Modulation by Phosphonium Ions of the Activity of Mitotropic Agents Based on the Chemiluminescence of Luminols

 , , ,
, , ,  and
and 
Abstract
:
1. Introduction
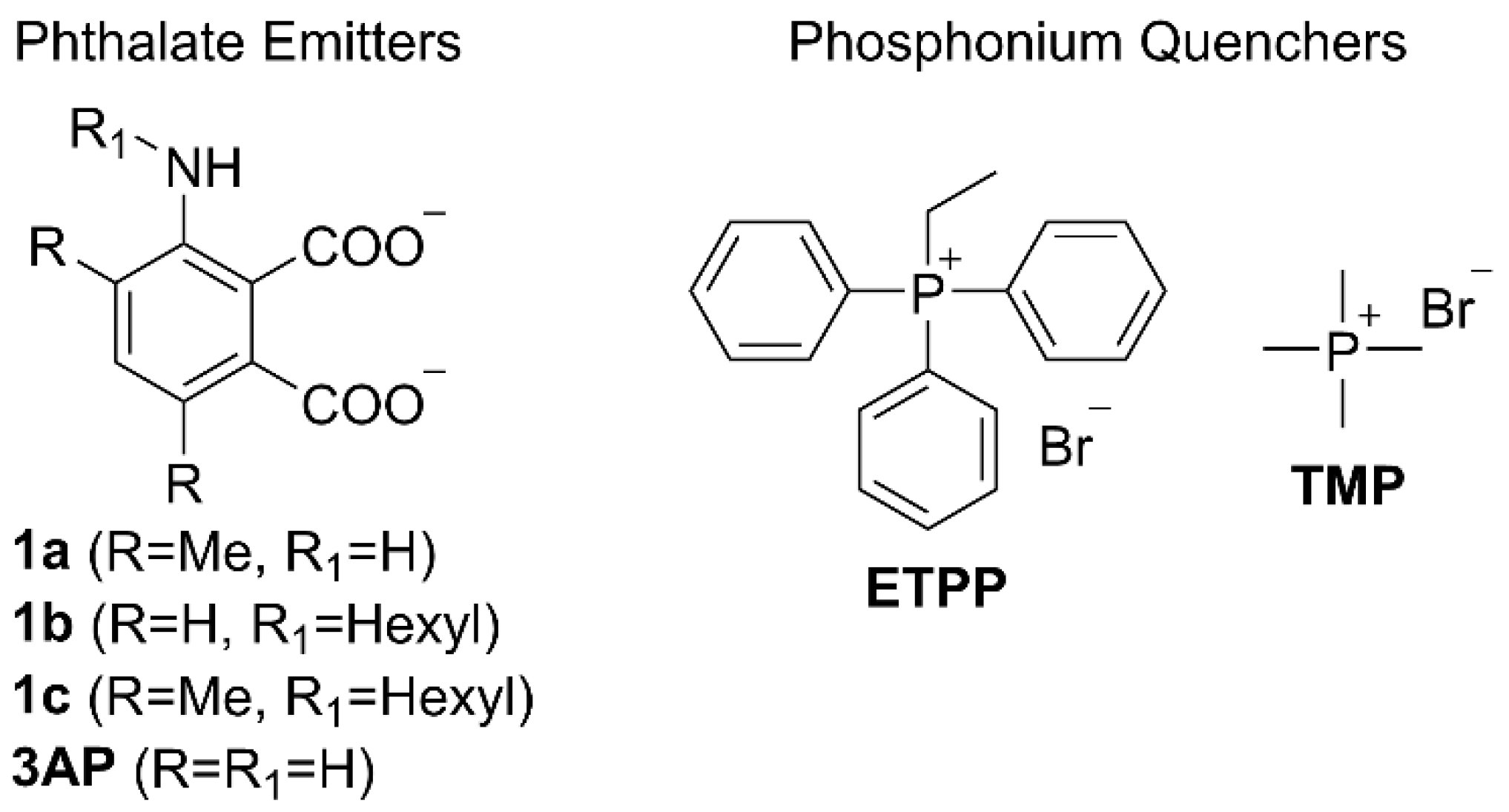
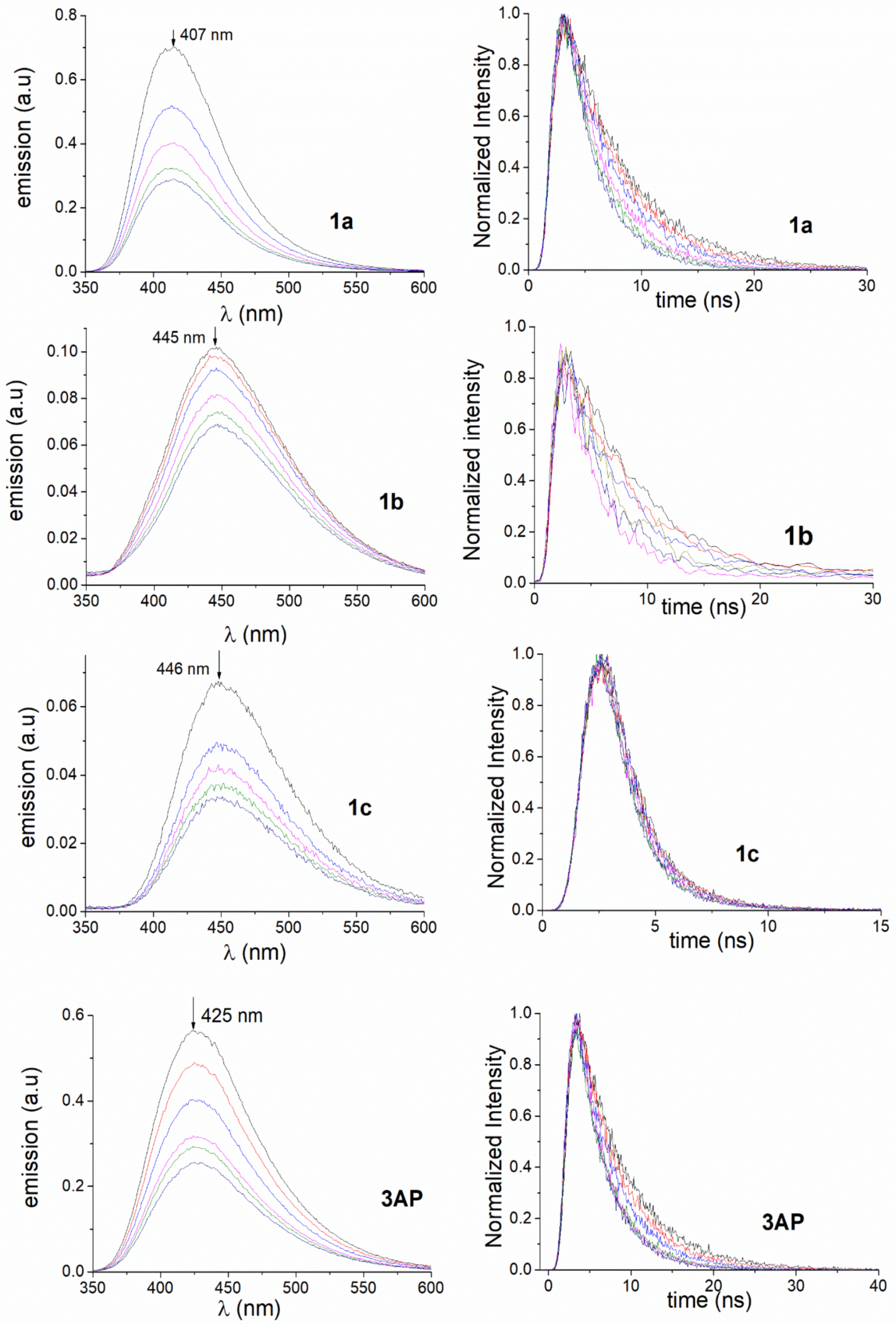
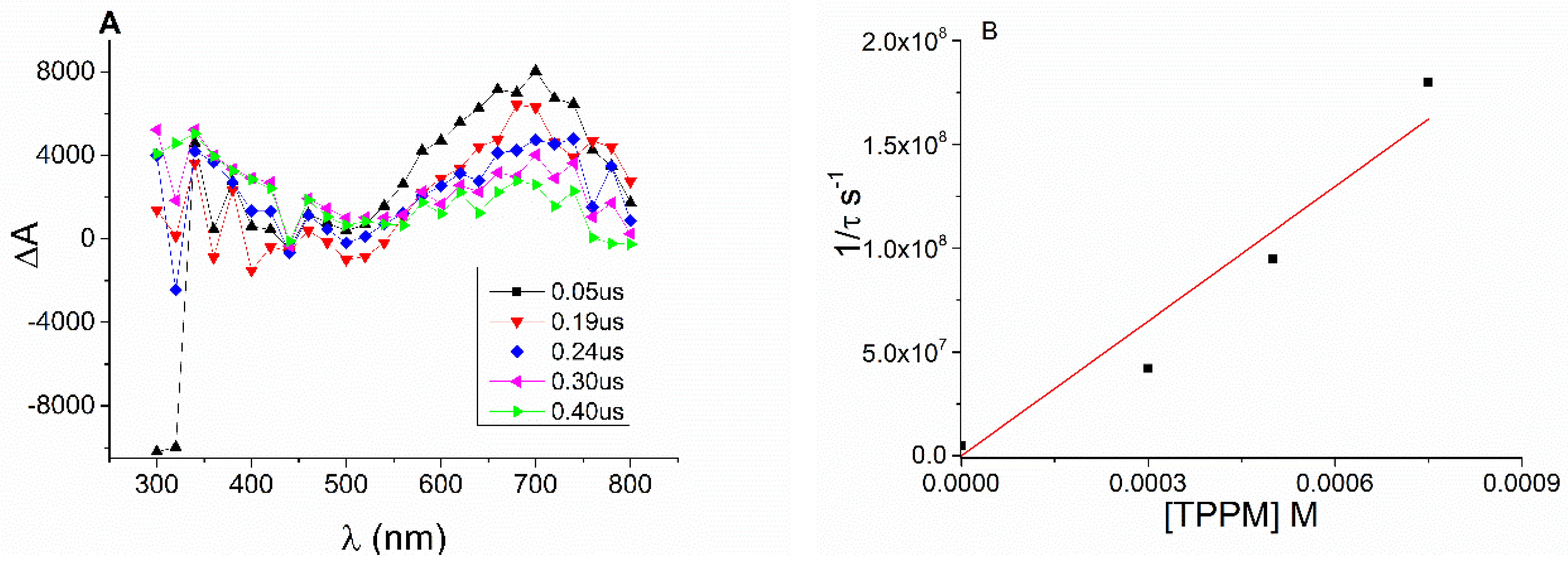
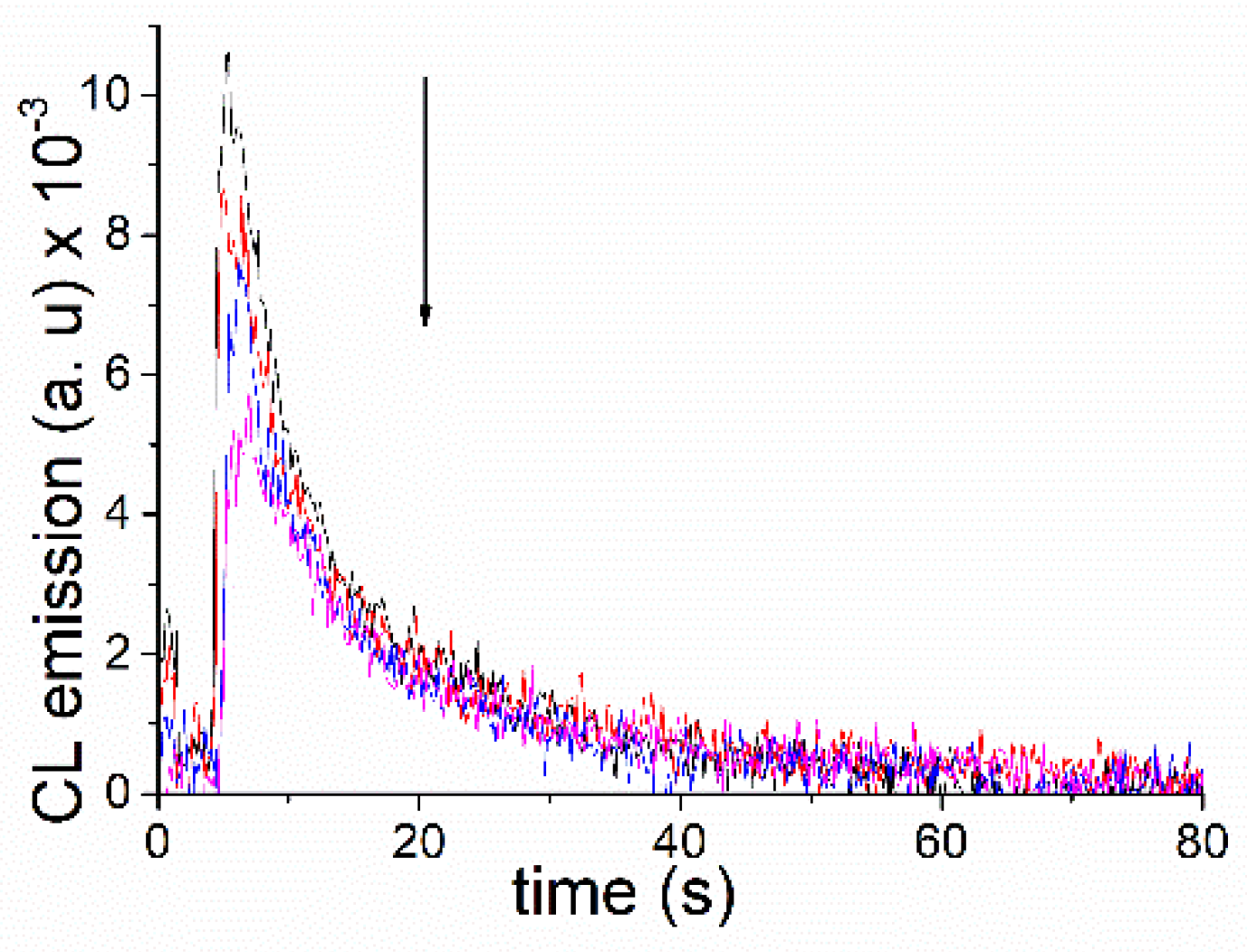
2. Results and Discussion
3. Materials and Methods
3.1. General Remarks
3.2. Photophysical Studies
3.3. Laser Flash Photolysis Studies
3.4. Electrochemical Studies
3.5. Synthesis Procedures
3.5.1. 4-Amino-5,7-dibromo-2-(sec-butyl)isoindoline-1,3-dione (3)
3.5.2. 4-Amino-5,7-dimethyl-2-(sec-butyl)isoindoline-1,3-dione (4)
3.5.3. General Procedure for the N-alkylation of 4-aminophthalimides (5a–5b)
3.5.4. General Procedure for the Synthesis of 3-Aminophthalic Anhydrides (6a–6c)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Green, D.R.; Reed, J.C. Mitochondria and Apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szewczyk, A.; Wojtczak, L. Mitochondria as a Pharmacological Target. Pharmacol. Rev. 2002, 54, 101–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019, 10, 1704. [Google Scholar] [CrossRef] [Green Version]
- Milane, L.; Trivedi, M.; Singh, A.; Talekar, M.; Amiji, M. Mitochondrial biology, targets, and drug delivery. J. Control. Release 2015, 207, 40–58. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Mukae, Y.; Harada, J.; Matsuda, T.; Mitsunari, K.; Matsuo, T.; Ohba, K.; Sakai, H. Pathological and Pharmacological Roles of Mitochondrial Reactive Oxygen Species in Malignant Neoplasms: Therapies Involving Chemical Compounds, Natural Products, and Photosensitizers. Molecules 2020, 25, 5252. [Google Scholar] [CrossRef]
- Smith, R.A.; Hartley, R.C.; Murphy, M.P. Mitochondria-Targeted Small Molecule Therapeutics and Probes. Antioxid. Redox Signal. 2011, 15, 3021–3038. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Hoye, A.T.; Davoren, J.E.; Wipf, P.; Fink, M.P.; Kagan, V.E. Targeting Mitochondria. Acc. Chem. Res. 2008, 41, 87–97. [Google Scholar] [CrossRef]
- Jean, S.R.; Tulumello, D.V.; Wisnovsky, S.P.; Lei, E.K.; Pereira, M.P.; Kelley, S.O. Molecular Vehicles for Mitochondrial Chemical Biology and Drug Delivery. ACS Chem. Biol. 2014, 9, 323–333. [Google Scholar] [CrossRef]
- Lu, P.; Bruno, B.J.; Rabenau, M.; Lim, C.S. Delivery of drugs and macromolecules to the mitochondria for cancer therapy. J. Control. Release 2016, 240, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tian, X.; Shin, I.; Yoon, J. Fluorescent and luminescent probes for detection of reactive oxygen and nitrogen species. Chem. Soc. Rev. 2011, 40, 4783–4804. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Srikun, D.; Chang, C.J. Mitochondrial-targeted fluorescent probes for reactive oxygen species. Curr. Opin. Chem. Biol. 2010, 14, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Liu, Y.; He, Y.; Wang, Y. Mitochondria and lysosome-targetable fluorescent probes for hydrogen peroxide. J. Mater. Chem. B 2021, 9, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Kishikawa, N.; Ohyama, K.; Ohba, Y.; Kohno, M.; Masuda, T.; Takadate, A.; Nakashima, K.; Kuroda, N. Evaluation of chemiluminescence reagents for selective detection of reactive oxygen species. Anal. Chim. Acta 2010, 665, 74–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodeigne, C.; Thunus, L.; Lejeune, R. Chemiluminescence as diagnostic tool. A review. Talanta 2000, 51, 415–439. [Google Scholar] [CrossRef]
- Khan, P.; Idrees, D.; Moxley, M.A.; Corbett, J.A.; Ahmad, F.; Von Figura, G.; Sly, W.S.; Waheed, A.; Hassan, M.I. Luminol-Based Chemiluminescent Signals: Clinical and Non-clinical Application and Future Uses. Appl. Biochem. Biotechnol. 2014, 173, 333–355. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Francis, K.P.; Prakash, A.; Ansaldi, D. Enhanced detection of myeloperoxidase activity in deep tissues through luminescent excitation of near-infrared nanoparticles. Nat. Med. 2013, 19, 500–505. [Google Scholar] [CrossRef]
- Gross, S.; Gammon, S.T.; Moss, B.L.; Rauch, D.; Harding, J.; Heinecke, J.W.; Ratner, L.; Piwnica-Worms, D. Bioluminescence imaging of myeloperoxidase activity in vivo. Nat. Med. 2009, 15, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivapackiam, J.; Liao, F.; Zhou, D.; Shoghi, K.I.; Gropler, R.J.; Gelman, A.E.; Sharma, V. Galuminox: Preclinical validation of a novel PET tracer for non-invasive imaging of oxidative stress in vivo. Redox Biol. 2020, 37, 101690. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Huang, J.; Fan, J.; Du, J.; Pu, K.; Peng, X. Chemiluminescence for bioimaging and therapeutics: Recent advances and challenges. Chem. Soc. Rev. 2020, 49, 6800–6815. [Google Scholar] [CrossRef] [PubMed]
- Pantelia, A.; Daskalaki, I.; Cuquerella, M.C.; Rotas, G.; Miranda, M.A.; Vougioukalakis, G.C. Synthesis and Chemiluminescent Properties of Amino-Acylated luminol Derivatives Bearing Phosphonium Cations. Molecules 2019, 24, 3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundrett, R.B.; White, E.H. Synthesis and chemiluminescence of derivatives of luminol and isoluminol. J. Am. Chem. Soc. 1974, 96, 7497–7502. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Díaz-Miara, Y.; Fichtler, R.; von Wangelin, A.J.; Pérez-Ruiz, R.; Sampedro, D. Steric Enhancement of the Chemiluminescence of Luminols. Chem. A Eur. J. 2015, 21, 9975–9979. [Google Scholar] [CrossRef]
- Mikroulis, T.; Cuquerella, M.C.; Giussani, A.; Pantelia, A.; Rodríguez-Muñiz, G.M.; Rotas, G.; Roca-Sanjuán, D.; Miranda, M.A.; Vougioukalakis, G.C. Building a Functionalizable, Potent Chemiluminescent Agent: A Rational Design Study on 6,8-Substituted Luminol Derivatives. J. Org. Chem. 2021, 86, 11388–11398. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Organic Chemicals in Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Burlington, MA, USA, 2003; pp. 380–388. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH 8 ETPP | pH 8 TMP | |||||
|---|---|---|---|---|---|---|
| Phthalate | ΦF pH 10 | τo (ns) | kq (M−1s−1) a | k′q (M−1s−1) b | kq (M−1s−1) a | k′q (M−1s−1) b |
| 3AP | 0.30 | 6.1 | 3.7 × 109 | 2.5 × 109 | 2.7 × 108 | 9.0 × 107 |
| 1a | 0.34 | 5.7 | 5.8 × 109 | 3.4 × 109 | - | - |
| 1b | 0.07 | 6.2 | 6.0 × 109 | 5.4 × 109 | - | - |
| 1c | 0.08 | 2.5 | 9.0 × 109 | 8.4 × 109 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Muñiz, G.M.; Mikroulis, T.; Pantelia, A.; Rotas, G.; Cuquerella, M.-C.; Vougioukalakis, G.C.; Miranda, M.A. Modulation by Phosphonium Ions of the Activity of Mitotropic Agents Based on the Chemiluminescence of Luminols. Molecules 2022, 27, 1245. https://doi.org/10.3390/molecules27041245
Rodríguez-Muñiz GM, Mikroulis T, Pantelia A, Rotas G, Cuquerella M-C, Vougioukalakis GC, Miranda MA. Modulation by Phosphonium Ions of the Activity of Mitotropic Agents Based on the Chemiluminescence of Luminols. Molecules. 2022; 27(4):1245. https://doi.org/10.3390/molecules27041245
Chicago/Turabian StyleRodríguez-Muñiz, Gemma M., Theodoros Mikroulis, Anna Pantelia, Georgios Rotas, Maria-Consuelo Cuquerella, Georgios C. Vougioukalakis, and Miguel A. Miranda. 2022. "Modulation by Phosphonium Ions of the Activity of Mitotropic Agents Based on the Chemiluminescence of Luminols" Molecules 27, no. 4: 1245. https://doi.org/10.3390/molecules27041245
APA StyleRodríguez-Muñiz, G. M., Mikroulis, T., Pantelia, A., Rotas, G., Cuquerella, M.-C., Vougioukalakis, G. C., & Miranda, M. A. (2022). Modulation by Phosphonium Ions of the Activity of Mitotropic Agents Based on the Chemiluminescence of Luminols. Molecules, 27(4), 1245. https://doi.org/10.3390/molecules27041245