
Kaempferol 3-Rhamnoside on Glutamate Release from Rat Cerebrocortical Nerve Terminals Involves P/Q-Type Ca2+ Channel and Ca2+/Calmodulin-Dependent Protein Kinase II-Dependent Pathway Suppression




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
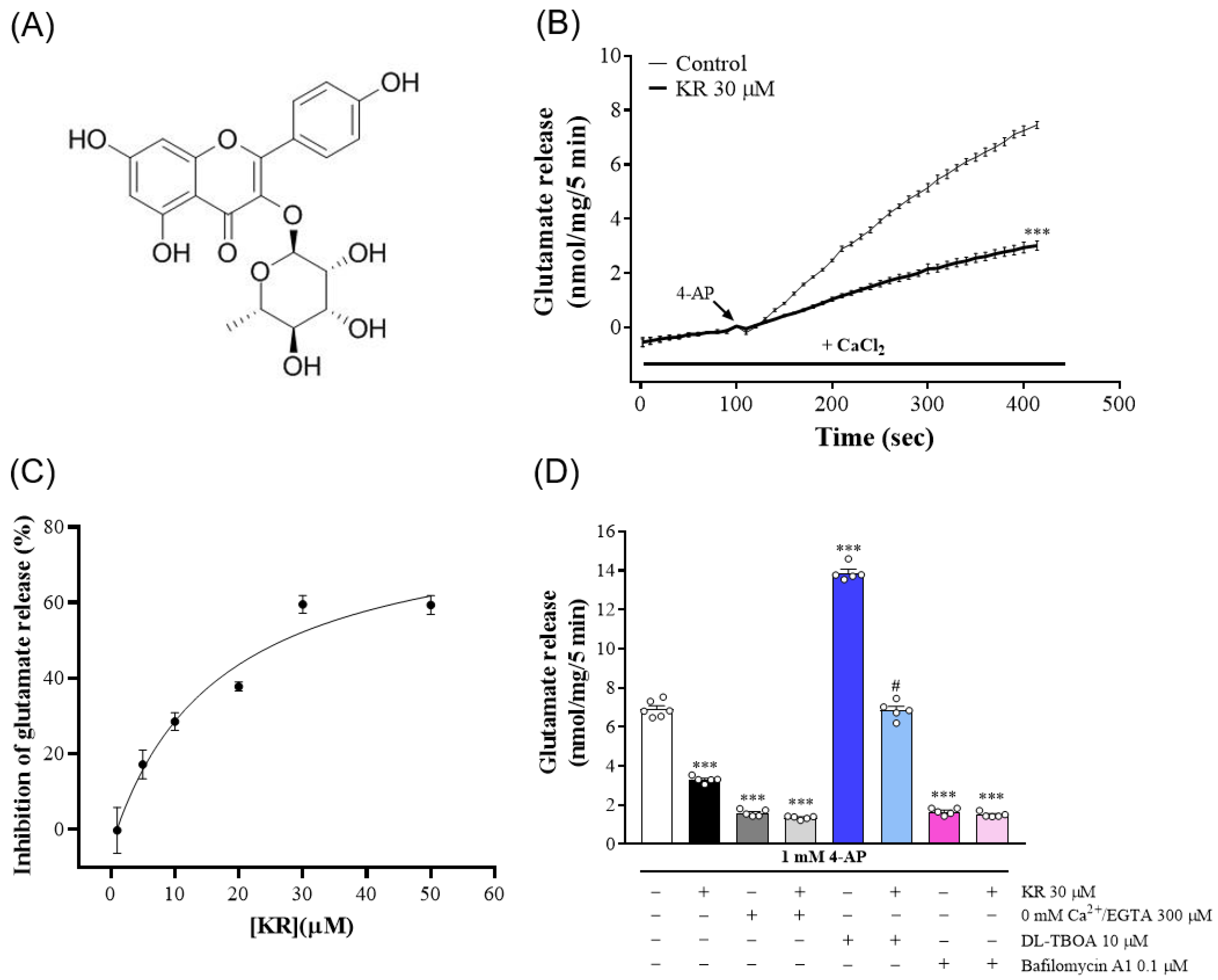
2.1. KR Inhibitsthe Release of Glutamate Evoked by 4-AP atRat Cerebrocortical Nerve Terminals
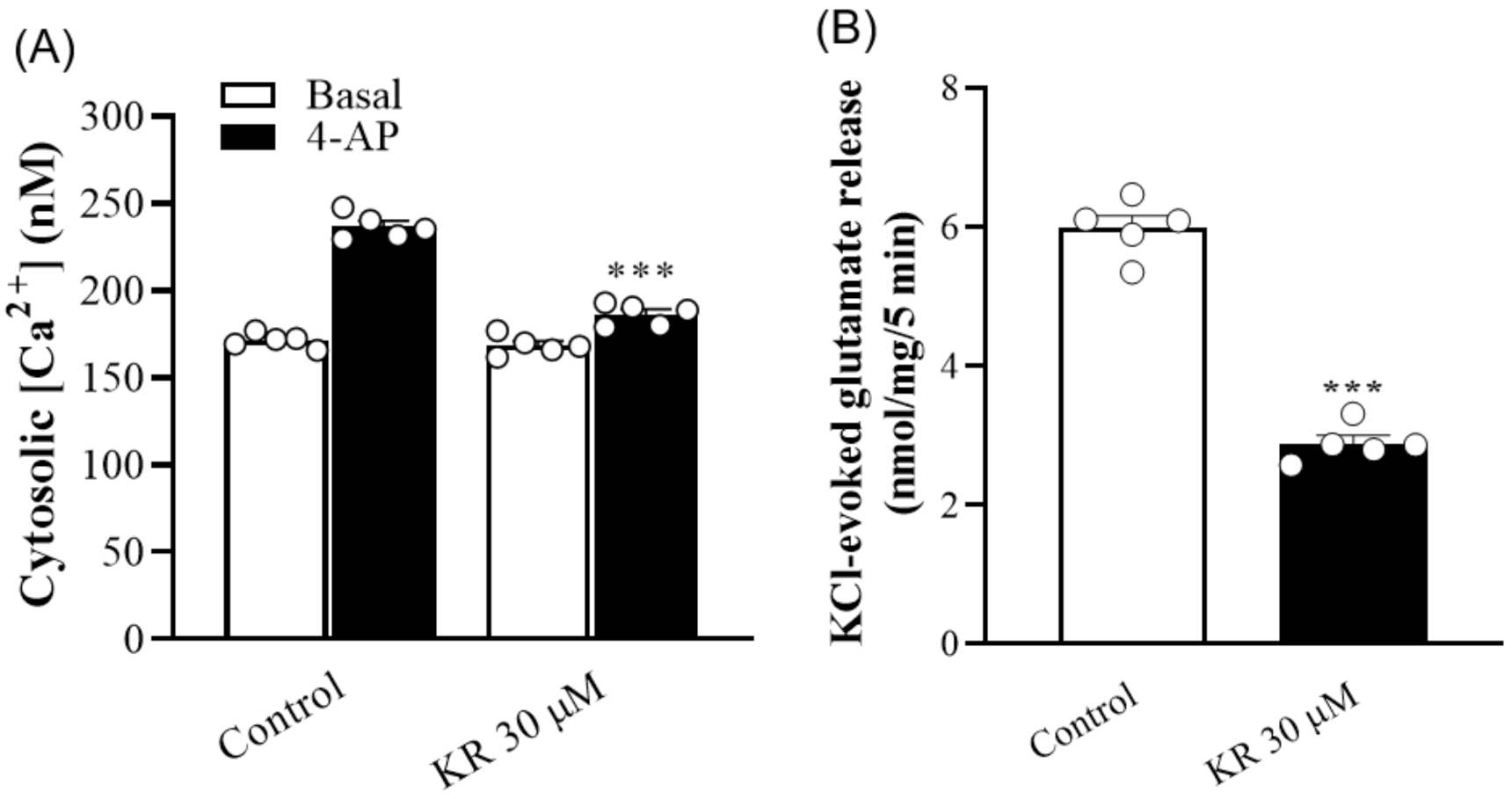
2.2. KR Reduces the 4-AP-Induced Increase in [Ca2+]C
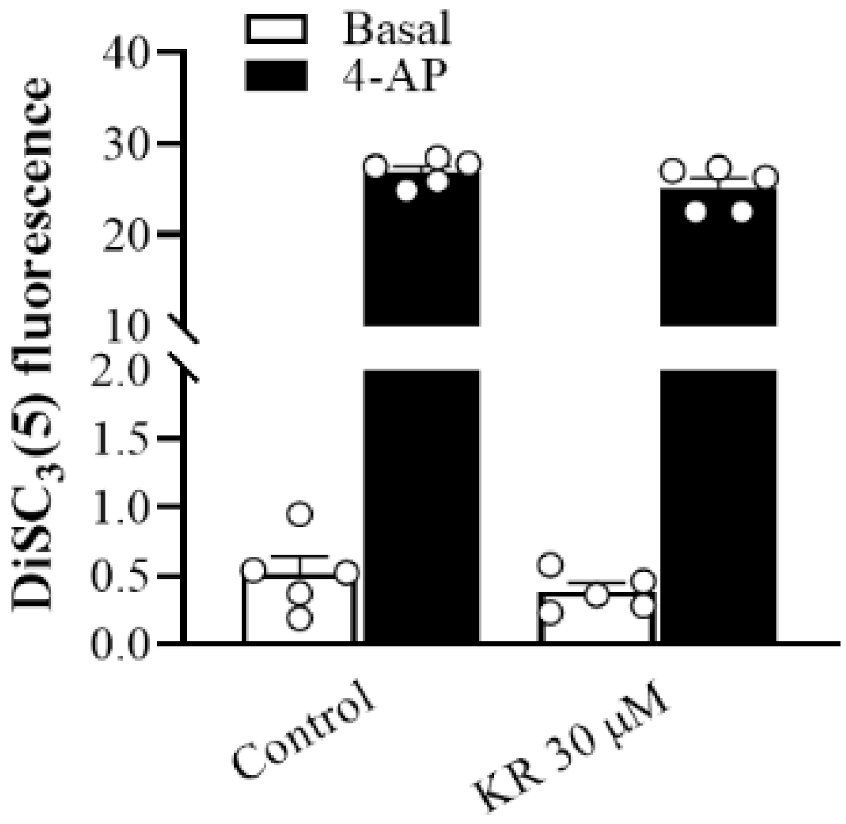
2.3. KR Does Not Affect Synaptosomal Membrane Potential
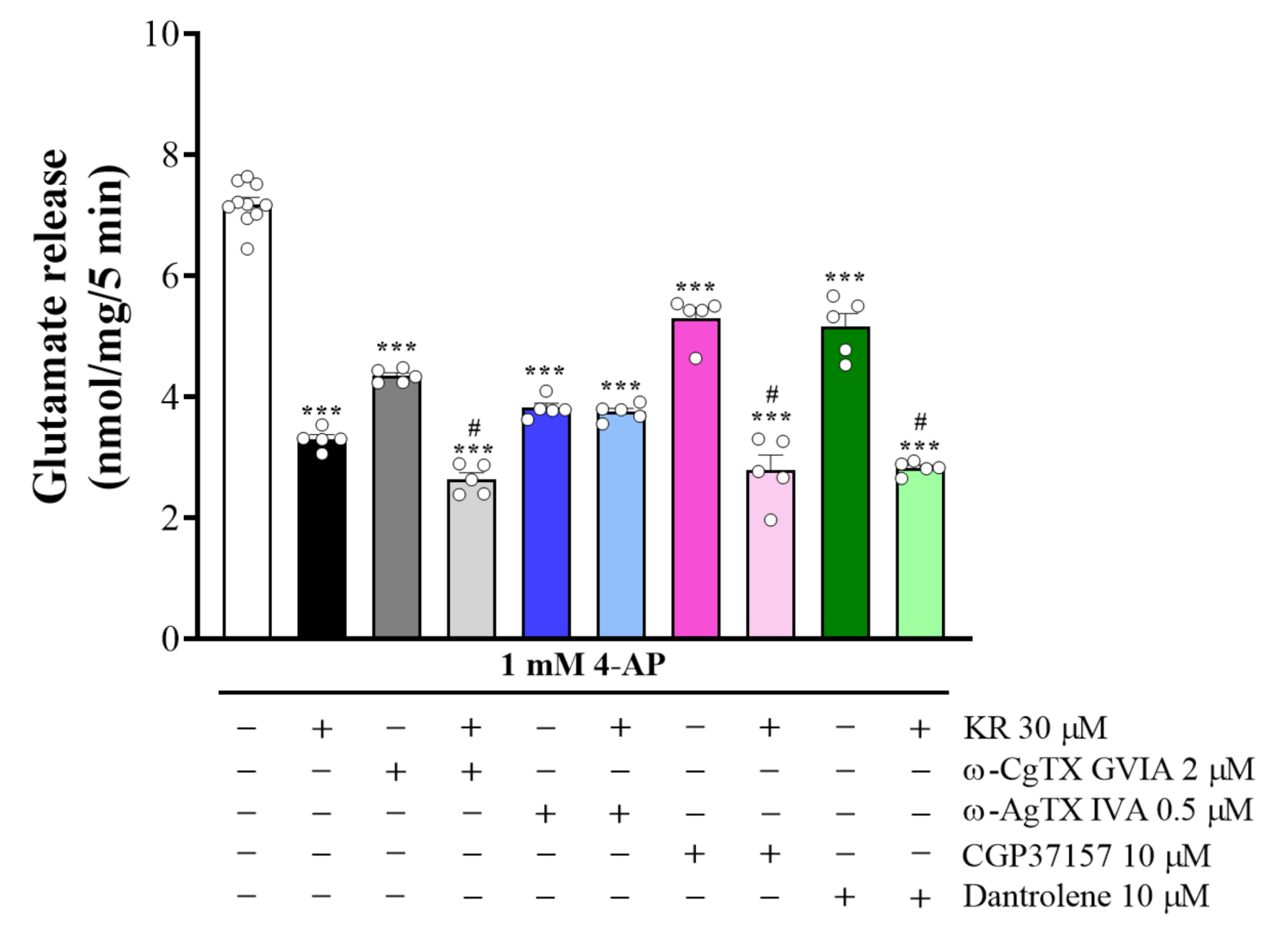
2.4. KR-Mediated Inhibition of 4-AP-Evoked Glutamate Release Is Abolished by P/Q-Type VDCCs Blockade
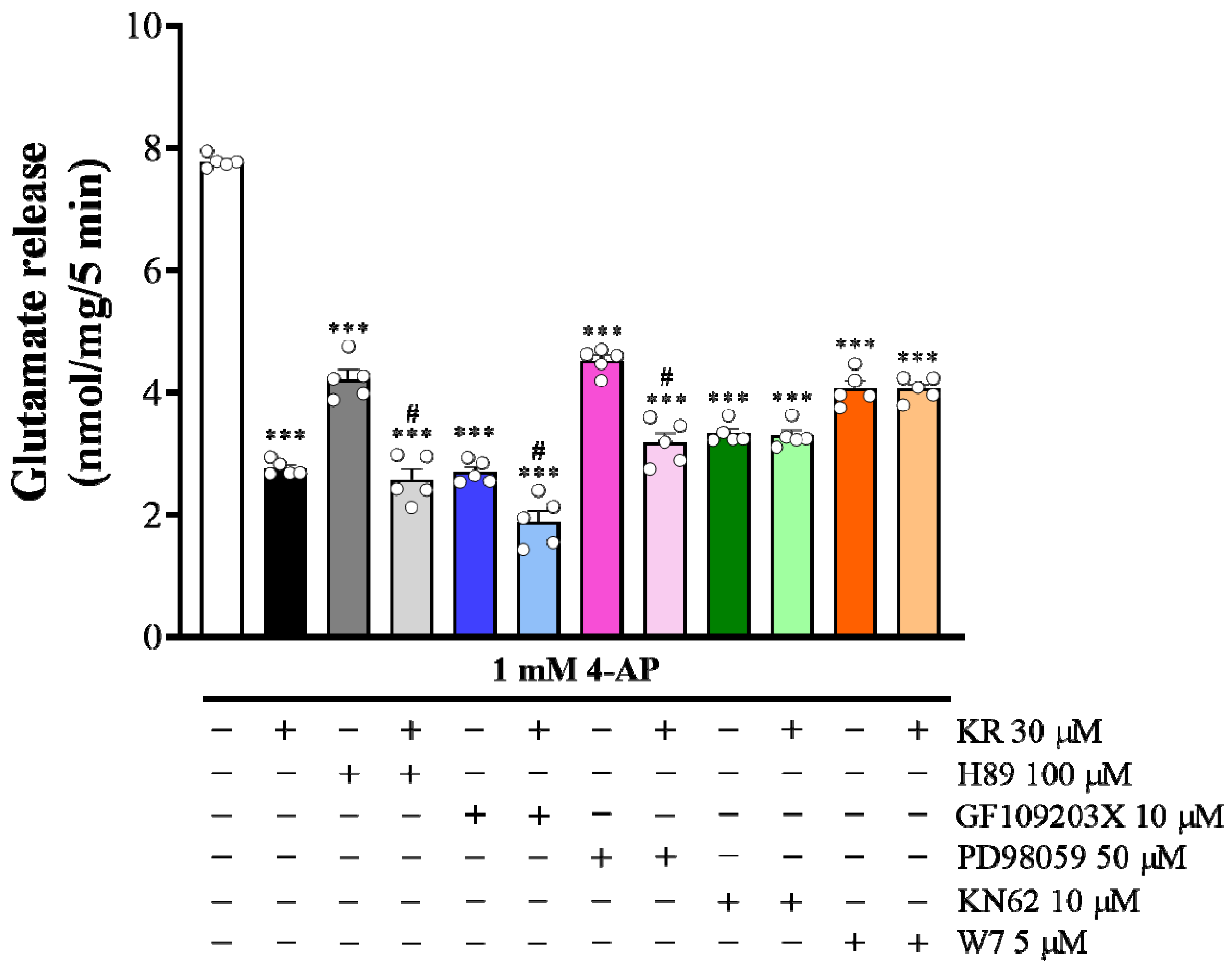
2.5. The Suppressed Ca2+/Calmodulin-Dependent Protein Kinase II Pathway Is Linked to KR-Mediated Inhibition of 4-AP-Evoked Glutamate Release
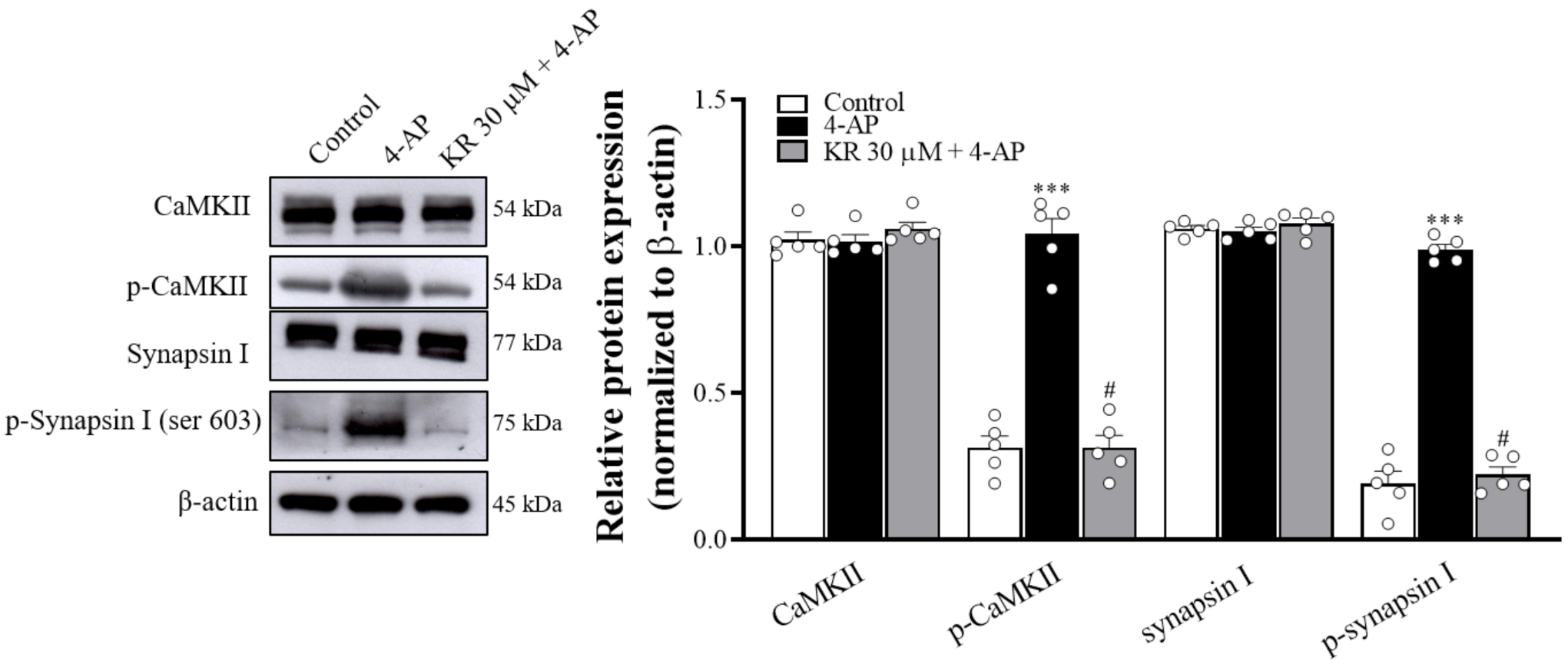
2.6. KR Reduces the Phosphorylation Levels of CaMKII and Synapsin I
3. Discussion
4. Materials and Methods
4.1. Isolation of KR
4.2. Chemicals
4.3. Animals
4.4. Preparation of Isolated Nerve Terminals
4.5. Endogenous Glutamate Release Assay
4.6. Cytosolic Ca2+Assay
4.7. Membrane Potential Assay
4.8. Western Blot Assay
4.9. Data and Statistical Analysis
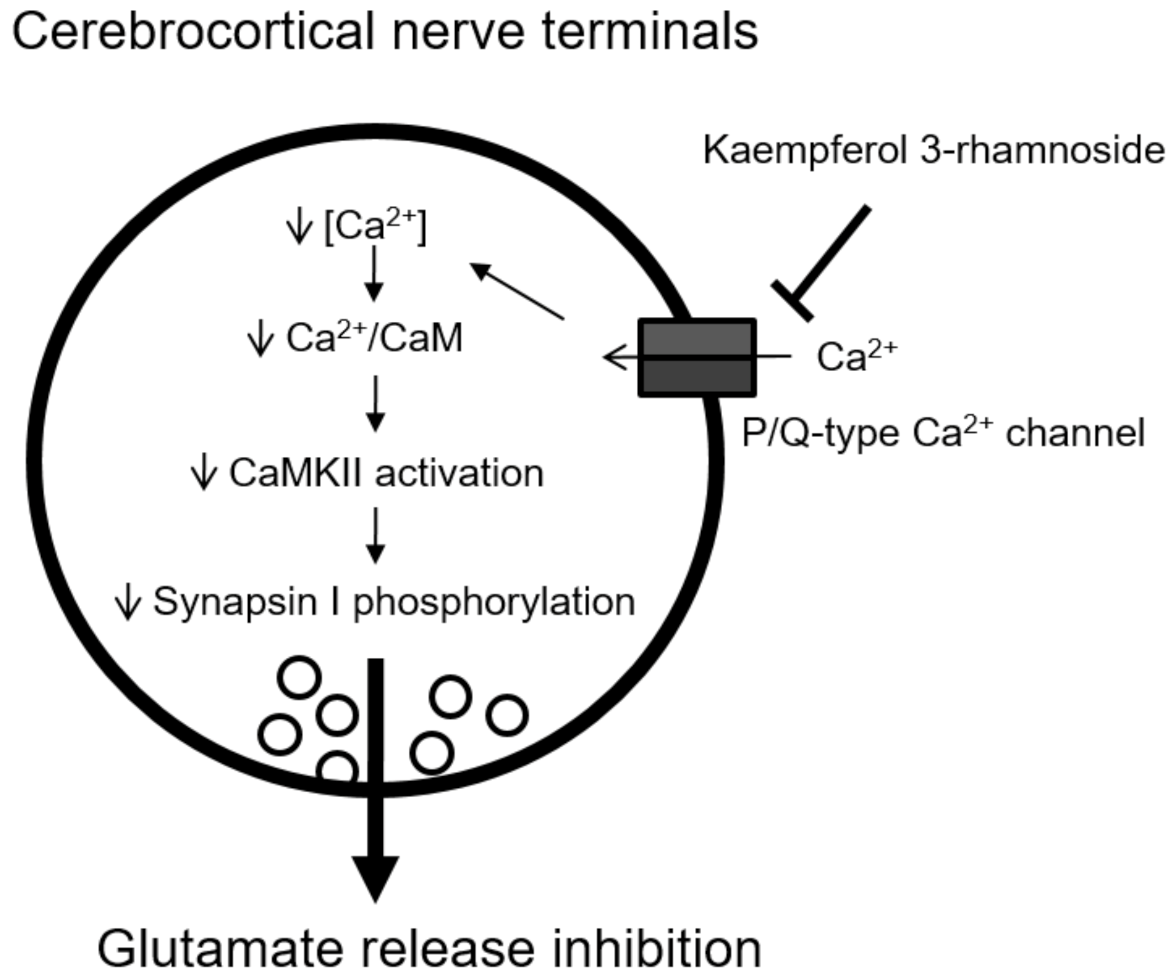
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Greenamyre, J.T.; Porter, R.H. Anatomy and physiology of glutamate in the CNS. Neurology 1994, 44, S7–S13. [Google Scholar] [PubMed]
- Headley, P.M.; Grillner, S. Excitatory amino acids and synaptic transmission: The evidence for a physiological function. Trends Pharmacol. Sci. 1990, 11, 205–211. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meldrum, B.; Garthwaite, J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 1990, 11, 379–387. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, J.C.; Egea, J.; Del Carmen Godino, M.; Fernandez-Gomez, F.J.; Sánchez-Prieto, J.; Gandía, L.; García, A.G.; Jordán, J.; Hernández-Guijo, J.M. Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca(2+) signalling in hippocampal neurons. Eur. J. Neurosci. 2007, 26, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Yang, Y.; Ivanova, D.; Fejtova, A.; Svenningsson, P. Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 2018, 143, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhao, B.; She, Y.; Song, X. Dexmedetomidine ameliorates lidocaine-induced spinal neurotoxicity via inhibiting glutamate release and the PKC pathway. Neurotoxicology 2018, 69, 77–83. [Google Scholar] [CrossRef]
- Chang, Y.; Lu, C.W.; Lin, T.Y.; Huang, S.K.; Wang, S.J. Baicalein, a Constituent of Scutellaria baicalensis, Reduces Glutamate Release and Protects Neuronal Cell Against Kainic Acid-Induced Excitotoxicity in Rats. Am. J. Chin. Med. 2016, 44, 943–962. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Kan, J.; Zhang, J.; Zhou, L.; Huang, Y.; Zhang, Y. Chinese Herbal Medicine Interventions in Neurological Disorder Therapeutics by Regulating Glutamate Signaling. Curr. Neuropharmacol. 2020, 18, 260–276. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Wang, S.J. 11-Keto-β-Boswellic Acid Attenuates Glutamate Release and Kainic Acid-Induced Excitotoxicity in the Rat Hippocampus. Planta Med. 2020, 86, 434–441. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Pan, T.L.; Wang, P.W.; Chiu, K.M.; Lee, M.Y.; Wang, S.J. Asiatic Acid Prevents Cognitive Deficits by Inhibiting Calpain Activation and Preserving Synaptic and Mitochondrial Function in Rats with Kainic Acid-Induced Seizure. Biomedicines 2021, 9, 284. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, G.; Yao, X. Chemical constituents in stems of Schima superba. Chin. Tradit. Herb. Drugs 2010, 41, 863–866. [Google Scholar]
- Lee, S.Y.; So, Y.J.; Shin, M.S.; Cho, J.Y.; Lee, J. Antibacterial effects of afzelin isolated from Cornus macrophylla on Pseudomonas aeruginosa, a leading cause of illness in immunocompromised individuals. Molecules 2014, 19, 3173–3180. [Google Scholar] [CrossRef]
- Lee, S.B.; Kang, J.W.; Kim, S.J.; Ahn, J.; Kim, J.; Lee, S.M. Afzelin ameliorates D-galactosamine and lipopolysaccharide-induced fulminant hepatic failure by modulating mitochondrial quality control and dynamics. Br. J. Pharmacol. 2017, 174, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, M.; Kim, J.M.; Lee, M.K.; Seo, S.J.; Park, K.Y. Afzelin suppresses proinflammatory responses inparticulate matter-exposed human keratinocytes. Int. J. Mol. Med. 2019, 43, 2516–2522. [Google Scholar] [CrossRef]
- Wu, C.; Wu, H.T.; Wang, Q.; Wang, G.H.; Yi, X.; Chen, Y.P.; Zhou, G.X. Anticandidal Potential of Stem Bark Extract from Schima superba and the Identification of Its Major Anticandidal Compound. Molecules 2019, 24, 1587. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, S.; Cho, E.J. Flavonoids from Acer okamotoanum Inhibit Adipocyte Differentiation and Promote Lipolysis in the 3T3-L1 Cells. Molecules 2020, 25, 1920. [Google Scholar] [CrossRef] [Green Version]
- Radziejewska, I.; Supruniuk, K.; Czarnomysy, R.; Buzun, K.; Bielawska, A. Anti-Cancer Potential of Afzelin towards AGS Gastric Cancer Cells. Pharmaceuticals 2021, 14, 973. [Google Scholar] [CrossRef]
- Oh, S.Y.; Jang, M.J.; Choi, Y.H.; Hwang, H.; Rhim, H.; Lee, B.; Choi, C.W.; Kim, M.S. Central administration of afzelin extracted from Ribes fasciculatum improves cognitive and memory function in a mouse model of dementia. Sci. Rep. 2021, 11, 9182. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Jarvie, P.E.; Heath, J.W.; Kidd, G.J.; Rostas, J.A. A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res. 1986, 372, 115–129. [Google Scholar] [CrossRef]
- Raiteri, L.; Raiteri, M. Synaptosomes still viable after 25 years of superfusion. Neurochem. Res. 2000, 25, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Presynaptic modulation of glutamate release. Prog. Brain Res. 1998, 116, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Sihra, T.S.; Sanchez-Prieto, J. The role of the plasma membrane and intracellular organelles in synaptosomal calcium regulation. Soc. Gen. Physiol. Ser. 1987, 42, 31–43. [Google Scholar] [PubMed]
- Vázquez, E.; Sánchez-Prieto, J. Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals. Eur. J. Neurosci. 1997, 9, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Millán, C.; Sánchez-Prieto, J. Differential coupling of N- and P/Q-type calcium channels to glutamate exocytosis in the rat cerebral cortex. Neurosci. Lett. 2002, 330, 29–32. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Parvez, M.K. Natural or Plant Products for the Treatment of Neurological Disorders: Current Knowledge. Curr. Drug Metab. 2018, 19, 424–428. [Google Scholar] [CrossRef]
- Rehman, M.U.; Wali, A.F.; Ahmad, A.; Shakeel, S.; Rasool, S.; Ali, R.; Rashid, S.M.; Madkhali, H.; Ganaie, M.A.; Khan, R. Neuroprotective Strategies for Neurological Disorders by Natural Products: An update. Curr. Neuropharmacol. 2019, 17, 247–267. [Google Scholar] [CrossRef]
- Sihra, T.S.; Nichols, R.A. Mechanisms in the regulation of neurotransmitter release from brain nerve terminals: Current hypotheses. Neurochem. Res. 1993, 18, 47–58. [Google Scholar] [CrossRef]
- Tibbs, G.R.; Barrie, A.P.; Van Mieghem, F.J.; McMahon, H.T.; Nicholls, D.G. Repetitive action potentials in isolated nerve terminals in the presence of 4-aminopyridine: Effects on cytosolic free Ca2+ and glutamate release. J. Neurochem. 1989, 53, 1693–1699. [Google Scholar] [CrossRef]
- Jarvis, S.E.; Zamponi, G.W. Interactions between presynaptic Ca2+ channels, cytoplasmic messengers and proteins of the synaptic vesicle release complex. Trends Pharmacol. Sci. 2001, 22, 519–525. [Google Scholar] [CrossRef]
- Benfenati, F.; Valtorta, F.; Rubenstein, J.L.; Gorelick, F.S.; Greengard, P.; Czernik, A.J. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature 1992, 359, 417–420. [Google Scholar] [CrossRef]
- Jovanovic, J.N.; Sihra, T.S.; Nairn, A.C.; Hemmings, H.C., Jr.; Greengard, P.; Czernik, A.J. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J. Neurosci. 2001, 21, 7944–7953. [Google Scholar] [CrossRef] [Green Version]
- Nichols, R.A.; Sihra, T.S.; Czernik, A.J.; Nairn, A.C.; Greengard, P. Calcium/calmodulin-dependent protein kinase II increases glutamate and noradrenaline release from synaptosomes. Nature 1990, 343, 647–651. [Google Scholar] [CrossRef]
- Abou-Shoer, M.; Ma, G.E.; Li, X.E.; Koonchanok, N.M.; Geahlen, R.L.; Chang, C.J. Flavonoids from Koelreuteriahenryi and other sources as protein-tyrosine kinase inhibitors. J. Nat. Prod. 1993, 56, 967–969. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Chiu, K.M.; Lee, M.Y.; Huang, J.H.; Wang, S.J. Silymarin Inhibits Glutamate Release and Prevents against Kainic Acid-Induced Excitotoxic Injury in Rats. Biomedicines 2020, 8, 486. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Sihra, T.S.; Bogonez, E.; Nicholls, D.G. Localized Ca2+ entry preferentially effects protein dephosphorylation, phosphorylation, and glutamate release. J. Biol. Chem. 1992, 267, 1983–1989. [Google Scholar] [CrossRef]
- Akerman, K.E.; Scott, I.G.; Heikkilä, J.E.; Heinonen, E. Ionic dependence of membrane potential and glutamate receptor-linked responses in synaptoneurosomes as measured with a cyanine dye, DiS-C2-(5). J. Neurochem. 1987, 48, 552–559. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-K.; Hung, C.-F.; Weng, J.-R.; Hsieh, T.-Y.; Wang, S.-J. Kaempferol 3-Rhamnoside on Glutamate Release from Rat Cerebrocortical Nerve Terminals Involves P/Q-Type Ca2+ Channel and Ca2+/Calmodulin-Dependent Protein Kinase II-Dependent Pathway Suppression. Molecules 2022, 27, 1342. https://doi.org/10.3390/molecules27041342
Lin T-K, Hung C-F, Weng J-R, Hsieh T-Y, Wang S-J. Kaempferol 3-Rhamnoside on Glutamate Release from Rat Cerebrocortical Nerve Terminals Involves P/Q-Type Ca2+ Channel and Ca2+/Calmodulin-Dependent Protein Kinase II-Dependent Pathway Suppression. Molecules. 2022; 27(4):1342. https://doi.org/10.3390/molecules27041342
Chicago/Turabian StyleLin, Tzu-Kang, Chi-Feng Hung, Jing-Ru Weng, Ting-Yang Hsieh, and Su-Jane Wang. 2022. "Kaempferol 3-Rhamnoside on Glutamate Release from Rat Cerebrocortical Nerve Terminals Involves P/Q-Type Ca2+ Channel and Ca2+/Calmodulin-Dependent Protein Kinase II-Dependent Pathway Suppression" Molecules 27, no. 4: 1342. https://doi.org/10.3390/molecules27041342
APA StyleLin, T.-K., Hung, C.-F., Weng, J.-R., Hsieh, T.-Y., & Wang, S.-J. (2022). Kaempferol 3-Rhamnoside on Glutamate Release from Rat Cerebrocortical Nerve Terminals Involves P/Q-Type Ca2+ Channel and Ca2+/Calmodulin-Dependent Protein Kinase II-Dependent Pathway Suppression. Molecules, 27(4), 1342. https://doi.org/10.3390/molecules27041342