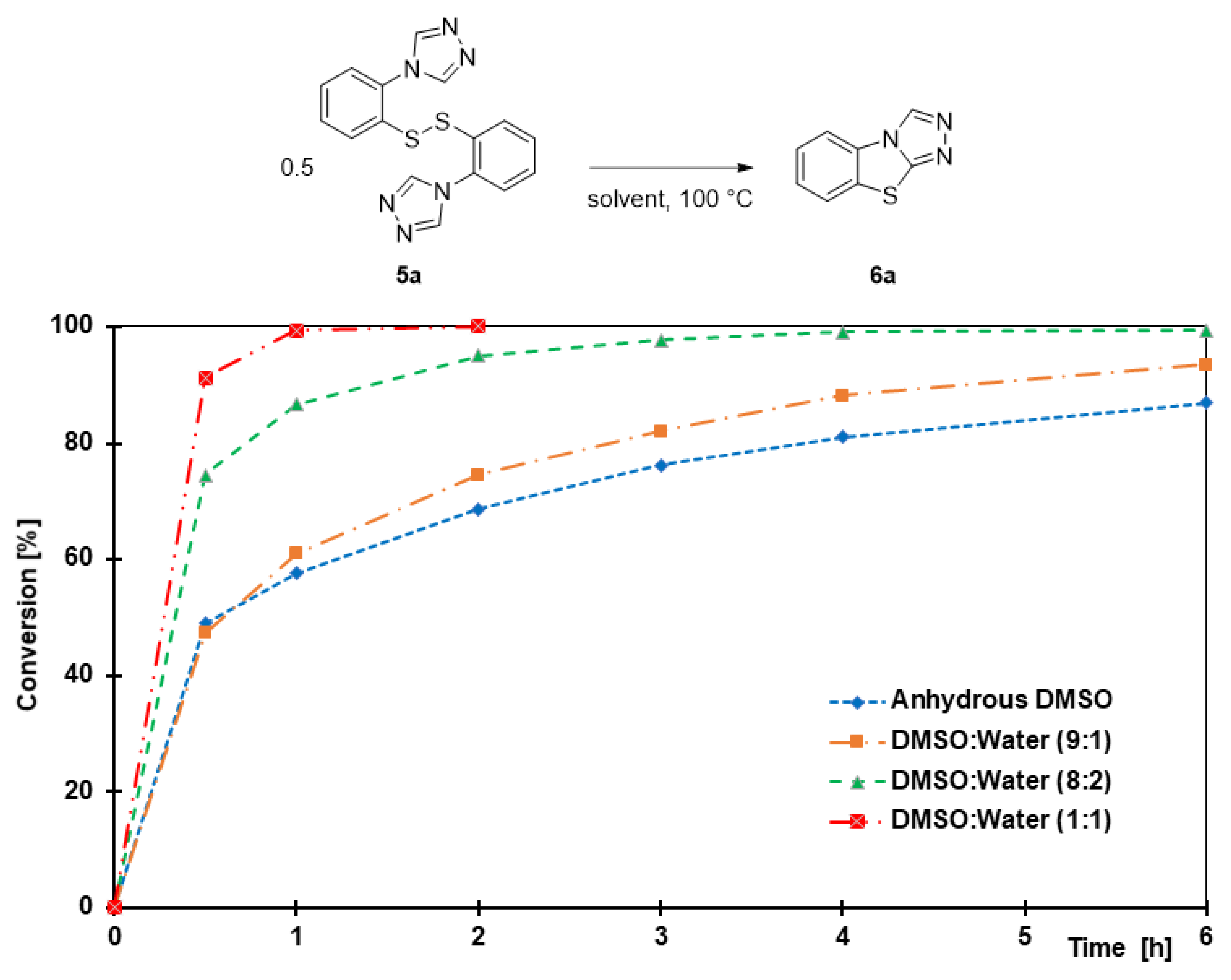
3.9.1. benzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6a)
The title compound was prepared twice according to general procedure 4 on a 297 mg (1.0 mmol) and a 1.49 g (5.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/acetone 4:1, Rf = 0.50) gave the product as a light yellow powder in 85% (149 mg, 0.85 mmol) and 90% (788 mg, 4.5 mmol) yield; m.p. 179–181 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.64 (s, 1H), 8.11 (dd, J = 8.0 Hz, J = 0.5 Hz, 1H), 8.05 (dd, J = 8.0 Hz, J = 0.4 Hz, 1H), 7.59 (td, J = 8.2 Hz, J = 1.0 Hz, 1H), and 7.49 (td, J = 8.3 Hz, J = 1.1 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 154.4, 136.8, 131.5, 129.0, 127.0, 126.6, 125.5, and 114.8. HRMS (ESI) m/z: [M + H] calculated for C8H5N3S, 176.0277; observed, 176.0276.
3.9.2. 8-Methylbenzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6b)
The title compound was prepared according to general procedure 4 on a 156 mg (0.50 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.48) gave the product as an off-white powder in 89% (38 mg, 0.445 mmol) yield; m.p. 185–187 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.62 (s, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.32 (d, J = 7.6 Hz, 1H), and 2.43 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 153.6, 136.9, 134.2, 131.0, 128.7, 127.1, 127.0, 122.3, and 19.2. HRMS (ESI) m/z: [M + H]+ calculated for C9H7N3S, 190.0434; observed, 190.0435.
3.9.3. 5-Methylbenzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6c)
The title compound was prepared according to general procedure 4 on a 311 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.46) gave the product as a light yellow powder in 82% (156 mg, 0.825 mmol) yield; m.p. 189–191 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.49 (s, 1H), 7.83–7.79 (m, 1H), 7.34 (t, J = 5.3 Hz, 1H), and 2.69 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 154.4, 138.0, 131.3, 128.6, 128.5, 126.1, 125.6, 122.6, and 18.7. HRMS (ESI) m/z: [M + H]+ calculated for C9H7N3S, 190.0434; observed, 190.0433.
3.9.4. Naphtho[2′,1′:4,5]thiazolo[2,3-c][1,2,4]triazole (6d)
The title compound was prepared according to general procedure 4 on a 174 mg (0.50 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.46) gave the product as a light yellow powder in 87% (98 mg, 0.433 mmol) yield; m.p. 238–239 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.72 (s, 1H), 8.25 (d, J = 8.8 Hz, 1H), 8.15 (t, J = 8.6 Hz, 2H), 8.00 (d, J = 8.2 Hz, 1H), 7.74 (td, J = 7.1 Hz, J = 0.9 Hz, 1H), and 7.67 (td, J = 8.1 Hz, J = 1.0 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 159.8, 136.8, 131.1, 129.4, 128.5, 128.1, 128.1, 127.2, 126.9, 126.5, 123.2, and 113.9. HRMS (ESI) m/z: [M + H]+ calculated for C12H7N3S, 226.0434; observed, 266.0433.
3.9.5. N-(benzo[4,5]thiazolo[2,3-c][1,2,4]triazol-6-Yl)acetamide (6e)
The title compound was prepared according to general procedure 4 on a 354 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.05) gave the product as a light yellow powder in 82% (190 mg, 0.82 mmol) yield; m.p. 313–315 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 10.43 (s, 1H), 9.69 (s, 1H), 8.56 (d, J = 1.8 Hz, 1H), 7.93 (d, J = 8.8 Hz, 1H), 7.44 (dd, J = 8.8 Hz, J = 2.0 Hz, 1H), and 2.11 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 168.8, 160.4, 151.4, 138.4, 136.7, 129.1, 125.5, 117.9, 105.3, and 24.0. HRMS (ESI) m/z: [M + H]+ calculated for C10H8N4OS, 233.0492; observed, 233.0492.
3.9.6. benzo[4,5]thiazolo[2,3-c][1,2,4]triazol-6-Amine (6f)
The title compound was prepared according to general procedure 4 on a 156 mg (0.50 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.10) gave the product as a light yellow powder in 60% (114 mg, 0.30 mmol) yield; m.p. 220–222 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.51 (s, 1H), 7.57 (d, J = 8.7 Hz, 1H), 7.15 (d, J = 2.1 Hz, 1H), 6.71 (dd, J = 8.7 Hz, J = 2.1 Hz, 1H), and 5.68 (s, 2H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 155.2, 148.7, 136.3, 129.8, 125.5, 115.7, 133.7, and 98.9. HRMS (ESI) m/z: [M + H]+ calculated for C8H6N4S, 191.0386; observed, 191.0387.
3.9.7. 6-(4H-1,2,4-Triazol-4-Yl)benzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6f′)
The title compound was prepared according to general procedure 4 on a 182 mg (0.50 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.09) gave the product as an off-white powder in 90% (375 mg, 0.471 mmol) yield. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.53 (s, 1H), 9.17 (s, 2H), 8.60 (d, J = 2.2 Hz, 1H), 8.27 (d, J = 8.8 Hz, 1H), and 7.86 (dd, J = 8.7 Hz, J = 2.2 Hz, 1H); 13C{1H} NMR was not obtained due to poor solubility. HRMS (ESI) m/z: [M + H]+ calculated for C10H7N6S, 243.0448; observed, 243.0447.
3.9.8. 6-Methoxybenzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6g)
The title compound was prepared according to general procedure 4 on an 82 mg (0.25 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.38) gave the product as an off-figurewhite powder in 81% (42 mg, 0.202 mmol) yield; m.p. 223–224 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.57 (s, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.82 (d, J = 2.4 Hz, 1H), 7.08 (dd, J = 8.9 Hz, J = 2.5 Hz, 1H), and 3.86 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 158.8, 155.4, 136.5, 129.7, 126.0, 122.1, 114.1, 100.4, and 55.9. HRMS (ESI) m/z: [M + H]+ calculated for C9H7N3OS, 206.0383; observed, 206.0382.
3.9.9. benzo[4,5]thiazolo[2,3-c][1,2,4]triazol-6-Ol (6h)
The title compound was prepared according to general procedure 4 on a 313 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 10:1, Rf = 0.05) gave the product as an off-white powder in 84% (159.7 mg, 0.84 mmol) yield; m.p. 217–219 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.57 (s, 1H), 7.78 (d, J = 8.8 Hz, 1H), 7.50 (d, J = 2.3 Hz, 1H), and 6.93 (dd, J = 8.8 Hz, J = 2.3 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 157.0, 155.3, 136.6, 129.8, 126.0, 120.2, 115.0, and 102.0. HRMS (ESI) m/z: [M + H]+ calculated for C8H4N3OS, 192.0226; observed, 192.0224.
3.9.10. [1,2,4]. triazolo[3′,4′:2,3]thiazolo[4,5-b]pyridine (6i)
The title compound was prepared according to general procedure 4 on a 75 mg (0.25 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.55) gave the product as a light orange powder in 76% (34 mg, 0.190 mmol) yield; m.p. 218–220 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.63 (s, 1H), 8.56–8.52 (m, 2H), and 7.56 (dd, J = 8.1 Hz, J = 4.9 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 153.8, 146.2, 141.4, 136.1, 135.1, 127.0, and 122.2. HRMS (ESI) m/z: [M + H]+ calculated for C7H4N4S, 177.0230; observed, 177.0230.
3.9.11. [1,2,4]. triazolo[3′,4′:2,3]thiazolo[5,4-b]pyridine (6j)
The title compound was prepared according to general procedure 4 on a 75 mg (0.25 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.55) gave the product as a light orange powder in 70% (31 mg, 0.176 mmol) yield; m.p. 220–221 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.69 (s, 1H), 8.58 (dd, J = 4.8 Hz, J = 1.4 Hz, 1H), 8.51 (dd, J = 8.2 Hz, J = 1.4 Hz, 1H), and 7.68 (dd, J = 8.2 Hz, J = 4.9 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 153.9, 151.1, 147.6, 137.7, 125.4, 122.7, and 122.1. HRMS (ESI) m/z: [M + H]+ calculated for C7H4N4S, 177.0230; observed, 177.0229.
3.9.12. 6-(Trifluoromethyl)benzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6k)
The title compound was prepared according to general procedure 4 on a 186 mg (0.50 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.38) gave the product as a light yellow powder in 86% (105 mg, 0.430 mmol) yield; m.p. 203–204 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.69 (s, 1H), 8.63 (s, 1H), 8.28 (d, J = 8.5 Hz, 1H), and 7.83 (d, J = 8.4 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 154.8, 137.2, 136.6, 129.3, 127.3 (q, JC–F = 32.6 Hz), 126.8, 123.9 (q, JC–F = 270.6 Hz), 122.9 (q, JC–F = 3.7 Hz), and 112. 3 (q, JC–F = 4.1 Hz). 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = −60.85. HRMS (ESI) m/z: [M + H]+ calculated for C9H4F3N3S, 244.0151; observed, 244.0151.
3.9.13. benzo[4,5]thiazolo[2,3-c][1,2,4]triazole-6-Carbonitrile (6l)
The title compound was prepared according to general procedure 4 on a 322 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 10:1, Rf = 0.38) gave the product as a light yellow powder in 80% (159 mg, 0.795 mmol) yield; m.p. 267–269 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.61 (s, 1H), 8.69 (s, 1H), 8.30 (d, J = 8.4 Hz, 1H), and 7.95 (d, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 154.7, 137.8, 137.1, 129.6, 129.3, 126.7, 118.6, 118.1, and 108.9. HRMS (ESI) m/z: [M + H]+ calculated for C9H4N4S, 201.0229; observed, 201.0230.
3.9.14. benzo[4,5]thiazolo[2,3-c][1,2,4]triazole-6-Carboxylic acid (6m)
The title compound was prepared according to general procedure 4 on a 341 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.38) gave the product as a white powder in 85% (186 mg, 0.847 mmol) yield; m.p. > 360 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.78 (s, 1H), 8.74 (d, J = 1.1 Hz, 1H), 8.16 (d, J = 8.4 Hz, 1H), and 8.03 (dd, J = 8.5 Hz, J = 1.6 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 167.1, 158.0, 152.9, 138.3, 131.8, 129.1, 125.7, 123.9, and 113.8. HRMS (ESI) m/z: [M + H]+ calculated for C9H5N3O2S, 220.0175; observed, 220.0176.
3.9.15. Ethyl benzo[4,5]thiazolo[2,3-c][1,2,4]triazole-6-Carboxylate (6m′)
The title compound was prepared according to general procedure 4 on a 341 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.38) gave the product as a light yellow powder in 86% (173 mg, 0.865 mmol) yield; m.p. 209–210 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.80 (s, 1H), 8.73 (s, 1H), 8.19 (d, J = 8.5 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 4.39 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), and 1.37 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 164.8, 137.2, 129.3, 128.5, 126.9, 125.7, 115.5, 61.4, and 14.2. HRMS (ESI) m/z: [M + H]+ calculated for C11H9N3O2S, 248.0488; observed, 248.0490.
3.9.16. 6-Bromobenzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6n)
The title compound was prepared according to general procedure 4 on a 118 mg (0.50 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.66) gave the product as an off-white powder in 88% (112 mg, 0.442 mmol) yield; m.p. 240–243 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.59 (s, 1H), 8.47 (s, 1H), 8.01 (d, J = 8.4 Hz, 1H), and 8.67 (d, J = 8.3 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 154.8, 136.9, 131.1, 130.0, 129.2, 127.2, 119.2, and 118.0. HRMS (ESI) m/z: [M + H]+ calculated for C8H4BrN3S, 253.9382; observed, 253.9381.
3.9.17. 7-Bromobenzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6o)
The title compound was prepared twice according to general procedure 4 on a 376 mg (1.0 mmol) scale and a 3.76 g (10 mmol) scale. The title compound was triturated with acetone to afford an off-white powder in 90% (229 mg, 0.90 mmol) and 92% (2.34 g, 9.2 mmol) yield; m.p. 273–275 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.65 (s, 1H), 8.35 (d, J = 1.9 Hz, 1H), 8.08 (t, J = 8.6 Hz, 1H), and 7.79 (dd, J = 8.6 Hz, J = 2.0 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 154.5, 134.0, 133.8, 129.9, 128.4, 127.8, 118.3, and 116.5. HRMS (ESI) m/z: [M + H]+ calculated for C8H4BrN3S, 253.9382; observed, 253.9381.
3.9.18. 6-Chlorobenzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6p)
The title compound was prepared according to general procedure 4 on a 166 mg (0.50 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.38) gave the product as an off-white powder in 91% (99 mg, 0.457 mmol) yield; m.p. 229–231 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.59 (s, 1H), 8.36 (d, J = 1.8 Hz, 1H), 8.08 (d, J = 8.6 Hz, 1H), and 7.56 (dd, J = 8.7 Hz, J = 2.0 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 154.7, 137.4, 131.8, 131.0, 130.3, 127.4, 127.0, and 115.7. HRMS (ESI) m/z: [M + H]+ calculated for C8H4ClN3S, 209.9887; observed, 209.9887.
3.9.19. 8-Chlorobenzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6q)
The title compound was prepared according to general procedure 4 on a 166 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.36) gave the product as an off-white powder in 90% (93.8 mg, 0.448 mmol) yield; m.p. 249–251 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.68 (s, 1H), 8.14–8.10 (m, 1H), and 7.66–7.64 (m, 2H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 153.0, 137.4, 130.8, 130.2, 128.6, 127.7, 126.3, and 113.8. HRMS (ESI) m/z: [M + H]+ calculated for C8H4ClN3S, 209.9887; observed, 209.9886.
3.9.20. 8-Fluorobenzo[4,5]thiazolo[2,3-c][1,2,4]triazole (6r)
The title compound was prepared according to general procedure 4 on a 315 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.40) gave the product as a light yellow powder in 98% (188 mg, 0.975 mmol) yield; m.p. 211–212 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.69 (s, 1H), 7.99 (d, J = 8.1 Hz, 1H), 7.69–7.63 (m, 1H), and 7.46 (t, J = 7.6 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 156.5 (d, JC–F = 244.7 Hz), 153.9, 137.3, 130.8 (d, JC–F = 6.8 Hz), 129.0 (d, JC–F = 7.8 Hz), 118.1 (d, JC–F = 22.3 Hz), 112.9 (d, JC–F = 18.1 Hz), and 111.4 (d, JC–F = 3.4 Hz). 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = −114.97. HRMS (ESI) m/z: [M + H]+ calculated for C8H4FN3S, 194.0183; observed, 194.0182.
3.9.21. 7-Fluorobenzo[[1,3]4,5]thiazolo[2,3-c][1,2,4]triazole (6s)
The title compound was prepared according to general procedure 4 on a 315 mg (1.00 mmol) scale. Purification by column chromatography (silica gel, 1. dichloromethane, and 2. dichloromethane/methanol 5:1, Rf = 0.38) gave the product as an off-white powder in 98% (188 mg, 0.976 mmol) yield; m.p. 255–258 °C. 1H NMR (400 MHz, DMSO-d6, 298 K): δ = 9.59 (s, 1H), 8.14 (dd, J = 8.9 Hz, J = 4.6 Hz, 1H), 8.01 (dd, J = 8.8 Hz, J = 2.6 Hz, 1H), and 7.47 (td, J = 9.0 Hz, J = 2.6 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ = 159.7 (d, JC–F = 242.0 Hz), 154.6, 136.8, 133.3 (q, JC–F = 11.0 Hz), 125.9 (q, JC–F = 2.1 Hz), 116.1 (d, JC–F = 9,4 Hz), 114.5 (d, JC–F = 24.7 Hz), and 112.5 (d, JC–F = 28.2 Hz). 19F{1H} NMR (376 MHz, DMSO-d6, 298 K, referenced to C6H5F): δ = −114.27. HRMS (ESI) m/z: [M + H]+ calculated for C8H4FN3S, 194.0183; observed, 194.0183.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}