
Synthesis, Characterization, and Catalytic Exploration of Mononuclear Mo(VI) Dioxido Complexes of (Z)-1-R-2-(4′,4′-Dimethyl-2′-oxazolin-2′-yl)-eth-1-en-1-ates †

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Synthesis and Characterisation
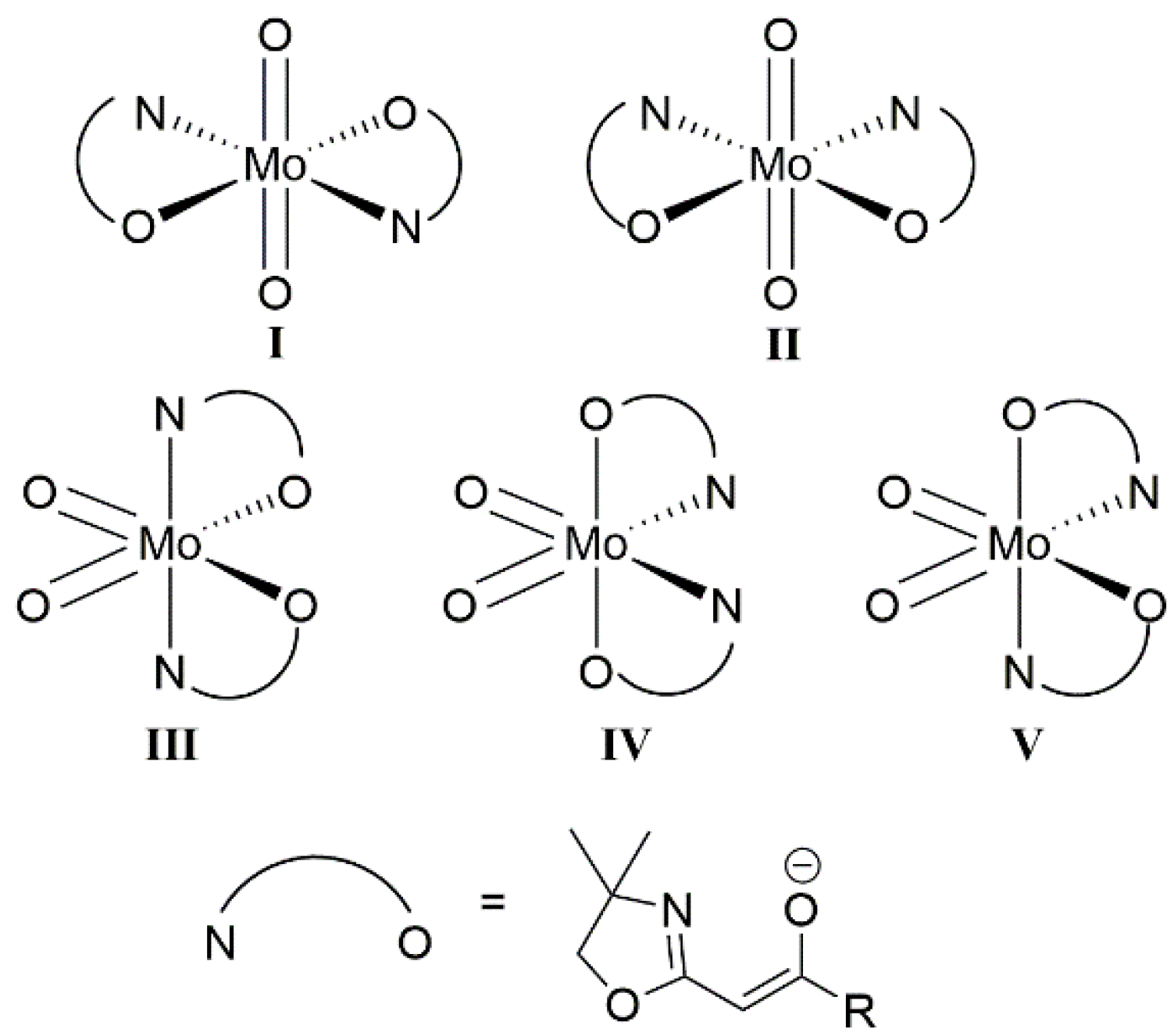
2.2. Semi-Empirical PM6(tm) Treatment of Hypothetical Complex 5
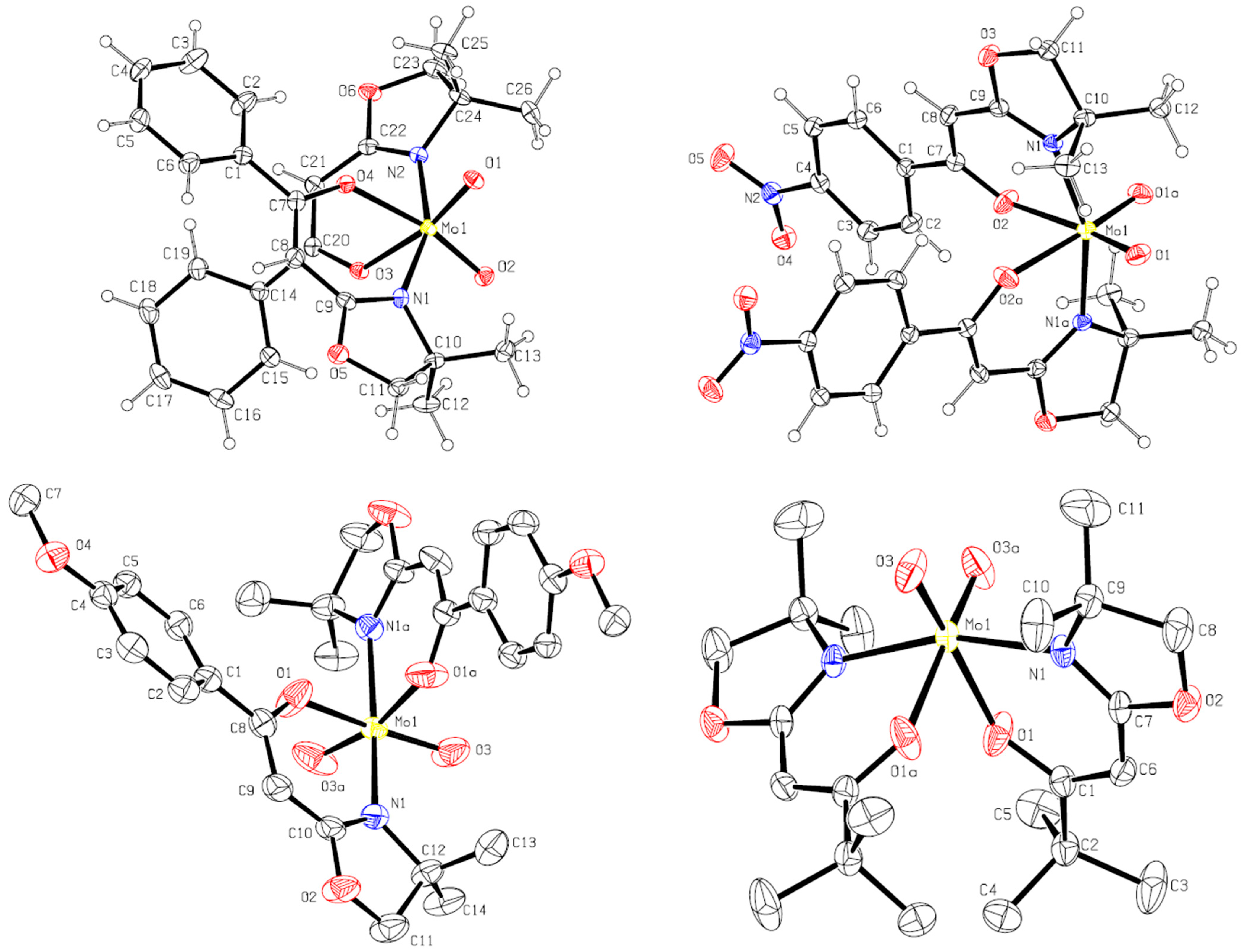
2.3. X-ray Structural Studies
2.4. Catalytic Explorations
2.4.1. Norbornene Polymerization
2.4.2. Benzoin Oxidation to Benzil
3. Discussion
3.1. Synthesis and Structural Investigations
3.2. Catalysis
4. Materials and Methods
4.1. Synthesis
4.1.1. General Information
4.1.2. Complex 1: MoO2(κ2-N,O-L1)2
4.1.3. Complex 2: MoO2(κ2-N,O-L2)2
4.1.4. Complex 3: MoO2(κ2-N,O-L3)2
4.1.5. Complex 4: MoO2(κ2-N,O-L4)2
4.2. Semi-Empirical PM6(tm) Calculations
4.3. X-ray Diffraction
4.4. Catalysis
4.4.1. Norbornene Polymerization
4.4.2. Benzoin Oxidation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Meyers, A.I.; Temple, D.L.; Nolen, R.L.; Mihelich, E.D. Oxazolines. IX. Synthesis of homologated acetic acids and esters. J. Org. Chem. 1974, 39, 2778–2783. [Google Scholar] [CrossRef]
- Tohda, Y.; Kawashima, T.; Ariga, M.; Akiyama, R.; Shudoh, H.; Mori, Y. A convenient synthesis of 2-acylmethyl-4,4-dimethyl-2-oxazolines. Useful reagents for β–keto ester synthesis. Bull. Chem. Soc. Jpn. 1984, 57, 2329–2330. [Google Scholar] [CrossRef]
- Tohda, Y.; Morikawa, M.; Kawashima, T.; Ariga, M.; Mori, Y. Crossed Claisen condensation of 2-alkyl-4,4-dimethyl-2-oxazolines with acid anhydrides by aluminum chloride and triethylamine. Chem. Lett. 1986, 273–274. [Google Scholar] [CrossRef]
- Castan, F.; Denonne, F.; Bigg, D.C.H. Preparation of 2-(β-oxo)-2-oxazolines and thiazolines by reaction of enamines with 2-chloroethyl iso(thio)cyanates. Synthesis 1993, 1081–1083. [Google Scholar] [CrossRef]
- Tohda, Y.; Yanagidani, T.; Hiramatsu, S.; Nishiwaki, N.; Tani, K.; Imagawa, K.; Ariga, M. Synthesis via 2-acylmethyl-2-oxazoline. I. A novel synthesis of 3-acyl-2-pyridones by Michael Addition of 2-acylmethyl-2-oxazoline to α,β–acetylenic ketones. Bull. Chem. Soc. Jpn. 1997, 70, 2781–2790. [Google Scholar] [CrossRef]
- Chen, X.; Wang, X.-L.; Lian, H.-Z.; Chen, J.-J.; Pan, Y.; Shi, Y.-Z. A new procedure to enols of 2-acylmethyl-4, 4-dimethyl-2-oxazolines under ultrasonically dispersed potassium system. Chin. J. Chem. 1999, 17, 80–83. [Google Scholar] [CrossRef]
- Junior, A.W.; Oliveira, A.R.M.; da Cunha, C.J.; Simonelli, F.; Marques, F.A. Synthesis of enaminones with stationary stereochemistry. J. Braz. Chem. Soc. 1999, 10, 369–374. [Google Scholar] [CrossRef]
- Song, Y.; de Silva, H.I.; Henry, W.P.; Ye, G.; Chatterjee, S.; Pittman, C.U., Jr. Regiochemistry of an ambident cyclic ketene-N,O-acetal nucleophile and its anion towards electrophiles. Tetrahedron Lett. 2011, 52, 4507–4511. [Google Scholar] [CrossRef]
- Jones, R.C.; Herasymchuk, K.; Mahdi, T.; Petrov, A.; Resanović, S.; Vaughan, D.G.; Lough, A.J.; Quail, J.W.; Koivisto, B.D.; Wylie, R.S.; et al. Tautomerism and metal complexation of 2-acylmethyl2-oxazolines: A combined synthetic, spectroscopic, crystallographic and theoretical treatment. Org. Biomol. Chem. 2013, 11, 3484–3493, Corrigendum in Org. Biomol. Chem. 2013, 11, 8509. [Google Scholar] [CrossRef] [Green Version]
- Tohda, Y.; Yanagidani, T.; Asaka, N. A novel abnormal Michael reaction of 2-Acyl-4,4-dimethyl-2-oxazolines with acetylenic ketones and esters. Bull. Chem. Soc. Jpn. 2016, 89, 810–822. [Google Scholar] [CrossRef]
- May, K.L.; Resanović, S.; Chojnacka, M.W.; Herasymchuk, K.; Vaughan, D.G.; Zhu, J.F.; Quail, J.W.; Lough, A.J.; Gossage, R.A. Divalent cobalt and copper coordination complexes of κ2-N, O-derivatives of (Z)-1-R-2-(2′-oxazolin-2′-yl)-eth-1-en-1-ates: Structure and reactivity patterns. Inorg. Chim. Acta 2021, 514, 119959. [Google Scholar] [CrossRef]
- Adjei, J.A.; Lough, A.J.; Gossage, R.A. Synthesis and characterization of κ2-N,O-oxazoline-enolate complexes of nickel(II): Explorations in coordination chemistry and metal-mediated polymerisation. RSC Adv. 2019, 9, 3956–3964. [Google Scholar] [CrossRef] [Green Version]
- May, K.L.; Clément, R.; Lough, A.J.; Gossage, R.A. Organometallic iridium complexes of (Z)-1-R-2-(2′-oxazolin-2′-yl)-eth-1-en-1-ates: Structural aspects, reactivity and applications in the catalytic dehydration of alkanes. Bull. Chem. Soc. Jpn. 2021, 94, 2043–2047. [Google Scholar] [CrossRef]
- Hill, M.C.; Lough, A.J.; Gossage, R.A. Heteroatom exchange chemistry in (Z)-1-R-2-(4′,4′-dimethyl)-2′-oxazolin-2′-yl)-eth-1-en-1-ols: Access to chelate-stabilized thioester analogues of dithiooxophospharanes. Chem. Lett. 2021; in press. [Google Scholar] [CrossRef]
- García-Martínez, J. Standing on the shoulders of giants—Your mentors and role models will shape your career. Chem. Eur. J. 2021, 27, 13664–13668. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.; Jones, R.C.; Vaughan, D.G.; Lough, A.J.; Gossage, R.A. The crystal and molecular structure of (Z)-2-[3-(4-Methoxybenzoyl)-4,4-dimethyl-1,2-oxazolidin-2-ylidene]-1-(4-methoxyphenyl)ethanone. Crystals 2011, 1, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Gehrke, H., Jr.; Veal, J. Acetylacetonate complexes of molybdenum (V) and molybdenum (VI). Inorg. Chim. Acta 1969, 3, 623–627. [Google Scholar] [CrossRef]
- Soptrajanov, B.; Nikolovski, A.; Petrov, I. Infra-red spectra of dioxobis(acetylacetonato) tungsten (VI) and dioxobis(acetylacetonato) molybdenum (VI). Spectrochim. Acta 1968, 24, 1617–1621. [Google Scholar] [CrossRef]
- Conte, M.; Hippler, M. Dynamic NMR and quantum-chemical study of the stereochemistry and stability of the chiral complex MoO2(acac)2 in solution. J. Phys. Chem. A 2016, 120, 6677–6687. [Google Scholar] [CrossRef]
- Minenkov, Y.; Sharapa, D.I.; Cavallo, L. Application of semiempirical methods to transition metal complexes: Fast results but hard-to-predict accuracy. J. Chem. Theory Comput. 2018, 14, 3428–3439. [Google Scholar] [CrossRef]
- Baerlocher, F.J.; Bucur, R.; Decken, A.; Eisnor, C.R.; Gossage, R.A.; Jackson, S.M.; Jolly, L.; Wheaton, S.L.; Wylie, R.S. Oxazoles XXII. The cobalt(II) coordination chemistry of 2-(ortho-anilinyl)-4,4-dimethyl-2-oxazoline: Synthesis, properties, and solid-state characterization. Aust. J. Chem. 2010, 63, 47–55. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for Windows—a version of ORTEP-III with a graphical user interface (GUI). J. Appl. Cryt. 1997, 30, 565. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. II 1987, S1–S19. [Google Scholar] [CrossRef]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d- and f-block metals. J. Chem. Soc. Dalton Trans. 1989, S1–S83. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization, 4th ed.; Wiley-Interscience: Hoboken, NJ, USA, 2004; pp. 103–105. [Google Scholar]
- Lehtonen, A.; Sillanpää, R. Dioxomolybdenum(VI) complexes with tri- and tetradentate aminobis(pheolate)s. Polyhedron 2005, 24, 257–265. [Google Scholar] [CrossRef]
- Barbaro, P.; Belderrain, T.R.; Bianchini, C.; Scapacci, G.; Masi, D. Dioxomolybdenum(VI) complexes with new enantiomerically pure amino diol ligands. Inorg. Chem. 1996, 35, 3362–3368. [Google Scholar] [CrossRef]
- Dinda, R.; Sengupta, P.; Ghosh, S.; Sheldrick, W.S. Synthesis, structure, and reactivity of a new molybdenum(VI) complex resembling the active center of molybdenum oxotransferases. Eur. J. Inorg. Chem. 2003, 363–369. [Google Scholar] [CrossRef]
- Maurya, M.R.; Dhaka, S.; Avecilla, F. Synthesis, characterization and catalytic activity of dioxomolybdenum(VI) complexes of tribasic pentadentate ligands. Polyhedron 2014, 67, 145–159. [Google Scholar] [CrossRef]
- Pasayat, S.; Dash, S.P.; Roy, S.; Dinda, R.; Dhaka, S.; Maurya, M.R.; Kaminsky, W.; Patil, Y.P.; Nethaji, M. Synthesis, structural studies and catalytic activity of dioxomolybdenum(VI) complexes with aroylhydrazones of napthol-derivative. Polyhedron 2014, 67, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ranjan Pramanik, N.; Ghosh, S.; Kumar Raychaudhuri, T.; Ray, S.; Butcher, R.J.; Mandal, S.S. Synthesis, characterization and crystal structure of oxomolybdenum(VI) and (IV) complexes of some tridentate ONS donor ligands. Polyhedron 2004, 23, 1595–1603. [Google Scholar] [CrossRef]
- Wong, Y.-L.; Yan, Y.; Chan, E.S.H.; Yang, Q.; Mak, T.C.W.; Ng, D.K.P. cis-Dioxo-tungsten(VI) and –molybdenum(VI) complexes with N2O2 tetradentate ligands: Synthesis, structure, electrochemistry and oxo-transfer properties. J. Chem. Soc. Dalton Trans. 1998, 3057–3064. [Google Scholar] [CrossRef]
- Wong, Y.-L.; Ma, J.-F.; Law, W.-F.; Yan, Y.; Wong, W.-T.; Zhang, Z.-Y.; Mak, T.C.W.; Ng, D.K.P. Synthesis, electrochemistry, and oxygen-atom transfer reactions of dioxotungsten(VI) and –molybdenum(VI) complexes with N2O2 and N2S2 tetradentate ligands. Eur. J. Inorg. Chem. 1999, 313–321. [Google Scholar] [CrossRef]
- Quintal, S.M.O.; Nogueira, H.I.S.; Carapuça, H.M.; Félix, V.; Drew, M.G.B. Polynuclear molybdenum and tungsten complexes of 3-hydroxypicolinic acid and the crystal structures of (nBu4N)2[Mo4O12(picOH)2] and (nHex4N)2[Mo2O6(picOH)2]. J. Chem. Soc. Dalton Trans. 2001, 3196–3201. [Google Scholar] [CrossRef]
- Quintal, S.M.O.; Nogueira, H.I.S.; Félix, V.; Drew, M.G.B. Coordination modes of 2-mercaptonicotinic acid: Synthesis and crystal structures of palladium(II), platinum(II), rhenium(III) and molybdenum(VI) complexes. J. Chem. Soc. Dalton Trans. 2002, 4479–4487. [Google Scholar] [CrossRef]
- Maurya, M.R.; Uprety, B.; Avecilla, F. Dioxidomolybdenum(VI) complexes of tripodal tetradentate ligands for catalytic oxygen atom transfer between benzoin and dimethyl sulfoxide and for oxidation of pyrogallol. Eur. J. Inorg. Chem. 2016, 4802–4813. [Google Scholar] [CrossRef]
- Hossain, M.K.; Schachner, J.A.; Haukka, M.; Lehtonen, A.; Mösch-Zanetti, N.C.; Nordlander, E. Dioxidomolybdenum(VI) and –tungsten(VI) complexes with tripodal amino bisphenolate ligands as epoxidation and oxo-transfer catalysts. Polyhedron 2017, 134, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Palanca, P.; Sanz, V.; Lahoz, L. Oxygen atom transfer involving oxomolybdenum complexes with sterically bulky thiocarboxylate ligands and biochemical interesting substrates in methanol at neutral pH. Inorg. Chim. Acta 1999, 285, 25–30. [Google Scholar] [CrossRef]
- Kumar Kurapati, S.; Pal, S. cis-Dioxomolybdeum(VI) complexes with unsymmetric linear tetradentate ligands: Syntheses, structures and bromoperoxidase activities. Appl. Organmoet. Chem. 2016, 30, 116–124. [Google Scholar] [CrossRef]
- Kumar Kurapati, S.; Maloth, S.; Pal, S. Complexes of cis-dioxomolydenum(VI) with unsymmetrical tripodal NO3-donor ligands: Synthesis, characterization and catalytic applications. Inorg. Chim. Acta 2015, 430, 66–73. [Google Scholar] [CrossRef]
- Majumber, S.; Pasayat, S.; Roy, S.; Dash, S.P.; Dhaka, S.; Maurya, M.R.; Reichelt, M.; Reuter, H.; Brzezinski, K.; Dinda, R. Dioxidomolybdenum(VI) complexes bearing sterically constrained aroylazine ligands: Synthesis, structural investigation and catalytic evaluation. Inorg. Chim. Acta 2018, 469, 366–378. [Google Scholar] [CrossRef]
- McMann, M.; Beaumont, A. Catalytic properties of novel polyoxomolybdate(VI) salts: The ring-opening metathesis polymerization (ROMP) of norbornene. J. Mol. Catal. A Chem. 1996, 108, 23–27. [Google Scholar] [CrossRef]
- Blank, F.; Janiak, C. Metal catalysts for the vinyl/additional polymerization of norbornene. Coord. Chem. Rev. 2009, 253, 827–861. [Google Scholar] [CrossRef]
- Janiak, C.; Lassahn, P.G. Metal catalysts for the vinyl polymerization of norbornene. J. Mol. Catal. A Chem. 2001, 166, 193–209. [Google Scholar] [CrossRef]
- Lehtonen, A.; Balcar, H.; Sedlacek, J.; Sillanpaa, R. Synthesis and ROMP activity of aminophenol-substituted tungsten (VI) and molybdenum (VI) complexes. J. Organomet. Chem. 2008, 693, 1171–1176. [Google Scholar] [CrossRef]
- Shi, Q.; Jie, S.; Zhang, S.; Yang, H.; Sun, W.-H. The advantages of metal complexes as catalysts towards polyolefins—The vinyl polymerization of norbornene. Macromol. Symp. 2007, 260, 74–79. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Fujita, A.; Suzuki, N.; Ito, T. Metathesis polymerization of norbornene and terminal acetylenes catalyzed by bis(acetonitrile) complexes of molybdenum and tungsten. J. Mol. Catal. A Chem. 2005, 240, 226–232. [Google Scholar] [CrossRef]
- Wong, Y.-L.; Yang, Q.; Zhou, Z.-Y.; Lee, H.K.; Mak, T.C.W.; Ng, D.K.P. Synthesis, structure and oxo-transfer properties of dioxotungsten(VI) complexes with pyridine-based NO- and NS-bidentate ligands. New J. Chem. 2001, 25, 353–357. [Google Scholar] [CrossRef]
- Gaussian 16.0; Gaussian Inc.: Wallingford, CT, USA, 2016.
- Nonius, B.V. Collect. Data Collection Software; Nonius BV: Delft, The Netherlands, 2002. [Google Scholar]
- Otwinowski, Z.; Minor, W. Macromolecular Crystallography. In Methods in Enzymology; Carter, C.W., Sweet, R.M., Eds.; Academic Press: London, UK, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Cryst. 2020, E76, 1–11. [Google Scholar] [CrossRef] [Green Version]

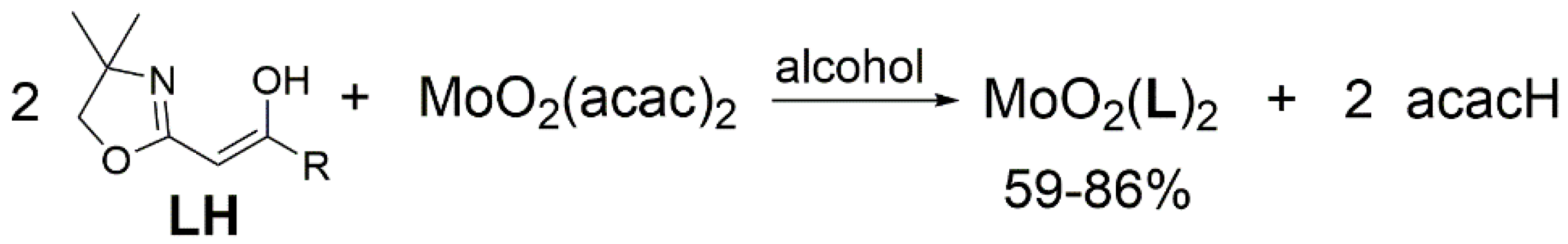


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Complex 1 | Complex 2 | Complex 3 | Complex 4 |
|---|---|---|---|---|
| Empirical Formula | C26H38N2O6Mo | C26H26N4O10Mo | C28H32N2O8Mo | C22H36N2O6Mo |
| Formula Weight (g/mol) | 560.44 | 650.45 | 620.50 | 520.47 |
| Temperature (K) | 150(2) | 150(2) | 150(2) | 150(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal System | monoclinic | monoclinic | orthorhombic | trigonal |
| Space group | P 21/n | C 2/c | P b c n | Pc1 |
| Unit Cell Dimensions (Å) | a = 11.1664(4) | a = 20.5793(6) | a =11.2202(2) | a = 19.9342(10) |
| (Å) | b = 11.1623(4) | b = 8.1384(3) | b = 12.9081(3) | b = 19.9342(10) |
| (Å) | c = 20.4516(4) | c = 17.5927(6) | c = 18.7510(4) | c = 11.5030(4) |
| (°) | α = 90 | α = 90 | α = 90 | α = 90 |
| (°) | β = 95.6150(19) | β = 113.6580(18) | β = 90 | β = 90 |
| (°) | γ = 90 | γ = 90 | γ = 90 | γ = 120 |
| Volume (Å3) | 2563.91(14) | 2698.84(16) | 2715.73(10) | 3958.6(3) |
| Z | 4 | 4 | 4 | 6 |
| Density (calcd; mg/m3) | 1.467 | 1.601 | 1.518 | 1.310 |
| Absorp. Coefficient (mm−1) | 0.560 | 0.551 | 0.536 | 0.532 |
| F(000) | 1152 | 1328 | 1280 | 1632 |
| Crystal Size (mm3) | 0.40 × 0.30 × 0.22 | 0.20 × 0.18 × 0.16 | 0.18 × 0.14 × 0.12 | 0.20 × 0.20 × 0.10 |
| θ range for Data Collection (°) | 2.59–27.48 | 2.58–27.51 | 2.64–27.48 | 2.70–27.48 |
| Index Ranges | −14 ≤ h ≤ 14 | −26 ≤ h ≤ 23 | −14 ≤ h ≤ 14 | −22 ≤ h ≤ 22 |
| −14 ≤ k ≤ 14 | −10 ≤ k ≤ 10 | −16 ≤ k ≤ 16 | −25 ≤ k ≤ 25 | |
| −21 ≤ l ≤ 26 | −22 ≤ l ≤ 22 | −24 ≤ l ≤ 24 | 0 ≤ l ≤ 14 | |
| Reflexions Collected | 16,606 | 12,858 | 19,994 | 3021 |
| Independent Reflexions | 5765 [R(int) = 0.0492] | 3089 [R(int) = 0.0391] | 3120 [R(int) = 0.0455] | 3021 [R(int) = 0.049] |
| Completeness to θ = x° (%) | x = 25.25 (99.6) | x = 27.51 (99.3) | x = 27.48 (99.9) | x = 27.48 (99.5) |
| Absorption Correction | Semi-empirical from equivalents | Semi-empirical from equivalents | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max./min. Transmission | 0.888/0.752 | 0.917/0.851 | 0.770/0.629 | 0.949/0.897 |
| Refinement Method | Full-matrix least squares on F2 | Full-matrix least squares on F2 | Full-matrix least squares on F2 | Full-matrix least squares on F2 |
| Data/Restraints/Parameters | 5765/0/320 | 3089/0/188 | 3120/0/180 | 3021/0/146 |
| Goodness of Fit on F2 | 1.069 | 1.107 | 1.071 | 1.041 |
| Final R indices [I > 2σ(I)] | R1 = 0.0441; wR2 = 0.0917 | R1 = 0.0404; wR2 = 0.0949 | R1 = 0.0449; wR2 = 0.1142 | R1 = 0.0522; wR2 = 0.1352 |
| R indices (all data) | R1 = 0.0689; wR2 = 0.1034 | R1 = 0.0521; wR2 = 0.1022 | R1 = 0.0702; wR2 = 0.1339 | R1 = 0.0852; wR2 = 0.1511 |
| Largest diff. peak and hole (eÅ−3) | 1.525 and 0.816 | 1.715 and 0.784 | 2.105 ad 0.975 | 1.437 and −0.742 |
| Observed Data | Complex 1 | Complex 2 | Complex 3 | Complex 4 |
| Mo=O | 1.705(2); 1.709(2) | 1.700(2) | 1.690(3) | 1.697(3) |
| Mo-O | 2.101(2); 2.119(2) | 2.103(2) | 2.096(2) | 2.072(3) |
| Mo-N | 2.117(3); 2.135(3) | 2.150(2) | 2.138(3) | 2.147(3) |
| C=N | 1.331(4); 1.314(4) | 1.310(4) | 1.324(4) | 1.311(4) |
| C=C | 1.369(5); 1.373(5) | 1.370(4) | 1.371(5) | 1.351(5) |
| O=Mo=O | 102.10(11) | 102.39(16) | 104.3(2) | 101.5(2) |
| N-Mo-N | 158.37(10) | 159.75(12) | 166.28(14) | 162.96(16) |
| O-Mo-O | 80.08(9) | 79.44(13) | 77.41(19) | 78.9(2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrov, A.; Adjei, J.A.; Lough, A.J.; Wylie, R.S.; Gossage, R.A. Synthesis, Characterization, and Catalytic Exploration of Mononuclear Mo(VI) Dioxido Complexes of (Z)-1-R-2-(4′,4′-Dimethyl-2′-oxazolin-2′-yl)-eth-1-en-1-ates. Molecules 2022, 27, 1309. https://doi.org/10.3390/molecules27041309
Petrov A, Adjei JA, Lough AJ, Wylie RS, Gossage RA. Synthesis, Characterization, and Catalytic Exploration of Mononuclear Mo(VI) Dioxido Complexes of (Z)-1-R-2-(4′,4′-Dimethyl-2′-oxazolin-2′-yl)-eth-1-en-1-ates. Molecules. 2022; 27(4):1309. https://doi.org/10.3390/molecules27041309
Chicago/Turabian StylePetrov, Anna, Jeanette A. Adjei, Alan J. Lough, R. Stephen Wylie, and Robert A. Gossage. 2022. "Synthesis, Characterization, and Catalytic Exploration of Mononuclear Mo(VI) Dioxido Complexes of (Z)-1-R-2-(4′,4′-Dimethyl-2′-oxazolin-2′-yl)-eth-1-en-1-ates" Molecules 27, no. 4: 1309. https://doi.org/10.3390/molecules27041309
APA StylePetrov, A., Adjei, J. A., Lough, A. J., Wylie, R. S., & Gossage, R. A. (2022). Synthesis, Characterization, and Catalytic Exploration of Mononuclear Mo(VI) Dioxido Complexes of (Z)-1-R-2-(4′,4′-Dimethyl-2′-oxazolin-2′-yl)-eth-1-en-1-ates. Molecules, 27(4), 1309. https://doi.org/10.3390/molecules27041309