
In Silico and Ex Vivo Studies on the Spasmolytic Activities of Fenchone Using Isolated Guinea Pig Trachea




Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Ethics Statement
2.3. Animals
2.4. Tissue Preparation
2.5. Determination of the Possible Mechanisms of Action
2.6. Molecular Docking Studies
2.7. ADMET Studies
2.8. Statistical Analysis
3. Results
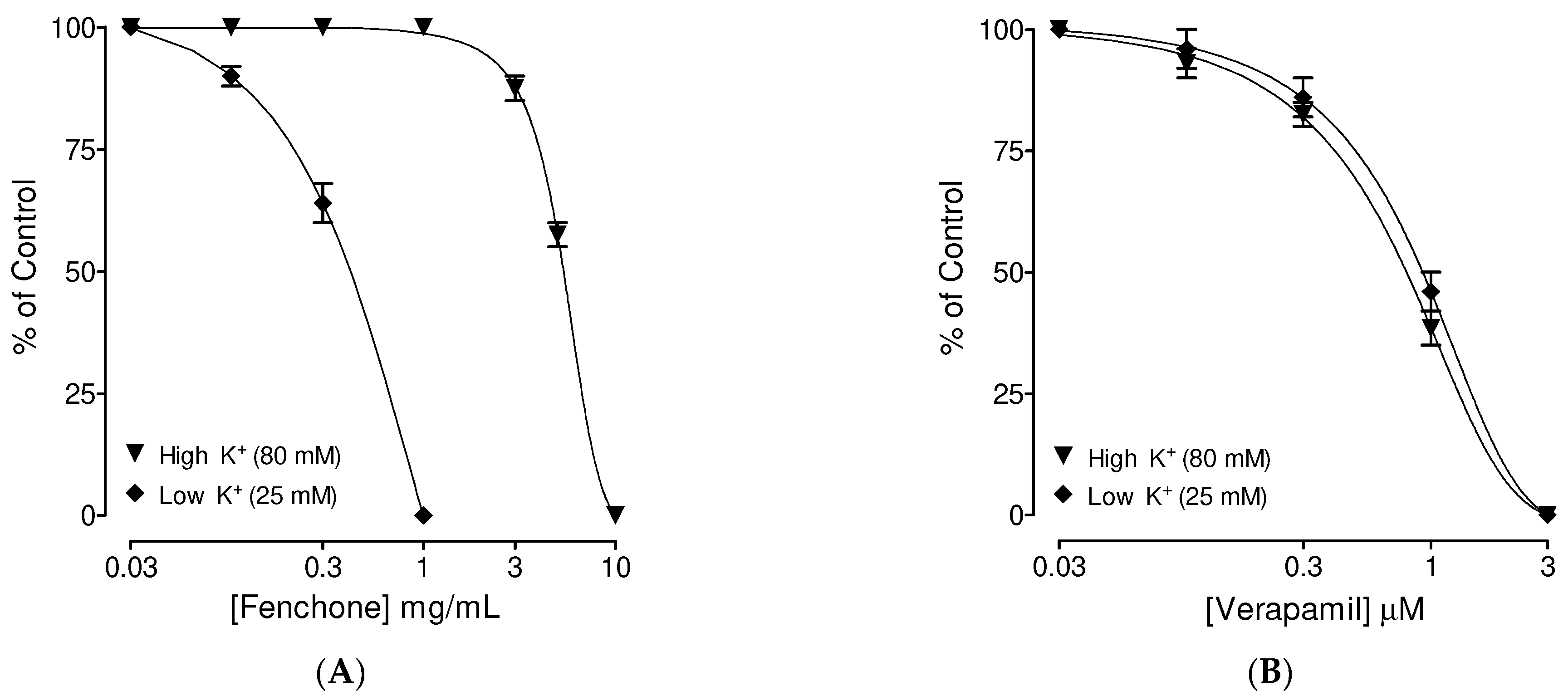
3.1. Preliminary Screening of Fenchone for Spasmolytic Effect
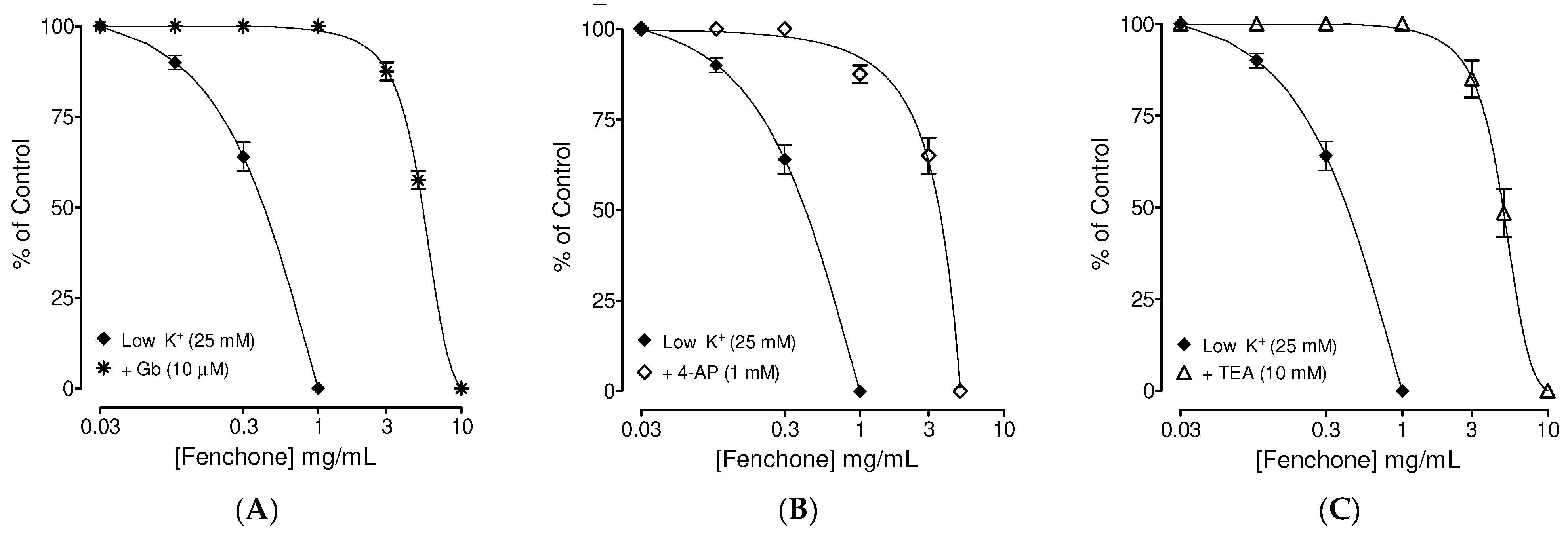
3.2. Determination of the Possible Effect of Fenchone on Activation of Subtype of K+ Channels
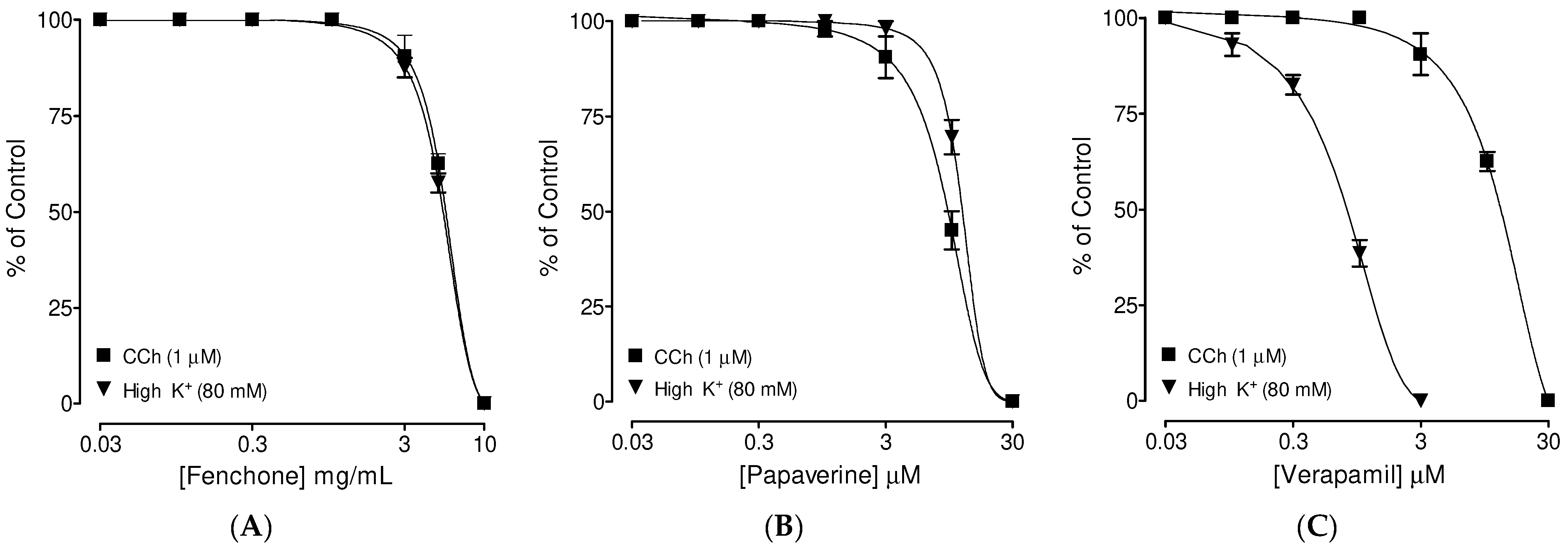
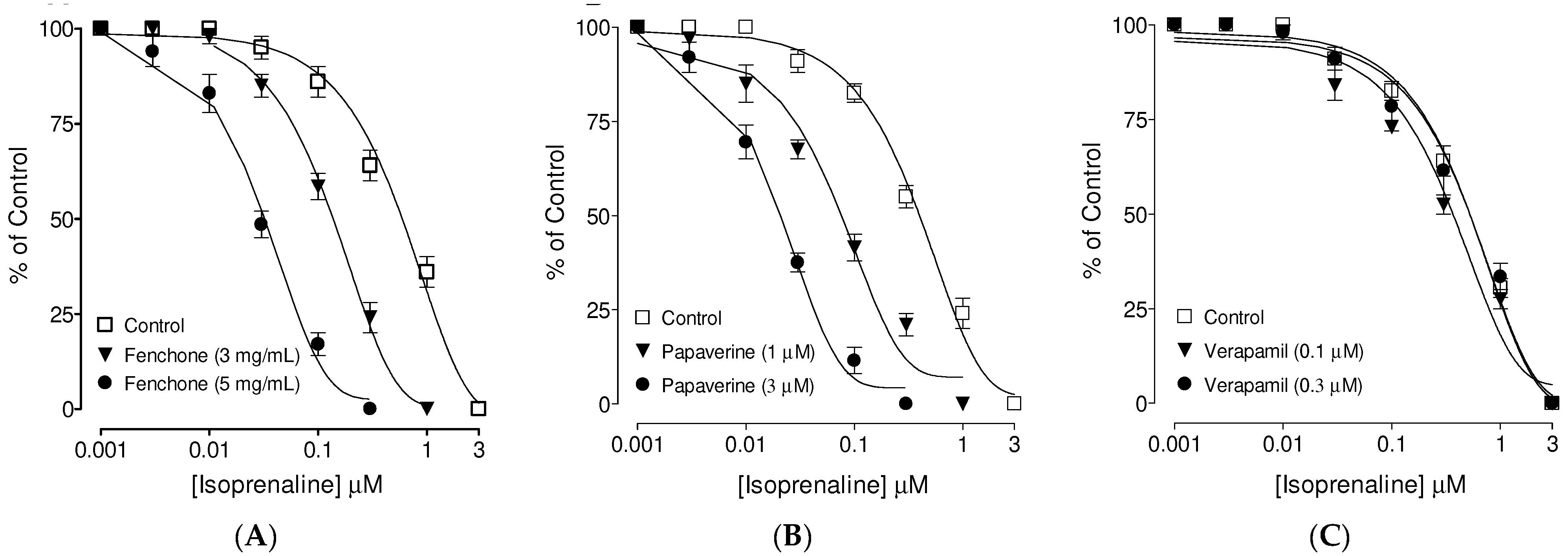
3.3. Confirmation of PDE Inhibitory-like Spasmolytic Effects of Fenchone
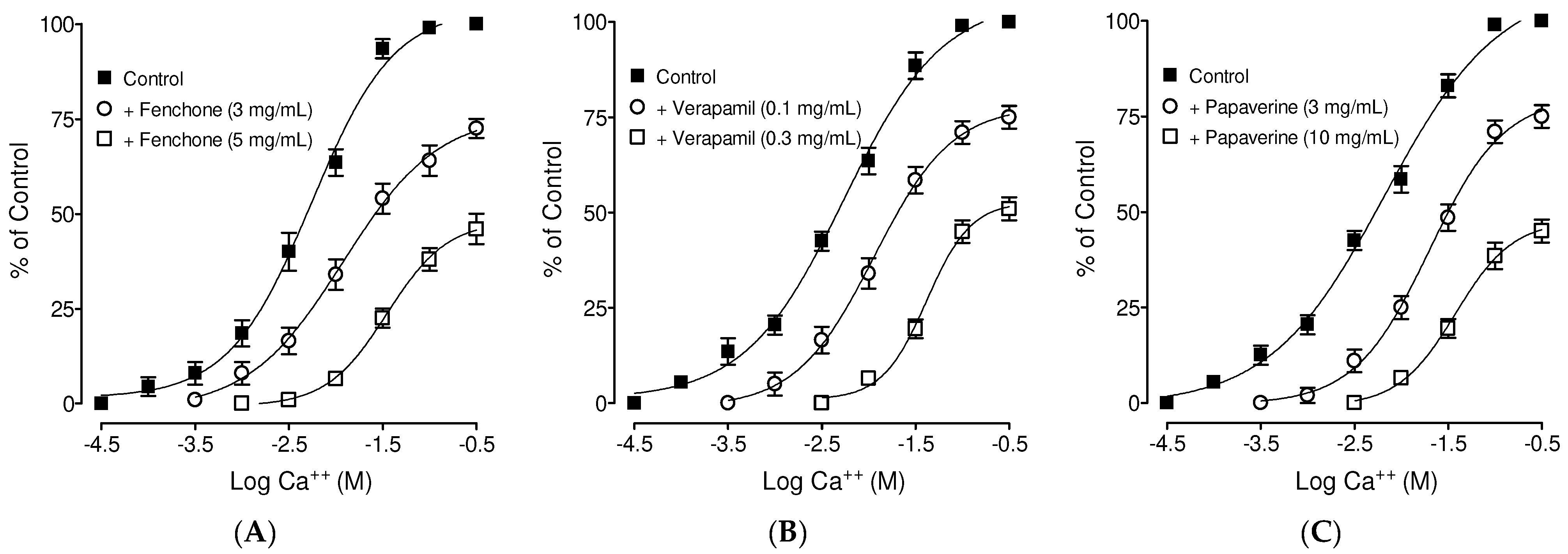
3.4. Confirmation of Ca++ Channel Inhibitory-like Spasmolytic Effects of Fenchone
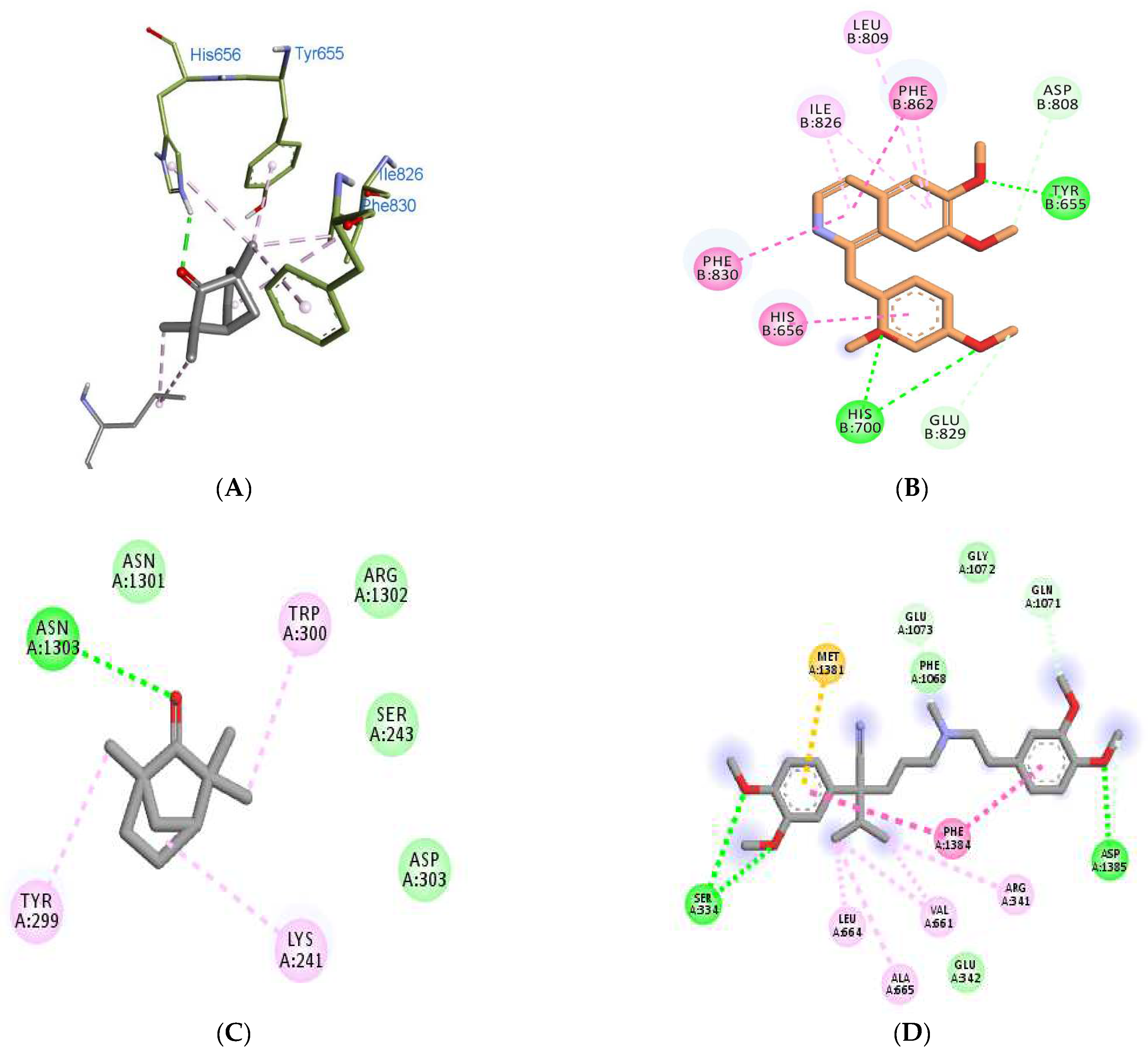
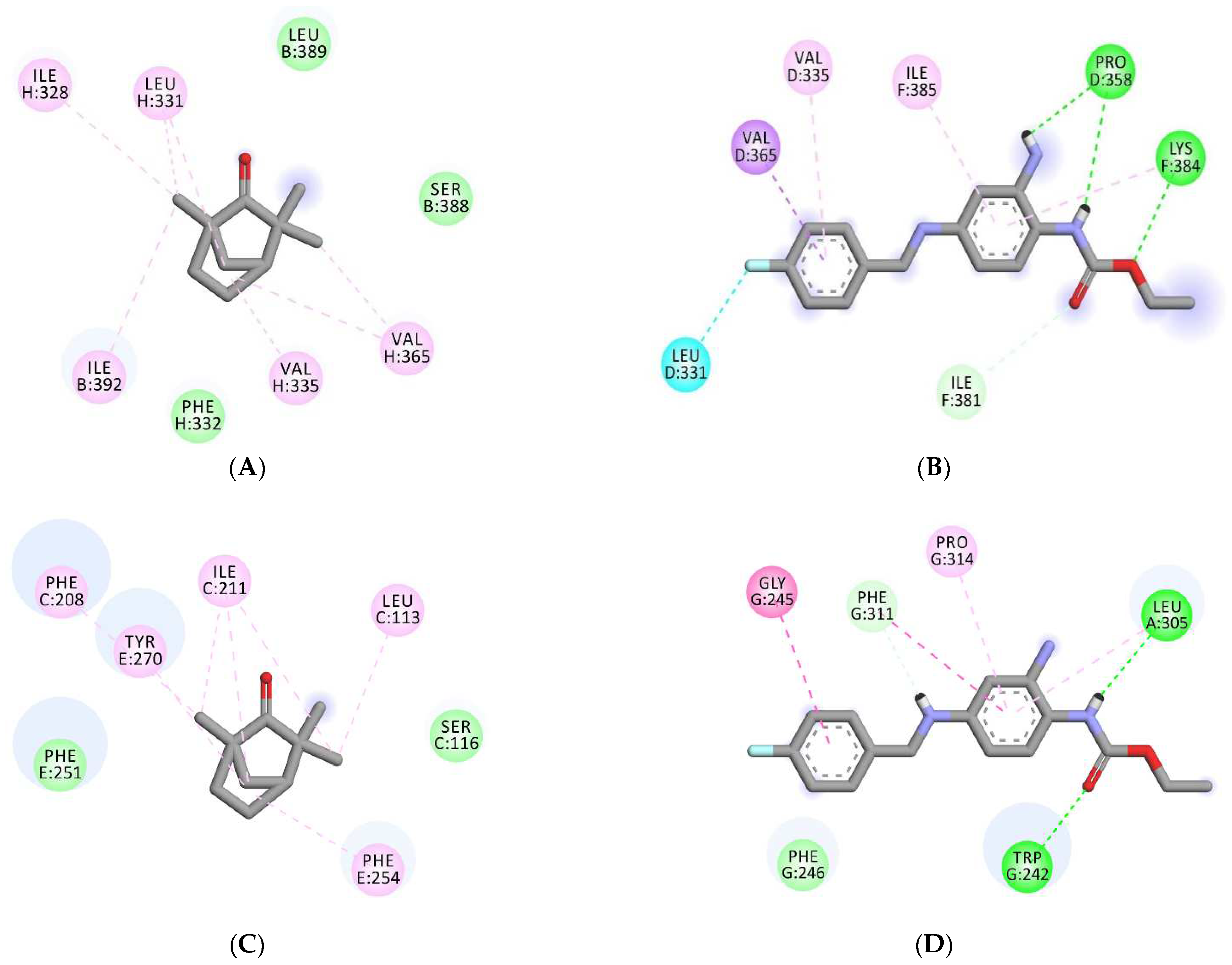
3.5. Molecular Docking Analyses
3.6. ADMET Profiling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Barnes, P.J. New therapies for chronic obstructive pulmonary disease. Med. Princ. Pract. 2010, 19, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Page, C.P.; Calzetta, L.; Matera, M.G. Pharmacology and therapeutics of bronchodilators. Pharmacol. Rev. 2012, 64, 450–504. [Google Scholar] [CrossRef] [PubMed]
- Kasirajan, B.; Maruthamuthu, R.; Gopalakrishnan, V.; Arumugam, K.; Asirvatham, H.; Murali, V.; Mohandass, R.; Bhaskar, A. A database for medicinal plants used in treatment of asthma. Bioinformation 2007, 2, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Canning, B.J. Animal model of asthma and chronic obstructive pulmonary diseases. Pulm. Pharmacol. Therapeut. 2008, 21, 695. [Google Scholar] [CrossRef]
- Gandevia, B. Historical review of the use of parasympatholytic agents in the treatment of respiratory disorders. Postgrad. Med. J. 1975, 51 (Suppl. 7), 13–20. [Google Scholar]
- Barnes, P.J.; Hansel, T.T. Prospects for new drugs for chronic obstructive pulmonary disease. Lancet 2004, 364, 985–996. [Google Scholar] [CrossRef]
- Wright, G.D. Unlocking the potential of natural products in drug discovery. Microb. Biotechnol. 2019, 12, 55–57. [Google Scholar] [CrossRef]
- Shen, B. A New Golden Age of Natural Products Drug Discovery. Cell 2015, 163, 1297–1300. [Google Scholar]
- Kim, T.; Song, B.; Cho, K.S.; Lee, I.S. Therapeutic Potential of Volatile Terpenes and Terpenoids from Forests for Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 2187. [Google Scholar] [CrossRef]
- Chi, T.; Ji, X.; Xia, M.; Rong, Y.; Qiu, F.Z.L. Effect of six extractions from Wuhu decoction on isolated tracheal smooth muscle in guinea pig. Zhong Guo Shi Yan Fang Ji Xue Za Zhi 2009, 15, 52–55. [Google Scholar]
- Pessoa, M.L.S.; Silva, L.M.O.; Araruna, M.E.C.; Serafim, C.A.L.; Júnior, E.B.A.; Silva, A.O.; Pessoa, M.M.B.; Neto, H.D.; Lima, E.O.; Batista, L.M. Antifungal activity and antidiarrheal activity via antimotility mechanisms of (-)-fenchone in experimental models. World J. Gastroenterol. 2020, 26, 6795–6809. [Google Scholar] [CrossRef] [PubMed]
- Küpeli Akkol, E.; İlhan, M.; Ayşe Demirel, M.; Keleş, H.; Tümen, I.; Süntar, İ. Thuja occidentalis L. and its active compound, α-thujone: Promising effects in the treatment of polycystic ovary syndrome without inducing osteoporosis. J. Ethnopharmacol. 2015, 168, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Algieri, F.; Rodriguez-Nogales, A.; Vezza, T.; Garrido-Mesa, J.; Garrido-Mesa, N.; Utrilla, M.P.; González-Tejero, M.R.; Casares-Porcel, M.; Molero-Mesa, J.; del Mar Contreras, M.; et al. Anti-inflammatory activity of hydroalcoholic extracts of Lavandula dentata L. and Lavandula stoechas L. J. Ethnopharmacol. 2016, 190, 142–158. [Google Scholar] [CrossRef] [PubMed]
- NRC (National Research Council). Guide for the Care and Use of Laboratory Animals; National Academy Press: Washington, WA, USA, 1996; pp. 1–7. [Google Scholar]
- Venkatasamy, R.; Spina, D. Novel relaxant effects of RPL554 on guinea pig tracheal smooth muscle contractility. Br. J. Pharmacol. 2016, 173, 2335–2351. [Google Scholar] [CrossRef] [PubMed]
- Holroyde, M. The influence of epithelium on the responsiveness of guinea-pig isolated trachea. Br. J. Pharmacol. 1986, 87, 501–507. [Google Scholar] [CrossRef]
- Van-Rossum, J.M. Cumulative dose response curves. II. Technique for making of dose-response curves in isolated organs and the evaluation of drug parameters. Arch. Int. Pharmacodyn. Therap. 1963, 143, 299–330. [Google Scholar]
- Farre, A.J.; Columbo, M.; Fort, M.; Gutierrez, B. Differential effects of various Ca++ antagonists. Gen. Pharmacol. 1991, 22, 177–181. [Google Scholar] [CrossRef]
- Cook, N.S. The pharmacology of potassium channels and their therapeutic potential. Trend Pharmacol. Sci. 1988, 9, 21–28. [Google Scholar] [CrossRef]
- Satake, N.; Shibata, M.; Shibata, S. The inhibitory effects of iberiotoxin and 4-aminopyridine on the relaxation induced by beta1- and beta2-adrenoceptor activation in rat aortic rings. Br. J. Pharmacol. 1996, 119, 505–510. [Google Scholar] [CrossRef]
- Frank, H.; Puschmann, A.; Schusdziarra, V.; Allescher, H.D. Functional evidence for a glibenclamide-sensitive K+ channel in rat ileal smooth muscle. Eur. J. Pharmacol. 1994, 271, 379–386. [Google Scholar] [CrossRef]
- Gilani, A.H.; Khan, A.U.; Jabeen, Q.; Subhan, F.; Ghafar, G. Antispasmodic and blood pressure lowering effects of Valeriana wallichii are mediated through K+ channel activation. J. Ethnopharmacol. 2005, 100, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Bolton, T.B. Mechanism of action of transmitters and other substances on smooth muscles. Physiol. Rev. 1979, 59, 606–718. [Google Scholar] [CrossRef] [PubMed]
- Karaki, H.; Ozaki, H.; Hori, M.; Mitsui-Saito, M.; Amano, K.I.; Harada, K.I.; Miyamoto, S.; Nakazawa, H.; Won, K.J.; Sato, K. Calcium movements, distribution, and functions in smooth muscle. Pharmacol. Rev. 1997, 49, 157–230. [Google Scholar] [PubMed]
- Godfraind, T.; Miller, R.; Wibo, M. Calcium antagonism and calcium entry blockade. Pharmacol. Rev. 1986, 38, 321–416. [Google Scholar]
- Rehman, N.U.; Ansari, M.N.; Hailea, T.; Karim, A.; Abujheisha, K.Y.; Ahamad, S.R.; Imam, F. Possible tracheal relaxant and antimicrobial effects of the essential oil of Ethiopian thyme specie (Thymus serrulatus Hoschst. Ex Benth.): A multiple mechanistic approach. Front. Pharmacol. 2021, 12, 615228. [Google Scholar] [CrossRef]
- Hamilton, T.C.; Weir, S.W.; Weston, A.H. Comparison of the effects of BRL 34915 and verapamil on electrical and mechanical activity in rat portal vein. Br. J. Pharmacol. 1986, 88, 103–111. [Google Scholar] [CrossRef]
- Blattner, R.; Classen, H.G.; Dehnert, H.; Doring, H.J. Experiments on Isolated Smooth Muscle Preparations; Hugo Sachs Elektronik KG: March, Germany, 1978; pp. 53–71. [Google Scholar]
- Lorenz, K.L.; Wells, J.N. Potentiation of the effects of sodium nitroprusside and isoproterenol by selective phosphodiesterase inhibitors. Mol. Pharmacol. 1983, 23, 424–430. [Google Scholar]
- Lis-Balchin, M.; Patel, J.; Hart, S. Studies on the mode of action of essential oils of scented-leaf Pelargonium (Geraniaceae). Phytother. Res. 1998, 12, 215–217. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment; Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Rehman, N.U.; Ansari, M.N.; Samad, A. In Silico, Ex Vivo and In Vivo Studies of Roflumilast as a Potential Antidiarrheal and Antispasmodic agent: Inhibition of the PDE-4 Enzyme and Voltage-gated Ca++ ion Channels. Molecules 2020, 25, 1008. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Gilani, A.H.; Khan, A.U.; Ghayur, M.N.; Ali, S.F.; Herzig, J.W. Antispasmodic effects of Rooibos tea (Aspalathus linearis) is mediated predominantly through K+-channel activation. Basic Clin. Pharmacol. Toxicol. 2006, 99, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Rehman, N.U.; Khan, A.U.; Alkharfy, K.M.; Gilani, A.H. Pharmacological Basis for the Medicinal Use of Lepidium sativum in Airways Disorders. Evidence-based complementary and alternative medicine. eCAM 2012, 2012, 596524. [Google Scholar] [PubMed]
- Bashir, S.; Memon, R.; Gilani, A.H. Antispasmodic and antidiarrheal activities of Valeriana hardwickii rhizome are putatively mediated through calcium channel blockade. Evid. Based Complement. Alternat. Med. 2011, 2011, 304960. [Google Scholar] [CrossRef]
- Shah, A.J.; Zaidi, M.A.; Hamidullah, S.H.; Gilani, A.H. Antidiarrheal and antispasmodic activities of Vincetoxicum stocksii are mediated through calcium channel blockade. Bangl. J. Pharmacol. 2011, 6, 46–50. [Google Scholar] [CrossRef][Green Version]
- Fleckenstein, A. Specific pharmacology of Ca++ in myocardium, cardiac pacemakers and vascular smooth muscle. Rev. Pharmacol. Toxicol. 1977, 17, 149–166. [Google Scholar] [CrossRef]
- Robertson, D.W.; Steinberg, M.I. Potassium channel modulators: Scientific applications and therapeutic promise. J. Med. Chem. 1990, 33, 1529–1541. [Google Scholar] [CrossRef]
- Cunha, J.; Campestrini, F.; Calixto, J.; Scremin, A.; Paulino, N. The mechanism of gentisic acid-induced relaxation of the guinea pig isolated trachea: The role of potassium channels and vasoactive intestinal peptide receptors. Braz. J. Med. Biol. Res. 2001, 34, 381–388. [Google Scholar] [CrossRef]
- Radulovic, M.; Anand, P.; Korsten, M.A.; Gong, B. Targeting Ion Channels: An Important Therapeutic Implication in Gastrointestinal Dysmotility in Patients with Spinal Cord Injury. J. Neurogastroenterol. Motil. 2015, 21, 494–502. [Google Scholar] [CrossRef]
- Groneberg, D.; Voussen, B.; Friebe, A. Integrative Control of Gastrointestinal Motility by Nitric Oxide. Curr. Med. Chem. 2016, 23, 2715–2735. [Google Scholar] [CrossRef]
- Quest, U. Potassium channel openers: Pharmacological and clinical aspects. Fund. Clin. Pharmacol. 1992, 6, 279–293. [Google Scholar] [CrossRef]
- Lenz, T.; Wagner, G. Potential role of potassium channel openers for the treatment of cardiovascular disease. In Hypertension: Pathophysiology, Diagnosis and Management; Laragh, J.H., Brenner, B.M., Eds.; Raven Press: New York, NY, USA, 1995; pp. 2953–2968. [Google Scholar]
- Rang, H.P.; Dale, M.M.; Ritter, J.M. Pharmacology, 4th ed.; Churchill Livingstone: New York, NY, USA, 1999; pp. 289–290. [Google Scholar]
- Smith, B.V.; Spina, D.; Page, C.P. Phosphodiesterase inhibitors. Br. J. Pharmacol. 2006, 47, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.A.E.; Naline, E.; Bakdach, H.; Advenier, C. β3-adrenoceptor agonists, BRL 37344 and SR 58611A, do not induce relaxation of human, sheep and Guinea-pig airway smooth muscle in vitro. Eur. Respir. J. 1994, 7, 1610–1615. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.T.; Liao, G.; Bi, X.; Oka, T.; Tamura, S.; Baudry, M. The PDE10A inhibitor, papaverine, differentially activates ERK in male and female rat striatal slices. Neuropharmacology 2011, 61, 1275–1281. [Google Scholar] [CrossRef]
- Van Den Brink, F.G. The model of functional interaction. I. Development and first check of a new model of functional synergism and antagonism. Eur. J. Pharmacol. 1973, 22, 270–278. [Google Scholar] [CrossRef]
- Nawrath, H. Action potential, membrane currents and force of contraction in cat ventricular heart muscle treated with papaverine. J. Pharmacol. Exp. Ther. 1981, 218, 544–549. [Google Scholar]
- Nicholas, J.G. Anticholinergic agents in asthma and COPD. Eur. J. Pharmacol. 2006, 533, 36–39. [Google Scholar]
- Roden, D.M. Antiarrhythmic drugs. In The Pharmacological Basis of Therapeutics, 11th ed.; Brunton, L.L., Lazo, J.S., Parker, K.L., Eds.; McGraw-Hill: New York, NY, USA, 2006; pp. 899–932. [Google Scholar]
- Gilani, A.H.; Rahman, A. Trends in Ethnopharmacology. J. Ethnopharmacol. 2005, 100, 43–49. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Scores | Interacting Residues | ||||
|---|---|---|---|---|---|
| Receptors/ Ligands | Fenchone | Co-Crystallized | Standard | Fenchone | Standard (PPV) |
| 3ITU | −5.2 | −6.8 | −8.3 (PPV) | His656, Tyr655, Ile826, Phe830, Leu770 | Tyr655, His656, His700, Asp808, Leu809, Ile826, Glu829, Phe830, Phe862 |
| 4NPW | −5.1 | −8.5 | −8.2 (PPV) | His223, His267, Leu292, Met336 | His223, His267, Gly269, Asp370, Gln395, Leu388 |
| 5LAQ | −5.3 | −8.3 | −8.4 (PPV) | His450, Asn455, Leu475, Asp447, Asp564, Thr517, Glu476 | Asn567, Leu565, Tyr405, Ser454, His450, His406, Gln615 |
| 6JPA | −6.3 | −7.6 | −7.8 (VPML) | Asn1303, Asn1301, Arg1302, Lys241, Ser243, Tyr299, Trp300, Asp303, | Glu 342, Asp1385, Ser334, Met1381, Phe1068, Gln1071, Gly1072, Glu1073. |
| Binding Scores | Interacting Residues | |||
|---|---|---|---|---|
| Receptors/ Ligands | Fenchone | Retigabine | Fenchone | Standard (RTG) |
| 7VNP | −4.6 | −7.3 | Gly E-319, Gly E-316, Gly G-316, Ser E-320, Ser G-320, Ser C-320, Gly C-316, Ser A-320, Gly A-319, Gly A-316, | Pro D-358, LeuD-331, Val D-335, Val D-365, Ile-F381, Lys F-384, Ile F-385, |
| 6EBM | −5.7 | −7.6 | Ser B-388, Leu B-389, Ile B-392, Ile H-328, Leu H-331, Phe H-332, Val H-335, Val H-365, | Trp G-242, Gly G-245, Phe G-246, Leu A-305, Phe G-311, Pro G-314, |
| Physicochemical Properties | |
|---|---|
| Formula | C10H16O |
| Molecular weight | 152.23 g/mol |
| Number of heavy atoms | 11 |
| Number of aromatic heavy atoms | 0 |
| Fraction Csp3 | 0.90 |
| Number of rotatable bonds | 0 |
| Number of H-bond acceptors | 1 |
| Number of H-bond donors | 0 |
| Molar Refractivity | 45.64 |
| TPSA | 17.07 Å2 |
| Drug-Likeness | Pass by Pfizer/Veber/Egan |
| Property | Model Name | Predicted Value | Unit |
|---|---|---|---|
| A | Water solubility | −2.668 | Numeric (log mol/L) |
| Caco2 permeability | 1.029 | Numeric (log Papp in 10−6 cm/s) | |
| Intestinal absorption (human) | 98.703 | Numeric (% Absorbed) | |
| Skin Permeability | −2.116 | Numeric (log Kp) | |
| P-glycoprotein substrate | No | Categorical (Yes/No) | |
| P-glycoprotein I inhibitor | No | Categorical (Yes/No) | |
| P-glycoprotein II inhibitor | No | Categorical (Yes/No) | |
| D | VDss (human) | 0.247 | Numeric (log L/kg) |
| Fraction unbound (human) | 0.421 | Numeric (Fu) | |
| BBB permeability | 0.636 | Numeric (log BB) | |
| CNS permeability | −2.067 | Numeric (log PS) | |
| M | CYP2D6 substrate | No | Categorical (Yes/No) |
| CYP3A4 substrate | No | Categorical (Yes/No) | |
| CYP1A2 inhibitor | No | Categorical (Yes/No) | |
| CYP2C19 inhibitor | No | Categorical (Yes/No) | |
| CYP2C9 inhibitor | No | Categorical (Yes/No) | |
| CYP2D6 inhibitor | No | Categorical (Yes/No) | |
| CYP3A4 inhibitor | No | Categorical (Yes/No) | |
| E | Total Clearance | 0.085 | Numeric (log ml/min/kg) |
| Renal OCT2 substrate | No | Categorical (Yes/No) | |
| T | AMES toxicity | No | Categorical (Yes/No) |
| Max. tolerated dose (human) | 0.751 | Numeric (log mg/kg/day) | |
| hERG I inhibitor | No | Categorical (Yes/No) | |
| hERG II inhibitor | No | Categorical (Yes/No) | |
| Oral Rat Acute Toxicity (LD50) | 1.836 | Numeric (mol/kg) | |
| Oral Rat Chronic Toxicity (LOAEL) | 1.982 | Numeric (log mg/kg/day) | |
| Hepatotoxicity | No | Categorical (Yes/No) | |
| Skin Sensitization | Yes | Categorical (Yes/No) | |
| T. Pyriformis toxicity | 0.292 | Numeric (log ug/L) | |
| Minnow toxicity | 1.218 | Numeric (log mM) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, N.U.; Ansari, M.N.; Samad, A.; Ahmad, W. In Silico and Ex Vivo Studies on the Spasmolytic Activities of Fenchone Using Isolated Guinea Pig Trachea. Molecules 2022, 27, 1360. https://doi.org/10.3390/molecules27041360
Rehman NU, Ansari MN, Samad A, Ahmad W. In Silico and Ex Vivo Studies on the Spasmolytic Activities of Fenchone Using Isolated Guinea Pig Trachea. Molecules. 2022; 27(4):1360. https://doi.org/10.3390/molecules27041360
Chicago/Turabian StyleRehman, Najeeb Ur, Mohd Nazam Ansari, Abdul Samad, and Wasim Ahmad. 2022. "In Silico and Ex Vivo Studies on the Spasmolytic Activities of Fenchone Using Isolated Guinea Pig Trachea" Molecules 27, no. 4: 1360. https://doi.org/10.3390/molecules27041360
APA StyleRehman, N. U., Ansari, M. N., Samad, A., & Ahmad, W. (2022). In Silico and Ex Vivo Studies on the Spasmolytic Activities of Fenchone Using Isolated Guinea Pig Trachea. Molecules, 27(4), 1360. https://doi.org/10.3390/molecules27041360