
The Role of Flavonoids as a Cardioprotective Strategy against Doxorubicin-Induced Cardiotoxicity: A Review

 ,
,
Abstract
1. Introduction
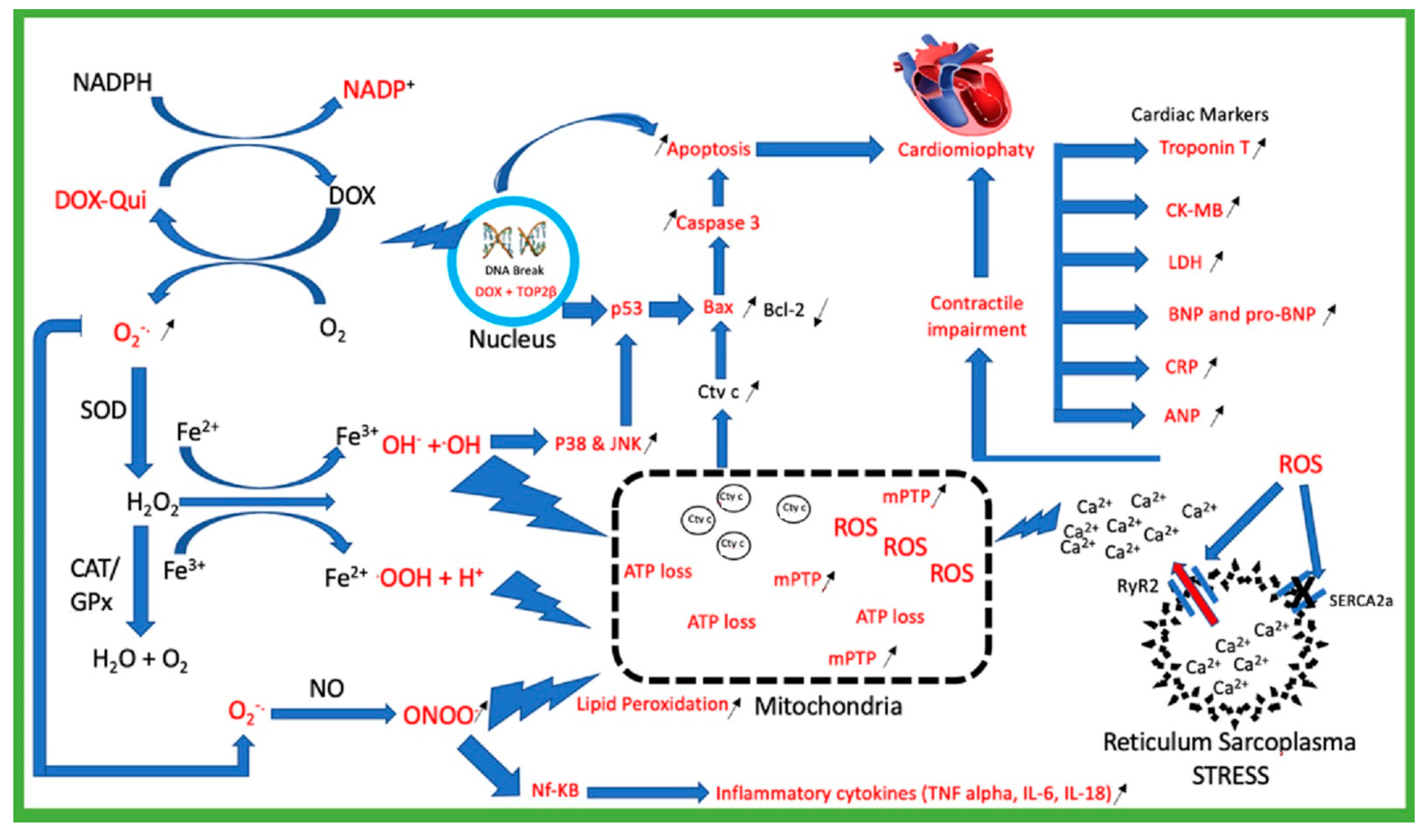
2. Doxorubicin Mechanism of Toxicity
2.1. Doxorubicin Generates Reactive Oxygen Species (ROS)
2.2. Mitochondria Injury
2.3. Topoisomerase 2β (TOP2β)
2.4. Calcium Homeostasis Dysregulation
2.5. Cardiac Biomarkers Injury
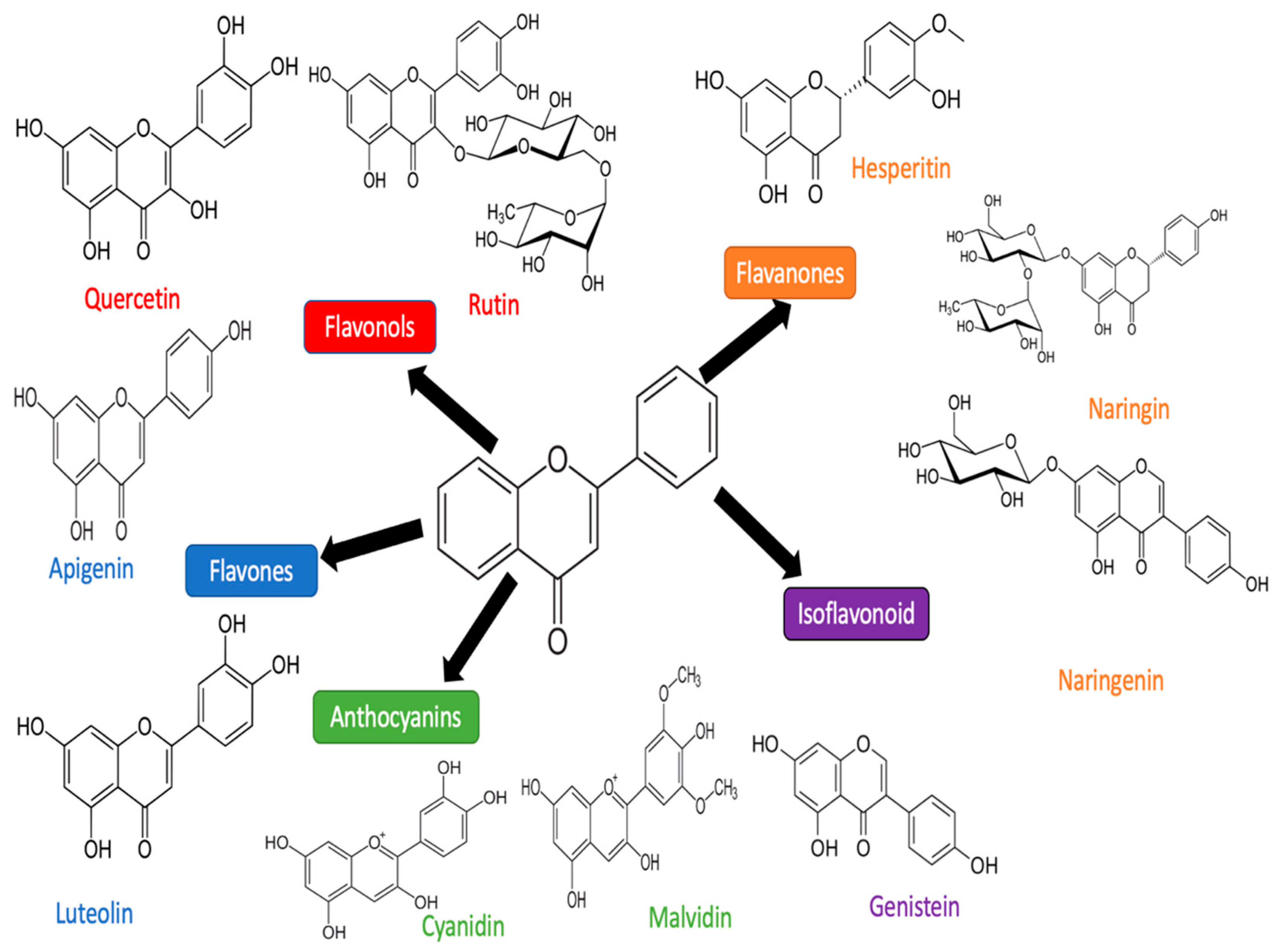
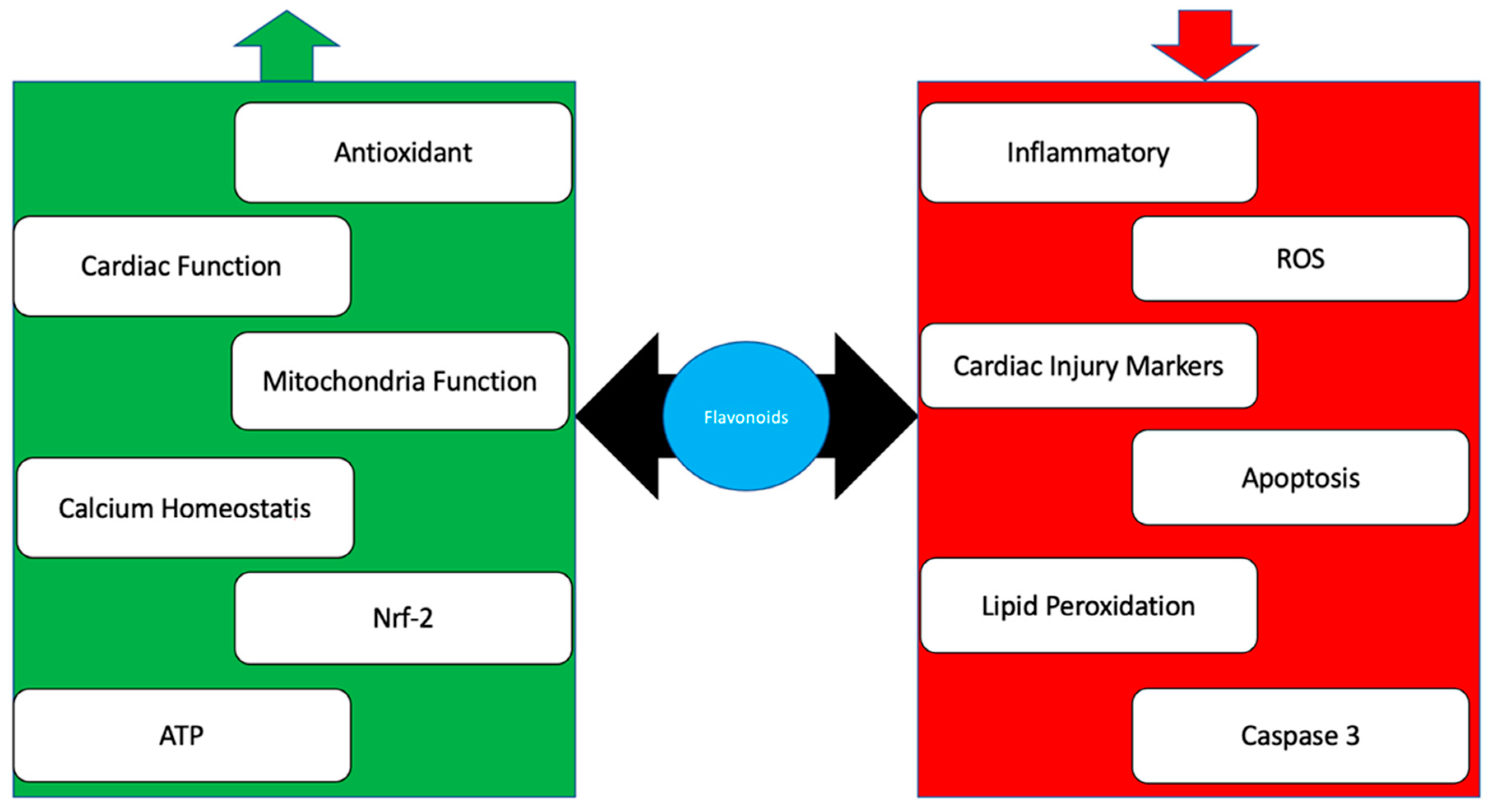
3. Flavonoid
3.1. Luteolin
3.2. Quercetin
3.3. Apigenin
3.4. Rutin
3.5. Cyanidin
3.6. Hesperidin
3.7. Chrysin
3.8. Naringenin and Narigin

{kind=link}
{kind=link}
{kind=link}
| Compound | Study Design | Flavonoid Dose | Doxorubicin Dose | Duration | Parameters | References |
|---|---|---|---|---|---|---|
| Luteolin | In vivo (rat) | 50 mg/kg 100 mg/kg (P.O 1 week in advance and gastric administration lasted for 5 weeks) | 16 mg/kg (Intraperitoneal injection once a week) | 5 weeks | ↓BNP, ↓CK-MB, ↓MDA, ↓LDH, ↑SOD, ↑Bcl2, ↓Bax, ↑p-AKT, ↓Caspase-3 | [48] |
| Luteolin-7-O-glucoside | In vitro (H9c2) | 10 and 20 µM (pre-treated for 24 h) | 10 µM (Incubated for 24 h) | 48 h | ↑Cell viability, ↓apoptosis, ↓ROS, ↑P-PTEN, ↓P-Akt, ↓P-ERK, ↓p-mTOR, ↓p-GSK-3bate | [114] |
| Luteolin | In vitro (H9c2) | 5, 10, 20 µM (pre-treated for 24 h) | 20 µM (Incubated for 24 h) | 48 h | ↑Cell viability, ↓CK, ↓LDH, ↓ROS, ↓ [Ca2+]i | [118] |
| Luteolin | In vitro (AMCMs) | 1, 10, 50 µM | 1 µM | 24 h | ↓LDH, ↓CK, ↓Apoptosis, ↓ROS, ↑Bcl-2, ↓Bax, ↓Caspase 9, ↑Bnip3, ↑Parkin, ↑Pink1, ↑LC3BII, ↑P62, ↓mTOR, ↑LAMP1, ↑TFEB, ↑Drp1 | [117] |
| Quercetin | In vivo (rat) | 10, 25, 50 mg/kg (P.O for 7 weeks) | 2 mg/kg (Intraperitoneal once a week until 4 weeks) | 7 weeks | ↓Blood pressure, ↓HR, ↓LVEDP, ↑coronary flow, ↑+(dp/dt) max, ↑-(dp/dt) max, ↓CK-MB, ↓LDH, ↓Na+, ↓K+, ↓MDA, ↑GSH, ↑SOD, ↑Catalase, ↑Nrf2 | [122] |
| Quercetin | In vivo (rat) | 2 mg/kg (P.O for 7 days) | 10 mg/kg (I.V on day 5) | 7 days | ↓AST, ↓LDH, ↑GSH, ↓BUN, ↓Creatinine, ↓TBRAS | [133] |
| Quercetin | In vitro (H9c2) | 100 µM (pre-treated for 48 h and 96 h) | 1 µM | 48 h and 96 h | ↑CR inhibition, ↓LDH, ↓iron chealting, ↓LPO IC50 | [163] |
| Quercetin | In vitro (H9c2) | 50 and 100 µM (Incubated 48 h) | 0–16μM (Incubated 48 h) | 48 h | ↑Cell viability, ↓apoptosis, ↑MMP, ↓ROS, ↑Bmi-1 | [128] |
| In vivo (Mice) | 100 mg/kg (P.O for 10 days) | 20 mg/kg (I.P) | 48 h | ↑LVEF, ↑LVFS, ↓LVEDD, ↓LVESD, ↓LDH, ↓MDA, ↑SOD, ↑Bmi-1 | ||
| Quercetin polymeric micelles | In vitro (H9c2) | µM | 0.01, 0.1, 1 µM | 48 h | ↓Caspase 3, ↓caspase 7, ↓ROS, ↓apoptosis | [164] |
| In vivo (mice) | 3.31 mg/kg (I.V every 3 days for 3 cycle) | 6 mg/kg (I.V every 3 days for 3 cycle) | 10 days | ↓AST, ↓ALT, ↓CK | ||
| Quercetin | In vitro (Neonatal Rat cardiomyocytes) | 10,20,40,80 µM (pre-treated for 22 h) | 1 µM (incubated 24 h) | 48 h (2 h normal condition) | ↑Cell viability, ↓LDH, ↓caspase 3, ↓apoptosis, ↑14-3-3γ, ↑MMP, ↑SOD, ↑Catalase, ↑Gpx, ↓MDA, ↑GSH, ↑GSSG | [129] |
| Quercetin | In vivo (rat) | 10 mg/kg (P.O for 6 weeks) | 2.5 mg/kg (I.P every 2 days for 2 weeks) | 6 weeks | ↓CK-MB, ↓LDH, ↓TNF, ↑SOD, ↑CAT, ↓MDA, ↓NO | [131] |
| Apigenin | In vivo (rat) | 25 mg/kg (P.O for 12 days) | 2 mg/kg (I.P every 2 days for 12 days) | 12 days | ↑%EF, ↑%FS, ↓LVIDd, ↓LVISd, ↓LDH, ↓CK-MB, ↓cTn-I, ↓ALT, ↓AST, ↓%Fibrosis, ↓MDA, ↑SOD, ↑Catalase, ↑Bcl-2, ↓Bax, ↓Caspase-3 | [137] |
| Apigenin | In vitro (Murine cardiomyocytes) | 20 µM (Incubated for 24 h) | 1 µM (incubated for 24 h) | 24 h | ↑Cell viability, ↓ROS, TBARS, ↑CAT, ↓Carbonyl protein, ↑SOD, ↑GST, ↑GPx, ↑GSH, ↑GR, ↓DNA fragmentation, ↓8-OHdG, ↓Cyt c, ↑Bcl-2, ↓Bax, ↓caspase 3, ↓caspase 9, ↓caspase 8, Apaf-1, FAS, t-Bid, ↓IκBα, ↓NF-κB, PKC-δ, ↓JNK, ↓p38, ↓p53, ↑PI3K, ↑Akt, mTOR, ↓iNOS, ↑HO-1, and ↑Nrf-2 | [136] |
| In vivo (rat) | 100 mg/kg (P.O 7 days) | 3 mg/kg (I.P on day 1,3,5) | 7 days | ↑Total erythrocytes, ↑Haemoglobin, Total leucocytes, ↓Total cholesterol, HDL, TGD, LDH, ↓CK, ↓AST, ↓Troponin I, ↓Troponin T, ↑SOD, ↓Protein carbonyl, ↓ROS, ↓TBARS, ↑CAT, ↑GPx, ↑GST, ↑GSH, ↓8-OHdG, ↑GR, ↓NADPH oxidase, ↓DNA fragmentation, ↓MMP, ↓Cyt C, ↑Bcl-2, ↓Bax, ↓Caspase 3, ↓caspase 9, ↓caspase 8, ↓FAS, ↓t-Bid, ↓IκBα, ↓NF-κB, ↓PKC-δ, ↓JNK, p38, ↓p53, ↑PI3K, ↑Akt, ↑mTOR, ↓iNOS, ↑HO-1, and ↑Nrf-2 | ||
| Apigenin | In vivo (mice) | 125 and 250 mg/kg (Gastric gavage for 17 days) | 3 mg/kg (I.P every 2 days for 16 days) | 17 days | ↓AST, ↓LDH, ↓CK, ↓Apoptosis, ↓Bax, ↑Bcl-2, ↓Beclin1, ↓LC3, ↑p-mTOR, ↑mTOR, ↑p-AKT, ↑AKT1/2/3, ↑PI3K | [138] |
| Rutin | In vivo (H9c2) | 10, 30, 50, or 70 μM (pre-treated for 1 h) | 5μM/pirarubicin (Incubated 24 h) | 24 h | ↑Cell viability, ↓ROS, ↓Apoptosis, ↓caspase 3, ↓caspase 7, ↓caspase 7, TGF-β1, p-p38 MAPK | [165] |
| Rutin | In vivo (mice) | 100 mg/kg (P.O for 11 weeks) | 3 mg/kg (I.P every 2 days for 2 weeks) | 11 weeks | ↑LVEF, ↑LVFS, ↓%fibrosis, | [166] |
| In vitro (cardiomyocytes) | 10 μM (pre-treated for 24 h) | 1 μM (incubated for 24 h) | 48 h | ↓Apoptosis, ↑Bcl-2, ↓Caspase 3, ↓P62, ↓LC3BI/II, ↓ATG5 | ||
| Rutin | In vivo (mice) | 100 μmol/kg (I.P for 5 days) | 15 mg/kg (I.P day 1) | 5 days | ↑GSHpx, ↓MDA, ↓CPK, ↓Total bone marrow, ↓NADPH IC50 | [146] |
| Rutin | In vivo (rat) | 50 mg/kg (P.O 3 times per week for 3 weeks) | 25 mg/kg | 3 weeks | ↓Total cholestrol, ↑HDL, ↓LDL, ↓CK, ↓LDH, ↓AST, ↑Glutathione, ↑GPx, ↑Glutathione-s-tranasferase, ↓MDA | [167] |
| Hesperidin | In vivo (rat) | 50 mg/kg (Gastric administration 3 times per week for 3 weeks) | 4 mg/kg (I.P 3 times per week for 2 weeks) | 3 weeks | ↓CK, ↓LDH, ↓NO, ↓MPO, ↓MDA, ↑GSH, ↑CAT, ↓Caspase 3 | [168] |
| Anthocyanin | In vitro (HL-1) | 0, 5, 25, 125, 250 μM | 0, 0.125, 0.25, 0.5, 1, 2, 4 μM | 48 h | ↑Cell viability, ↓RAS | [148] |
| Anthocyanin | In vitro (H9c2) | 20 and 40 μg/mL (post-treated for 24 h) | 1 μM (treated for 6 and 12 h) | 36 h | ↑Cell viability, ↓apoptosis, ↓CHIP, ↑HSF1, ↓IGF-IIR, ↓caspase 3, p-NFκB, ↑p-Akt, ↑ERα, ↑ERβ | [149] |
| Chrysin | In vivo (rat) | 25 and 50 mg/kg (P.O for 12 days) | 15 mg/kg (I.P on day 12) | 12 days | ↓CK-MB, ↓LDH, ↓MDA, ↓NF-κB, ↓iNOS, ↓COX-2, ↓Bax, ↑Bcl2, ↓TNF-α, COX-2, ↑SOD, ↑CAT, ↓NO, ↓Apoptosis, ↑GSH, ↑Cyc C | [155] |
| Chrysin | In vivo (rat) | 50 mg/kg (P.O 4 times per week for 5 weeks) | 5 mg/kg (I.P once a week for 4 weeks) | 4 weeks | ↓VEGF, ↑AKT, ↑PTEN, ↓NF-κB, ↓Bax, Bcl-2, ↓P53, ↓MAPK, GSH, ↓MDA, ↑CAT, ↑SOD, ↑Gpx, ↑GR | [155] |
| Hesperidin | In vivo (rat) | 25, 50, 100 mg/kg (P.O 5 times per weeks for 5 weeks) | 4 mg/kg (I.P once a week for 5 weeks) | 5 weeks | ↓MDA, ↑GSH, ↓NF-kB, ↓p38, ↓Caspase-3, ↓apoptosis, ↓% demaged cell | [151] |
| Hesperidin solid nano particle | In vivo (rat) | 20 mg/kg (P.O for 7 days) | 15 mg/kg (I.P on day 5) | 7 days | ↓CK-MB, ↓Troponin I, ↓MDA, ↑SOD, ↑CAT, ↓Apoptosis, ↓Caspase 3 | [152] |
| Anthocyanin | In vitro (H9c2) | 100–800 μg/mL | 3 μmol/L for 12 h | 12 h | ↓NO, ↓TNF-α, ↓TMAO, ↓LDH, ↓CK | [169] |
| In vivo (mice) | 100 and 200 mg/kg (P.O for 25 days) | 13 mg/kg injected on day 26, 27, and 18 | 28 days | ↓NO, ↓LDH, ↓CK, ↓TNF-α ↓TMAO | ||
| Naringenin | In vivo (rat) | 25 mg/kg (P.O for 7 days) | 15 mg/kg (I.P on day 7) | 7 days | ↓LDH, ↓CPK, ↓MDA, ↑SOD, ↑GSH, ↑CAT, ↑GST | [170] |
| Naringenin | In vivo (rat) | 100 mg/kg (P.O for 2 weeks) | 15 mg/kg (I.P on day 14) | 2 weeks | ↓CK-MB, ↓Creatinine, ↓AST, ↓ALT, ↓Urea, ↓LDH, ↓TNF-α, ↓IL-6, ↓IL-1β, ↓TBARS, ↑GSH, ↑CAT, ↑SOD, ↑GST, ↑GPx | [160] |
| naringenin-7-O-glucoside | In vitro (H9c2) | 5, 10, 20, 40, and 80 μM (pre-treated for 24 h) | 10 μM (Incubated 24 h) | 48 h | ↓Cell viability, ↓ROS, ↓LDH, ↓CK, ↑GSH, ↑GPx, ↓ [Ca2+]I | [171] |
| Naringenin | In vivo (rat) | 15 mg/kg (P.O for 30 days) | 15 mg/kg (I.P on day 30) | 30 days | ↑SOD, ↑CAT, ↑GSH | [159] |
| Naringin | In vivo (rat) | 50 and 100 mg/kg (I.P for 14 days) | 15 mg/kg (I.P on day 10) | 14 days | ↑GSH, ↑SOD, ↑CAT, ↓MDA, ↓NADH, ↓Cyt-C, | [161] |
| Naringin | In vivo (rat) | 50 mg/kg (P.O for 10 weeks) | 3 mg/kg (I.P on week 1,3,5,7,9) | 10 weeks | ↓LDH, ↓Troponin T, ↓MDA, ↑CAT, ↑SOD, ↑GPx, ↓TGFβ1, ↓TNF-α, ↓IL-6, ↓IL-10 | [162] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Zanden, S.Y.; Qiao, X.; Neefjes, J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. 2021, 288, 6095–6111. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, Y.; Wang, S.; He, X.; Liu, M.; Bai, B.; Tian, C.; Sun, R.; Yu, T.; Chu, X. Role of acetylation in doxorubicin-induced cardiotoxicity. Redox Biol. 2021, 46, 102089. [Google Scholar] [CrossRef]
- De Oliveira, B.L.; Niederer, S. A biophysical systems approach to identifying the pathways of acute and chronic doxorubicin mitochondrial cardiotoxicity. PLoS Comput. Biol. 2016, 12, e1005214. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D. Anthracycline Cardiotoxicity: Worrisome Enough to Have You Quaking? Circ. Res. 2018, 122, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Lefrak, E.A.; Piťha, J.; Rosenheim, S.; Gottlieb, J.A. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 1973, 32, 302–314. [Google Scholar] [CrossRef]
- Reinbolt, R.E.; Patel, R.; Pan, X.; Timmers, C.D.; Pilarski, R.; Shapiro, C.L.; Lustberg, M.B. Risk factors for anthracycline-associated cardiotoxicity. Support. Care Cancer 2016, 24, 2173–2180. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.B.; Ewer, M.; et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.B.; Davis, M.K.; Law, A.; Sulpher, J. Shared risk factors for cardiovascular disease and cancer: Implications for preventive health and clinical care in oncology patients. Can. J. Cardiol. 2016, 32, 900–907. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Patel, D.; Wu, X. The role of topoisomerase IIβ in the mechanisms of action of the doxorubicin cardioprotective agent dexrazoxane. Cardiovasc. Toxicol. 2020, 20, 312–320. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Wang, L.; Qiao, Y.; Zhou, Q.; Li, H.; Chen, S.; Yin, D.; Huang, Q.; He, M. Doxorubicin induces endotheliotoxicity and mitochondrial dysfunction via ROS/eNOS/NO pathway. Front. Pharmacol. 2020, 10, 1531. [Google Scholar] [CrossRef] [PubMed]
- Agustini, F.D.; Arozal, W.; Louisa, M.; Siswanto, S.; Soetikno, V.; Nafrialdi, N.; Suyatna, F. Cardioprotection mechanism of mangiferin on doxorubicin-induced rats: Focus on intracellular calcium regulation. Pharm. Biol. 2016, 54, 1289–1297. [Google Scholar] [CrossRef]
- Qin, Y.; Guo, T.; Wang, Z.; Zhao, Y. The role of iron in doxorubicin-induced cardiotoxicity: Recent advances and implication for drug delivery. J. Mater. Chem. B 2021, 9, 4793–4803. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef]
- Shabalala, S.; Muller, C.J.; Louw, J.; Johnson, R. Polyphenols, autophagy and doxorubicin-induced cardiotoxicity. Life Sci. 2017, 180, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhong, L.; Roush, S.F.; Pentassuglia, L.; Peng, X.; Samaras, S.; Davidson, J.M.; Sawyer, D.B.; Lim, C.C. Disruption of a GATA4/Ankrd1 signaling axis in cardiomyocytes leads to sarcomere disarray: Implications for anthracycline cardiomyopathy. PLoS ONE 2012, 7, e35743. [Google Scholar] [CrossRef]
- Vejpongsa, P.; Yeh, E.T. Prevention of anthracycline-induced cardiotoxicity: Challenges and opportunities. J. Am. Coll. Cardiol. 2014, 64, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Gyöngyösi, M.; Lukovic, D.; Zlabinger, K.; Spannbauer, A.; Gugerell, A.; Pavo, N.; Traxler, D.; Pils, D.; Maurer, G.; Jakab, A.; et al. Liposomal doxorubicin attenuates cardiotoxicity via induction of interferon-related DNA damage resistance. Cardiovasc. Res. 2020, 116, 970–982. [Google Scholar] [CrossRef]
- Reichardt, P.; Tabone, M.D.; Mora, J.; Morland, B.; Jones, R.L. Risk–benefit of dexrazoxane for preventing anthracycline-related cardiotoxicity: Re-evaluating the European labeling. Future Oncol. 2018, 14, 2663–2676. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, H.; Sebag, I.A.; Plana, J.C.; Januzzi, J.L.; Ky, B.; Cohen, V.; Gosavi, S.; Carver, J.R.; Wiegers, S.E.; Martin, R.P.; et al. Early detection and prediction of cardiotoxicity in chemotherapy-treated patients. Am. J. Cardiol. 2011, 107, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Dillard, C.J.; German, J.B. Phytochemicals: Nutraceuticals and human health. J. Sci. Food Agric. 2000, 80, 1744–1756. [Google Scholar] [CrossRef]
- Sadzuka, Y.; Sugiyama, T.; Shimoi, K.; Kinae, N.; Hirota, S. Protective effect of flavonoids on doxorubicin-induced cardiotoxicity. Toxicol. Lett. 1997, 92, 1–7. [Google Scholar] [CrossRef]
- Bast, A.; Kaiserová, H.; Den Hartog, G.J.; Haenen, G.R.; Van Der Vijgh, W.J. Protectors against doxorubicin-induced cardiotoxicity: Flavonoids. Cell Biol. Toxicol. 2007, 23, 39–47. [Google Scholar] [CrossRef]
- Feliciano, R.P.; Pritzel, S.; Heiss, C.; Rodriguez-Mateos, A. Flavonoid intake and cardiovascular disease risk. Curr. Opin. Food Sci. 2015, 2, 92–99. [Google Scholar] [CrossRef]
- Bartlett, J.J.; Trivedi, P.C.; Pulinilkunnil, T. Autophagic dysregulation in doxorubicin cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 104, 1–8. [Google Scholar] [CrossRef]
- Alkuraishy, H.M.; Al-Gareeb, A.I.; Al-hussaniy, H.A. Doxorubicin-induced cardiotoxicity: Molecular mechanism and protection by conventional drugs and natural products. Int. J. Clin. Oncol. Cancer Res. 2017, 2, 31–44. [Google Scholar]
- Pilco-Ferreto, N. and Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef]
- Carvalho, F.S.; Burgeiro, A.; Garcia, R.; Moreno, A.J.; Carvalho, R.A.; Oliveira, P.J. Doxorubicin-induced cardiotoxicity: From bioenergetic failure and cell death to cardiomyopathy. Med. Res. Rev. 2014, 34, 106–135. [Google Scholar] [CrossRef]
- Yan, Y.; Finkel, T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic. Biol. Med. 2017, 109, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, Y.; Zheng, D.; Wei, M.; Xu, H.; Peng, T. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc. Res. 2013, 97, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kalivendi, S.V.; Kotamraju, S.; Zhao, H.; Joseph, J.; Kalyanaraman, B. Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase: Effect of antiapoptotic antioxidants and calcium. J. Biol. Chem. 2001, 276, 47266–47276. [Google Scholar] [CrossRef] [PubMed]
- Beinert, H.; Kennedy, M.C.; Stout, C.D. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 1996, 96, 2335–2374. [Google Scholar] [CrossRef] [PubMed]
- Panjrath, G.S.; Patel, V.; Valdiviezo, C.I.; Narula, N.; Narula, J.; Jain, D. Potentiation of doxorubicin cardiotoxicity by iron loading in a rodent model. J. Am. Coll. Cardiol. 2007, 49, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. IJC Heart Vasc. 2016, 10, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef]
- Gilleron, M.; Marechal, X.; Montaigne, D.; Franczak, J.; Neviere, R.; Lancel, S. NADPH oxidases participate to doxorubicin-induced cardiac myocyte apoptosis. Biochem. Biophys. Res. Commun. 2009, 388, 727–731. [Google Scholar] [CrossRef]
- Šimůnek, T.; Štěrba, M.; Popelová, O.; Adamcová, M.; Hrdina, R.; Geršl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef]
- Spallarossa, P.; Garibaldi, S.; Altieri, P.; Fabbi, P.; Manca, V.; Nasti, S.; Rossettin, P.; Ghigliotti, G.; Ballestrero, A.; Patrone, F.; et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J. Mol. Cell. Cardiol. 2004, 37, 837–846. [Google Scholar] [CrossRef]
- Dorn, G.W., II. Mitochondrial dynamics in heart disease. Biochim. Biophys. Acta—Mol. Cell Res. 2013, 1833, 233–241. [Google Scholar] [CrossRef]
- Dos Santos, D.S.; dos Santos Goldenberg, R.C. Doxorubicin-induced cardiotoxicity: From mechanisms to development of efficient therapy. In Cardiotoxicity; Tan, W., Ed.; IntechOpen: London, UK, 2018; pp. 3–24. [Google Scholar]
- Heller, B.I.; Jacobson, W.E. Renal hemodynamics in heart disease. Am. Heart J. 1950, 39, 188–204. [Google Scholar] [CrossRef]
- Guven, C.; Sevgiler, Y.; Taskin, E. Mitochondrial dysfunction associated with doxorubicin. In Mitochondrial Diseases; Taskin, E., Guven, C., Sevgiler, Y., Eds.; IntechOpen: London, UK, 2018; p. 323. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. The definition and measurement of antioxidants in biological systems. Free Radic. Biol. Med. 1995, 18, 125–126. [Google Scholar] [CrossRef]
- Dudek, J.; Hartmann, M.; Rehling, P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim. Biophys. Acta—Mol. Basis Dis. 2019, 1865, 810–821. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Corrigendum to “Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib”. Oxid. Med. Cell. Longev. 2019, 2019, 9601435. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef]
- Feijen, E.A.; Leisenring, W.M.; Stratton, K.L.; Ness, K.K.; Van Der Pal, H.J.; Van Dalen, E.C.; Armstrong, G.T.; Aune, G.J.; Green, D.M.; Hudson, M.M.; et al. Derivation of anthracycline and anthraquinone equivalence ratios to doxorubicin for late-onset cardiotoxicity. JAMA Oncol. 2019, 5, 864–871. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Yang, J.J.; Zhang, H.S. Carvedilol (CAR) combined with carnosic acid (CAA) attenuates doxorubicin-induced cardiotoxicity by suppressing excessive oxidative stress, inflammation, apoptosis and autophagy. Biomed. Pharmacother. 2019, 109, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Birari, L.; Wagh, S.; Patil, K.R.; Mahajan, U.B.; Unger, B.; Belemkar, S.; Goyal, S.N.; Ojha, S.; Patil, C.R. Aloin alleviates doxorubicin-induced cardiotoxicity in rats by abrogating oxidative stress and pro-inflammatory cytokines. Cancer Chemother. Pharmacol. 2020, 86, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Yarmohammadi, F.; Rezaee, R.; Karimi, G. Natural compounds against doxorubicin-induced cardiotoxicity: A review on the involvement of Nrf2/ARE signaling pathway. Phytother. Res. 2021, 35, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Abo-Salem, O.M. The protective effect of aminoguanidine on doxorubicin-induced nephropathy in rats. J. Biochem. Mol. Toxicol. 2012, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Kim, D.S.; Yadav, R.K.; Kim, H.R.; Chae, H.J. Sulforaphane prevents doxorubicin-induced oxidative stress and cell death in rat H9c2 cells. Int. J. Mol. Med. 2015, 36, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Q.; Zhang, L.; Zhao, G.X.; Chen, Y.; Sun, K.L.; Wang, B. Fucoxanthin attenuates doxorubicin-induced cardiotoxicity via anti-oxidant and anti-apoptotic mechanisms associated with p38, JNK and p53 pathways. J. Funct. Foods 2019, 62, 103542. [Google Scholar] [CrossRef]
- Zhang, Y.; Ahmad, K.A.; Khan, F.U.; Yan, S.; Ihsan, A.U.; Ding, Q. Chitosan oligosaccharides prevent doxorubicin-induced oxidative stress and cardiac apoptosis through activating p38 and JNK MAPK mediated Nrf2/ARE pathway. Chem.-Biol. Interact. 2019, 305, 54–65. [Google Scholar] [CrossRef] [PubMed]
- El Btaouri, H.; Morjani, H.; Greffe, Y.; Charpentier, E.; Martiny, L. Role of JNK/ATF-2 pathway in inhibition of thrombospondin-1 (TSP-1) expression and apoptosis mediated by doxorubicin and camptothecin in FTC-133 cells. Biochim. Biophys. Acta—Mol. Cell Res. 2011, 1813, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Tocchetti, C.; Molinaro, M.; Angelone, T.; Lionetti, V.; Madonna, R.; Mangiacapra, F.; Moccia, F.; Penna, C.; Sartiani, L.; Quaini, F.; et al. Nitroso-redox balance and modulation of basal myocardial function: An update from the Italian Society of Cardiovascular Research (SIRC). Curr. Drug Targets 2015, 16, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem.-Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Revis, N.W.; Marušić, N. Effects of doxorubicin and its aglycone metabolite on calcium sequestration by rabbit heart, liver, and kidney mitochondria. Life Sci. 1979, 25, 1055–1063. [Google Scholar] [CrossRef]
- Sag, C.M.; Köhler, A.C.; Anderson, M.E.; Backs, J.; Maier, L.S. CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 51, 749–759. [Google Scholar] [CrossRef]
- Jean, S.R.; Ahmed, M.; Lei, E.K.; Wisnovsky, S.P.; Kelley, S.O. Peptide-Mediated Delivery of Chemical Probes and Therapeutics to Mitochondria. Acc Chem Res. 2016, 49, 1893–1902. [Google Scholar] [CrossRef]
- Cui, N.; Wu, F.; Lu, W.J.; Bai, R.; Ke, B.; Liu, T.; Li, L.; Lan, F.; Cui, M. Doxorubicin-induced cardiotoxicity is maturation dependent due to the shift from topoisomerase IIα to IIβ in human stem cell-derived cardiomyocytes. J. Cell. Mol. Med. 2019, 23, 4627–4639. [Google Scholar] [CrossRef]
- Rodrigo, R.S.; Nathalie, A.; Elodie, T.; Gonzalo, G.A.; Philippe, T.; Françoise, D.; Julien, D.; Angela, C.; Bérénice, B.; Jean-Yves, B.; et al. Topoisomerase II-alpha protein expression and histological response following doxorubicin-based induction chemotherapy predict survival of locally advanced soft tissues sarcomas. Eur. J. Cancer 2011, 47, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as topoisomerase II poisons: From early studies to new perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta—Rev. Cancer 2014, 1845, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, G.R.; Tulumello, D.V.; Kelley, S.O. Targeted delivery of doxorubicin to mitochondria. ACS Chem. Biol. 2013, 8, 1389–1395. [Google Scholar] [CrossRef]
- Timm, K.N.; Tyler, D.J. The role of AMPK activation for cardio protection in doxorubicin-induced cardiotoxicity. Cardiovasc. Drugs Ther. 2020, 34, 255–269. [Google Scholar]
- Fouad, A.A.; Albuali, W.H.; Al-Mulhim, A.S.; Jresat, I. Cardioprotective effect of cannabidiol in rats exposed to doxorubicin toxicity. Environ. Toxicol. Pharmacol. 2013, 36, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.D.; Lam, A.; Tham, S.; Dulhunty, A.F.; Beard, N.A. Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol. Pharmacol. 2014, 86, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Mattila, M.; Koskenvuo, J.; Söderström, M.; Eerola, K.; Savontaus, M. Intramyocardial injection of SERCA2a-expressing lentivirus improves myocardial function in doxorubicin-induced heart failure. J. Gene Med. 2016, 18, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Shati, A.A.; Dallak, M. Acylated ghrelin protects the hearts of rats from doxorubicin-induced Fas/FasL apoptosis by stimulating SERCA2a mediated by activation of PKA and Akt. Cardiovasc. Toxicol. 2019, 19, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Preau, S.; Baccouch, R.; Modine, T.; Fayad, G.; Lancel, S.; Neviere, R. Doxorubicin induces mitochondrial permeability transition and contractile dysfunction in the human myocardium. Mitochondrion 2011, 11, 22–26. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Carpi, A.; Coppola, C.; Quintavalle, C.; Rea, D.; Campesan, M.; Arcari, A.; Piscopo, G.; Cipresso, C.; Monti, M.G.; et al. Ranolazine protects from doxorubicin-induced oxidative stress and cardiac dysfunction. Eur. J. Heart Fail. 2014, 16, 358–366. [Google Scholar] [CrossRef]
- Asensio-López, M.C.; Soler, F.; Sánchez-Más, J.; Pascual-Figal, D.; Fernández-Belda, F.; Lax, A. Early oxidative damage induced by doxorubicin: Source of production, protection by GKT137831 and effect on Ca2+ transporters in HL-1 cardiomyocytes. Arch. Biochem. Biophys. 2016, 594, 26–36. [Google Scholar] [CrossRef]
- Upadhyay, S.; Gupta, K.B.; Mantha, A.K.; Dhiman, M. A short review: Doxorubicin and its effect on cardiac proteins. J. Cell. Biochem. 2021, 122, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.; Morris, P.; Fazlanie, A.L.; Vijayan, S.; Dancso, B.; Dastidar, A.G.; Plein, S.; Mueller, C.; Haaf, P. Cardiac biomarkers of the acute coronary syndrome: From history to high-sensitivity cardiac troponin. Intern. Emerg. Med. 2017, 12, 147–155. [Google Scholar] [CrossRef]
- Ladenson, J.H. Reflections on the evolution of cardiac biomarkers. Clin. Chem. 2012, 58, 21–24. [Google Scholar] [CrossRef][Green Version]
- Allahham, M.; Singh, M.; Jneid, H. Cardiac Biomarkers in Acute Myocardial Infarction. In Biomarkers in Cardiovascular Disease; Elsevier: Amsterdam, The Netherlands, 2019; pp. 109–114. [Google Scholar]
- Ahmad, M.I.; Sharma, N. Biomarkers in acute myocardial infarction. J. Clin. Exp. Cardiol. 2012, 3, 222. [Google Scholar] [CrossRef]
- Aldous, S.J. Cardiac biomarkers in acute myocardial infarction. Int. J. Cardiol. 2013, 164, 282–294. [Google Scholar] [CrossRef]
- Zhang, G.J.; Luo, Z.H.; Huang, M.J.; Ang, J.J.; Kang, T.G.; Ji, H. An integrated chip for rapid, sensitive, and multiplexed detection of cardiac biomarkers from fingerprick blood. Biosens. Bioelectron. 2011, 28, 459–463. [Google Scholar] [CrossRef]
- Fathil, M.F.; Arshad, M.M.; Gopinath, S.C.; Hashim, U.; Adzhri, R.; Ayub, R.M.; Ruslinda, A.R.; Nuzaihan, M.; Azman, A.H.; Zaki, M.; et al. Diagnostics on acute myocardial infarction: Cardiac troponin biomarkers. Biosens. Bioelectron. 2015, 70, 209–220. [Google Scholar] [CrossRef]
- Bjurman, C.; Petzold, M.; Venge, P.; Farbemo, J.; Fu, M.L.; Hammarsten, O. High-sensitive cardiac troponin, NT-proBNP, hFABP and copeptin levels in relation to glomerular filtration rates and a medical record of cardiovascular disease. Clin. Biochem. 2015, 48, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Atas, E.; Kismet, E.; Kesik, V.; Karaoglu, B.; Aydemir, G.; Korkmazer, N.; Demirkaya, E.; Karslioglu, Y.; Yurttutan, N.; Unay, B.; et al. Cardiac troponin-I, brain natriuretic peptide and endothelin-1 levels in a rat model of doxorubicin-induced cardiac injury. J. Cancer Res. Ther. 2015, 11, 882. [Google Scholar] [CrossRef]
- Reagan, W.J.; York, M.; Berridge, B.; Schultze, E.; Walker, D.; Pettit, S. Comparison of cardiac troponin I and T, including the evaluation of an ultrasensitive assay, as indicators of doxorubicin-induced cardiotoxicity. Toxicol. Pathol. 2013, 41, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, R.P.; Neves, A.L.; Guimarães, H. Cardiac Biomarkers in Neonatology: BNP/NT pro-BNP, Troponin I/T, CKMB and Myoglobin, a systematic review. J. Pediatr. Neonatal Individ. Med. 2017, 6, e060219. [Google Scholar]
- Khiati, S.; Dalla Rosa, I.; Sourbier, C.; Ma, X.; Rao, V.A.; Neckers, L.M.; Zhang, H.; Pommier, Y. Mitochondrial topoisomerase I (top1mt) is a novel limiting factor of doxorubicin cardiotoxicity. Clin. Cancer Res. 2014, 20, 4873–4881. [Google Scholar] [CrossRef]
- Johnson, N.A.; Slack, G.W.; Savage, K.J.; Connors, J.M.; Ben-Neriah, S.; Rogic, S.; Scott, D.W.; Tan, K.L.; Steidl, C.; Sehn, L.H.; et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 2012, 30, 3452–3459. [Google Scholar] [CrossRef] [PubMed]
- Santaguida, P.L.; Don-Wauchope, A.C.; Oremus, M.; McKelvie, R.; Ali, U.; Hill, S.A.; Balion, C.; Booth, R.A.; Brown, J.A.; Bustamam, A.; et al. BNP, and NT-proBNP as prognostic markers in persons with acute decompensated heart failure: A systematic review. Heart Fail. Rev. 2014, 19, 453–470. [Google Scholar] [CrossRef]
- Farnsworth, C.W.; Bailey, A.L.; Jaffe, A.S.; Scott, M.G. Diagnostic concordance between NT-proBNP and BNP for suspected heart failure. Clin. Biochem. 2018, 59, 50–55. [Google Scholar] [CrossRef]
- Fertin, M.; Hennache, B.; Hamon, M.; Ennezat, P.V.; Biausque, F.; Elkohen, M.; Nugue, O.; Tricot, O.; Lamblin, N.; Pinet, F.; et al. Usefulness of serial assessment of B-type natriuretic peptide, troponin I, and C-reactive protein to predict left ventricular remodeling after acute myocardial infarction (from the REVE-2 study). Am. J. Cardiol. 2010, 106, 1410–1416. [Google Scholar] [CrossRef]
- Terahara, N. Flavonoids in foods: A review. Nat. Prod. Commun. 2015, 10, 521–528. [Google Scholar] [CrossRef]
- Agrawal, A.D. Pharmacological activities of flavonoids: A review. Int. J. Pharm. Sci. Nanotechnol. 2011, 4, 1394–1398. [Google Scholar] [CrossRef]
- Mulvihill, E.E.; Huff, M.W. Antiatherogenic properties of flavonoids: Implications for cardiovascular health. Can. J. Cardiol. 2010, 26, 17A–21A. [Google Scholar] [CrossRef]
- Chiva-Blanch, G.; Badimon, L. Effects of polyphenol intake on the metabolic syndrome: Current evidence from human trials. Oxid. Med. Cell. Longev. 2017, 2017, 5812401. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.; Peluso, I.; Raguzzini, A. Flavonoids as anti-inflammatory agents. Proc. Nutr. Soc. 2010, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Treml, J.; Šmejkal, K. Flavonoids as potent scavengers of hydroxyl radicals. Compr. Rev. Food Sci. Food Saf. 2016, 15, 720–738. [Google Scholar] [CrossRef]
- Zaragozá, C.; Monserrat, J.; Mantecón, C.; Villaescusa, L.; Zaragozá, F.; Álvarez-Mon, M. Antiplatelet activity of flavonoid and coumarin drugs. Vasc. Pharmacol. 2016, 87, 139–149. [Google Scholar] [CrossRef]
- Vazhappilly, C.G.; Ansari, S.A.; Al-Jaleeli, R.; Al-Azawi, A.M.; Ramadan, W.S.; Menon, V.; Hodeify, R.; Siddiqui, S.S.; Merheb, M.; Matar, R.; et al. Role of flavonoids in thrombotic, cardiovascular, and inflammatory diseases. Inflammopharmacology 2019, 27, 863–869. [Google Scholar] [CrossRef]
- Ojeda, D.; Jiménez-Ferrer, E.; Zamilpa, A.; Herrera-Arellano, A.; Tortoriello, J.; Alvarez, L. Inhibition of angiotensin-converting enzyme (ACE) activity by the anthocyanins delphinidin-and cyanidin-3-O-sambubiosides from Hibiscus sabdariffa. J. Ethnopharmacol. 2010, 127, 7–10. [Google Scholar] [CrossRef]
- Mishra, P.K.; Raghuram, G.V.; Bhargava, A.; Ahirwar, A.; Samarth, R.; Upadhyaya, R.; Jain, S.K.; Pathak, N. In vitro and in vivo evaluation of the anticarcinogenic and cancer chemopreventive potential of a flavonoid-rich fraction from a traditional Indian herb Selaginella bryopteris. Br. J. Nutr. 2011, 106, 1154–1168. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Shang, P.; Li, D. Luteolin: A flavonoid that has multiple cardioprotective effects and its molecular mechanisms. Front. Pharmacol. 2017, 8, 692. [Google Scholar] [CrossRef]
- Putri, N.L.; Elya, B.; Puspitasari, N. Antioxidant activity and lipoxygenase inhibition test with total flavonoid content from Garcinia kydia Roxburgh leaves extract. Pharmacogn. J. 2017, 9, 280–284. [Google Scholar] [CrossRef]
- Brodowska, K.M. Natural flavonoids: Classification, potential role, and application of flavonoid analogues. Eur. J. Biol. Res. 2017, 7, 108–123. [Google Scholar]
- Manzoor, M.F.; Ahmad, N.; Ahmed, Z.; Siddique, R.; Zeng, X.A.; Rahaman, A.; Muhammad Aadil, R.; Wahab, A. Novel extraction techniques and pharmaceutical activities of luteolin and its derivatives. J. Food Biochem. 2019, 43, e12974. [Google Scholar] [CrossRef]
- Aziz, N.; Kim, M.Y.; Cho, J.Y. Anti-inflammatory effects of luteolin: A review of in vitro, in vivo, and in silico studies. J. Ethnopharmacol. 2018, 225, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Linn, B.S.; Zhang, Y.; Ren, J. A review on the antioxidative and prooxidative properties of luteolin. React. Oxyg. Species 2019, 7, 136–147. [Google Scholar] [CrossRef]
- Arai, Y.; Endo, S.; Miyagi, N.; Abe, N.; Miura, T.; Nishinaka, T.; Terada, T.; Oyama, M.; Goda, H.; El-Kabbani, O.; et al. Structure-activity relationship of flavonoids as potent inhibitors of carbonyl reductase 1 (CBR1). Fitoterapia 2015, 101, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Schaupp, C.M.; White, C.C.; Merrill, G.F.; Kavanagh, T.J. Metabolism of doxorubicin to the cardiotoxic metabolite doxorubicinol is increased in a mouse model of chronic glutathione deficiency: A potential role for carbonyl reductase 3. Chem.-Biol. Interact. 2015, 234, 154–161. [Google Scholar] [CrossRef]
- Yu, D.; Li, M.; Tian, Y.; Liu, J.; Shang, J. Luteolin inhibits ROS-activated MAPK pathway in myocardial ischemia/reperfusion injury. Life Sci. 2015, 122, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Xu, T.; Wu, P.; Pan, D.; Chen, J.; Chen, J.; Zhang, B.; Zhu, H.; Li, D. Luteolin improves cardiac dysfunction in heart failure rats by regulating sarcoplasmic reticulum Ca 2+-ATPase 2a. Sci. Rep. 2017, 7, 41017. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zheng, L.; Yang, B.; Wang, X.; Ying, Y. Luteolin attenuates atherosclerosis via modulating signal transducer and activator of transcription 3-mediated inflammatory response. Drug Des. Dev. Ther. 2019, 13, 3899–3911. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, C.; Liu, C.; Wei, F. Luteolin attenuates doxorubicin-induced cardiotoxicity by modulating the PHLPP1/AKT/Bcl-2 signaling pathway. PeerJ 2020, 8, e8845. [Google Scholar] [CrossRef]
- Yao, H.; Shang, Z.; Wang, P.; Li, S.; Zhang, Q.; Tian, H.; Ren, D.; Han, X. Protection of luteolin-7-O-glucoside against doxorubicin-induced injury through PTEN/Akt and ERK pathway in H9c2 cells. Cardiovasc. Toxicol. 2016, 16, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Gharanei, M.; Hussain, A.; Janneh, O.; Maddock, H.L. Doxorubicin-induced myocardial injury is exacerbated following ischaemic stress via an opening of the mitochondrial permeability transition pore. Toxicol. Appl. Pharmacol. 2013, 268, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Q.; Li, G.; Ke, D. Ghrelin stimulates angiogenesis via GHSR1a-dependent MEK/ERK and PI3K/Akt signal pathways in rat cardiac microvascular endothelial cells. Peptides 2012, 33, 92–100. [Google Scholar] [CrossRef]
- Xu, H.; Yu, W.; Sun, S.; Li, C.; Zhang, Y.; Ren, J. Luteolin attenuates doxorubicin-induced cardiotoxicity through promoting mitochondrial autophagy. Front. Physiol. 2020, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Han, X.Z.; Li, X.; Ren, D.M.; Wang, X.N.; Lou, H.X. Flavonoids from Dracocephalum tanguticum and their cardioprotective effects against doxorubicin-induced toxicity in H9c2 cells. Bioorg. Med. Chem. Lett. 2010, 20, 6411–6415. [Google Scholar] [CrossRef] [PubMed]
- Syahputra, R.A.; Harahap, U.; Dalimunthe, A.; Pandapotan, M.; Satria, D. Protective effect of Vernonia amygdalina Delile against doxorubicin-induced cardiotoxicity. Heliyon 2021, 7, e07434. [Google Scholar] [CrossRef] [PubMed]
- Azarabadi, S.; Abdollahi, H.; Torabi, M.; Salehi, Z.; Nasiri, J. ROS generation, oxidative burst and dynamic expression profiles of ROS-scavenging enzymes of superoxide dismutase (SOD), catalase (CAT), and ascorbate peroxidase (APX) in response to Erwinia amylovora in pear (Pyrus communis L). Eur. J. Plant Pathol. 2017, 147, 279–294. [Google Scholar] [CrossRef]
- Razavi-Azarkhiavi, K.; Iranshahy, M.; Sahebkar, A.; Shirani, K.; Karimi, G. The protective role of phenolic compounds against doxorubicin-induced cardiotoxicity: A comprehensive review. Nutr. Cancer 2016, 68, 892–917. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, C.; Zhu, H.; Wang, S.; Zhou, Y.; Zhao, J.; Xia, Y.; Li, D. Luteolin modulates SERCA2a via Sp1 upregulation to attenuate myocardial ischemia/reperfusion injury in mice. Sci. Rep. 2020, 10, 15407. [Google Scholar] [CrossRef]
- Kelly, G.S. Quercetin. Altern. Med. Rev. 2011, 16, 172–195. [Google Scholar]
- Sultana, B.; Anwar, F. Flavonols (kaempferol, quercetin, myricetin) contents of selected fruits, vegetables, and medicinal plants. Food Chem. 2008, 108, 879–884. [Google Scholar] [CrossRef]
- Batiha, G.E.; Beshbishy, A.M.; Ikram, M.; Mulla, Z.S.; El-Hack, M.E.; Taha, A.E.; Algammal, A.M.; Elewa, Y.H. The pharmacological activity, biochemical properties, and pharmacokinetics of the major natural polyphenolic flavonoid: Quercetin. Foods 2020, 9, 374. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Zhang, Z.Y.; Wang, R.X. Protective mechanisms of quercetin against myocardial ischemia-reperfusion injury. Front. Physiol. 2020, 11, 956. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Hu, R.Y.; Chou, H.C. Quercetin-induced cardioprotection against doxorubicin cytotoxicity. J. Biomed. Sci. 2013, 20, 95. [Google Scholar] [CrossRef]
- Dong, Q.; Chen, L.; Lu, Q.; Sharma, S.; Li, L.; Morimoto, S.; Wang, G. Quercetin attenuates doxorubicin cardiotoxicity by modulating B mi-1 expression. Br. J. Pharmacol. 2014, 171, 4440–4454. [Google Scholar] [CrossRef]
- Chen, X.; Peng, X.; Luo, Y.; You, J.; Yin, D.; Xu, Q.; He, H.; He, M. Quercetin protects cardiomyocytes against doxorubicin-induced toxicity by suppressing oxidative stress and improving mitochondrial function via 14-3-3γ. Toxicol. Mech. Methods 2019, 29, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.A.; Abtar, A.N.; Othman, H.H.; Aziz, T.A. Effects of quercetin, sitagliptin alone or in combination in testicular toxicity induced by doxorubicin in rats. Drug Des. Dev. Ther. 2019, 13, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Matouk, A.I.; Taye, A.; Heeba, G.H.; El-Moselhy, M.A. Quercetin augments the protective effect of losartan against chronic doxorubicin cardiotoxicity in rats. Environ. Toxicol. Pharmacol. 2013, 36, 443–450. [Google Scholar] [CrossRef]
- Al-Oanzi, Z.H.; Elasbali, A.M.; Alruwaili, N.K.; Alotaibi, N.H.; Alharbi, K.S.; Alzarea, A.I.; Alsuwayt, B.H.; Al-Enazi, M.M. Protective effect of baicalein alone and losartan-baicalein combination therapy on doxorubicin-induced hepatotoxicity in rats. Toxicol. Environ. Health Sci. 2020, 12, 45–54. [Google Scholar] [CrossRef]
- Sharma, A.; Parikh, M.; Shah, H.; Gandhi, T. Modulation of Nrf2 by quercetin in doxorubicin-treated rats. Heliyon 2020, 6, e03803. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The therapeutic potential of apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Rahul; Naz, F.; Ali, F.; Jyoti, S.; Siddique, Y.H. Health functionality of apigenin: A review. Int. J. Food Prop. 2017, 20, 1197–1238. [Google Scholar] [CrossRef]
- Sahu, R.; Dua, T.K.; Das, S.; De Feo, V.; Dewanjee, S. Wheat phenolics suppress doxorubicin-induced cardiotoxicity via inhibition of oxidative stress, MAP kinase activation, NF-κB pathway, PI3K/Akt/mTOR impairment, and cardiac apoptosis. Food Chem. Toxicol. 2019, 125, 503–519. [Google Scholar] [CrossRef]
- Zare, M.F.; Rakhshan, K.; Aboutaleb, N.; Nikbakht, F.; Naderi, N.; Bakhshesh, M.; Azizi, Y. Apigenin attenuates doxorubicin-induced cardiotoxicity via reducing oxidative stress and apoptosis in male rats. Life Sci. 2019, 232, 116623. [Google Scholar] [CrossRef]
- Yu, W.; Sun, H.; Zha, W.; Cui, W.; Xu, L.; Min, Q.; Wu, J. Apigenin attenuates adriamycin-induced cardiomyocyte apoptosis via the PI3K/AKT/mTOR pathway. Evid.-Based Complement. Altern. Med. 2017, 2017, 2590676. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Ma, S.; Zhu, Y.; Shao, Q.; Hou, J.; Li, X. Apigenin-7-O-β-d-(6″-p-coumaroyl)-glucopyranoside reduces myocardial ischaemia/reperfusion injury in an experimental model via regulating the inflammation response. Pharm. Biol. 2020, 58, 80–88. [Google Scholar] [CrossRef]
- Al-Dhabi, N.A.; Arasu, M.V.; Park, C.H.; Park, S.U. An up-to-date review of rutin and its biological and pharmacological activities. EXCLI J. 2015, 14, 59–63. [Google Scholar] [PubMed]
- Dagnon, S.; Novkova, Z.; Bojilov, D.; Nedialkov, P.; Kouassi, C. Development of surrogate standards approach for the determination of polyphenols in Vernonia amygdalina Del. J. Food Compos. Anal. 2019, 82, 103231. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Hsiu, S.-L.; Wen, K.-C.; Lin, S.-P.; Tsai, S.-Y.; Hou, Y.-C.; Chao, P.-D. Bioavailability and metabolic pharmacokinetics of rutin and quercetin in rats. J. Food Drug Anal. 2005, 13, 5. [Google Scholar] [CrossRef]
- Panchal, S.K.; Poudyal, H.; Arumugam, T.V.; Brown, L. Rutin attenuates metabolic changes, nonalcoholic steatohepatitis, and cardiovascular remodeling in high-carbohydrate, high-fat diet-fed rats. J. Nutr. 2011, 141, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.G.; Miranda-Silva, D.; Costa, S.M.; Barros, C.; Hamdani, N.; Moura, C.; Mendes, M.J.; Sousa-Mendes, C.; Trindade, F.; Fontoura, D.; et al. Early myocardial changes induced by doxorubicin in the nonfailing dilated ventricle. Am. J. Physiol.—Heart Circ. Physiol. 2019, 316, H459–H475. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cho, H.; Lee, S.; Woo, J.S.; Cho, B.H.; Kang, J.H.; Jeong, Y.M.; Cheng, X.W.; Kim, W. Enhanced-autophagy by exenatide mitigates doxorubicin-induced cardiotoxicity. Int. J. Cardiol. 2017, 232, 40–47. [Google Scholar] [CrossRef]
- Dirks-Naylor, A.J. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci. 2013, 93, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tang, N.; Liu, F.Y.; Yang, Z.; Ma, S.Q.; An, P.; Wu, H.M.; Fan, D.; Tang, Q.Z. TLR9 deficiency alleviates doxorubicin-induced cardiotoxicity via the regulation of autophagy. J. Cell. Mol. Med. 1091, 24, 10913–10923. [Google Scholar] [CrossRef] [PubMed]
- Petroni, K.; Trinei, M.; Fornari, M.; Calvenzani, V.; Marinelli, A.; Micheli, L.A.; Pilu, R.; Matros, A.; Mock, H.P.; Tonelli, C.; et al. Dietary cyanidin 3-glucoside from purple corn ameliorates doxorubicin-induced cardiotoxicity in mice. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.C.; Kuo, W.W.; Shen, C.Y.; Chen, Y.F.; Lin, Y.M.; Ho, T.J.; Padma, V.V.; Lo, J.F.; Huang, C.Y. Anthocyanin attenuates doxorubicin-induced cardiotoxicity via estrogen receptor-α/β and stabilizes HSF1 to inhibit the IGF-IIR apoptotic pathway. Int. J. Mol. Sci. 2016, 17, 1588. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, W.J.; Chang, S.E.; Lee, G.Y. Hesperidin, a popular antioxidant inhibits melanogenesis via Erk1/2 mediated MITF degradation. Int. J. Mol. Sci. 2015, 16, 18384–18395. [Google Scholar] [CrossRef]
- Trivedi, P.P.; Kushwaha, S.; Tripathi, D.N.; Jena, G.B. Cardioprotective effects of hesperetin against doxorubicin-induced oxidative stress and DNA damage in rats. Cardiovasc. Toxicol. 2011, 11, 215–225. [Google Scholar] [CrossRef]
- Saad, S.; Ahmad, I.; Kawish, S.M.; Khan, U.A.; Ahmad, F.J.; Ali, A.; Jain, G.K. Improved cardioprotective effects of hesperidin solid lipid nanoparticles prepared by supercritical antisolvent technology. Colloids Surf. B Biointerfaces 2020, 187, 110628. [Google Scholar] [CrossRef] [PubMed]
- Doerr, V.; Montalvo, R.N.; Kwon, O.S.; Talbert, E.E.; Hain, B.A.; Houston, F.E.; Smuder, A.J. Prevention of doxorubicin-induced autophagy attenuates oxidative stress and skeletal muscle dysfunction. Antioxidants 2020, 9, 263. [Google Scholar] [CrossRef]
- Mani, R.; Natesan, V. Chrysin: Sources, beneficial pharmacological activities, and molecular mechanism of action. Phytochemistry 2018, 145, 187–196. [Google Scholar] [CrossRef]
- Mantawy, E.M.; Esmat, A.; El-Bakly, W.M.; ElDin, R.A.; El-Demerdash, E. Mechanistic clues to the protective effect of chrysin against doxorubicin-induced cardiomyopathy: Plausible roles of p53, MAPK and AKT pathways. Sci. Rep. 2017, 7, 4795. [Google Scholar] [CrossRef]
- Mantawy, E.M.; El-Bakly, W.M.; Esmat, A.; Badr, A.M.; El-Demerdash, E. Chrysin alleviates acute doxorubicin cardiotoxicity in rats via suppression of oxidative stress, inflammation and apoptosis. Eur. J. Pharmacol. 2014, 728, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Fokou, P.V.; Sharifi-Rad, M.; Zucca, P.; Pezzani, R.; Martins, N.; Sharifi-Rad, J. The therapeutic potential of naringenin: A review of clinical trials. Pharmaceuticals 2019, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T. Adriamycin (doxorubicin) cardiotoxicity: A review. West. J. Med. 1979, 131, 364. [Google Scholar]
- Shiromwar, S.S.; Chidrawar, V.R. Combined effects of p-coumaric acid and naringenin against doxorubicin-induced cardiotoxicity in rats. Pharmacogn. Res. 2011, 3, 214–219. [Google Scholar] [CrossRef]
- Abd-Allah, A.R.; Al-Majed, A.A.; Mostafa, A.M.; Al-Shabanah, O.A.; Din, A.G.E.; Nagi, M.N. Protective effect of arabic gum against cardiotoxicity induced by doxorubicin in mice: A possible mechanism of protection. J. Biochem. Mol. Toxicol. 2002, 16, 254–259. [Google Scholar] [CrossRef]
- Kwatra, M.; Kumar, V.; Jangra, A.; Mishra, M.; Ahmed, S.; Ghosh, P.; Vohora, D.; Khanam, R. Ameliorative effect of naringin against doxorubicin-induced acute cardiac toxicity in rats. Pharm. Biol. 2016, 54, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Subburaman, S.; Ganesan, K.; Ramachandran, M. Protective role of naringenin against doxorubicin-induced cardiotoxicity in a rat model: Histopathology and mRNA expression profile studies. J. Environ. Pathol. Toxicol. Oncol. 2014, 33, 363–376. [Google Scholar] [CrossRef]
- Kaiserová, H.; Šimůnek, T.; van der Vijgh, W.J.; Bast, A.; Kvasničková, E. Flavonoids as protectors against doxorubicin cardiotoxicity: Role of iron chelation, antioxidant activity and inhibition of carbonyl reductase. Biochim. Biophys. Acta—Mol. Basis Dis. 2007, 1772, 1065–1074. [Google Scholar] [CrossRef]
- Cote, B.; Carlson, L.J.; Rao, D.A.; Alani, A.W. Combinatorial resveratrol and quercetin polymeric micelles mitigate doxorubicin-induced cardiotoxicity in vitro and in vivo. J. Control. Release 2015, 213, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Sun, B.; Tong, Q.; Ren, L. Rutin protects against pirarubicin-induced cardiotoxicity through the TGF-β1-p38 MAPK signaling pathway. Evid.-Based Complement. Altern. Med. 2017, 2017, 1759385. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, L.; Ma, J.; Lu, L.; Wang, X.; Ren, J.; Yang, J. Rutin attenuates doxorubicin-induced cardiotoxicity via regulating autophagy and apoptosis. Biochim. Biophys. Acta—Mol. Basis Dis. 2017, 1863, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Hozayen, W.G.; Abou Seif, H.S. Protective effects of rutin and hesperidin against doxorubicin-induced lipodystrophy and cardiotoxicity in albino rats. J. Am. Sci. 2011, 7, 765–775. [Google Scholar]
- Tang, S.; Kan, J.; Sun, R.; Cai, H.; Hong, J.; Jin, C.; Zong, S. Anthocyanins from purple sweet potato alleviate doxorubicin-induced cardiotoxicity in vitro and in vivo. J. Food Biochem. 2021, 45, e13869. [Google Scholar] [CrossRef] [PubMed]
- Donia, T.I.; Gerges, M.N.; Mohamed, T.M. Amelioration effect of Egyptian sweet orange hesperidin on Ehrlich ascites carcinoma (EAC) bearing mice. Chem.-Biol. Interact. 2018, 285, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Arafa, H.M.; Abd-Ellah, M.F.; Hafez, H.F. Abatement by naringenin of doxorubicin-induced cardiac toxicity in rats. J. Egypt. Natl. Cancer Inst. 2005, 17, 291–300. [Google Scholar]
- Han, X.; Gao, S.; Cheng, Y.; Sun, Y.; Liu, W.; Tang, L.; Ren, D. Protective effect of naringenin-7-O-glucoside against oxidative stress induced by doxorubicin in H9c2 cardiomyocytes. Biosci. Trends 2012, 6, 19–25. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syahputra, R.A.; Harahap, U.; Dalimunthe, A.; Nasution, M.P.; Satria, D. The Role of Flavonoids as a Cardioprotective Strategy against Doxorubicin-Induced Cardiotoxicity: A Review. Molecules 2022, 27, 1320. https://doi.org/10.3390/molecules27041320
Syahputra RA, Harahap U, Dalimunthe A, Nasution MP, Satria D. The Role of Flavonoids as a Cardioprotective Strategy against Doxorubicin-Induced Cardiotoxicity: A Review. Molecules. 2022; 27(4):1320. https://doi.org/10.3390/molecules27041320
Chicago/Turabian StyleSyahputra, Rony Abdi, Urip Harahap, Aminah Dalimunthe, M. Pandapotan Nasution, and Denny Satria. 2022. "The Role of Flavonoids as a Cardioprotective Strategy against Doxorubicin-Induced Cardiotoxicity: A Review" Molecules 27, no. 4: 1320. https://doi.org/10.3390/molecules27041320
APA StyleSyahputra, R. A., Harahap, U., Dalimunthe, A., Nasution, M. P., & Satria, D. (2022). The Role of Flavonoids as a Cardioprotective Strategy against Doxorubicin-Induced Cardiotoxicity: A Review. Molecules, 27(4), 1320. https://doi.org/10.3390/molecules27041320