
Kinetic and Molecular Docking Studies to Determine the Effect of Inhibitors on the Activity and Structure of Fused G6PD::6PGL Protein from Trichomonas vaginalis

 ,
,  , ,
, ,  , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Purification of the Recombinant Fused TvG6PD::6PGL Enzyme
2.2. Functional Assays
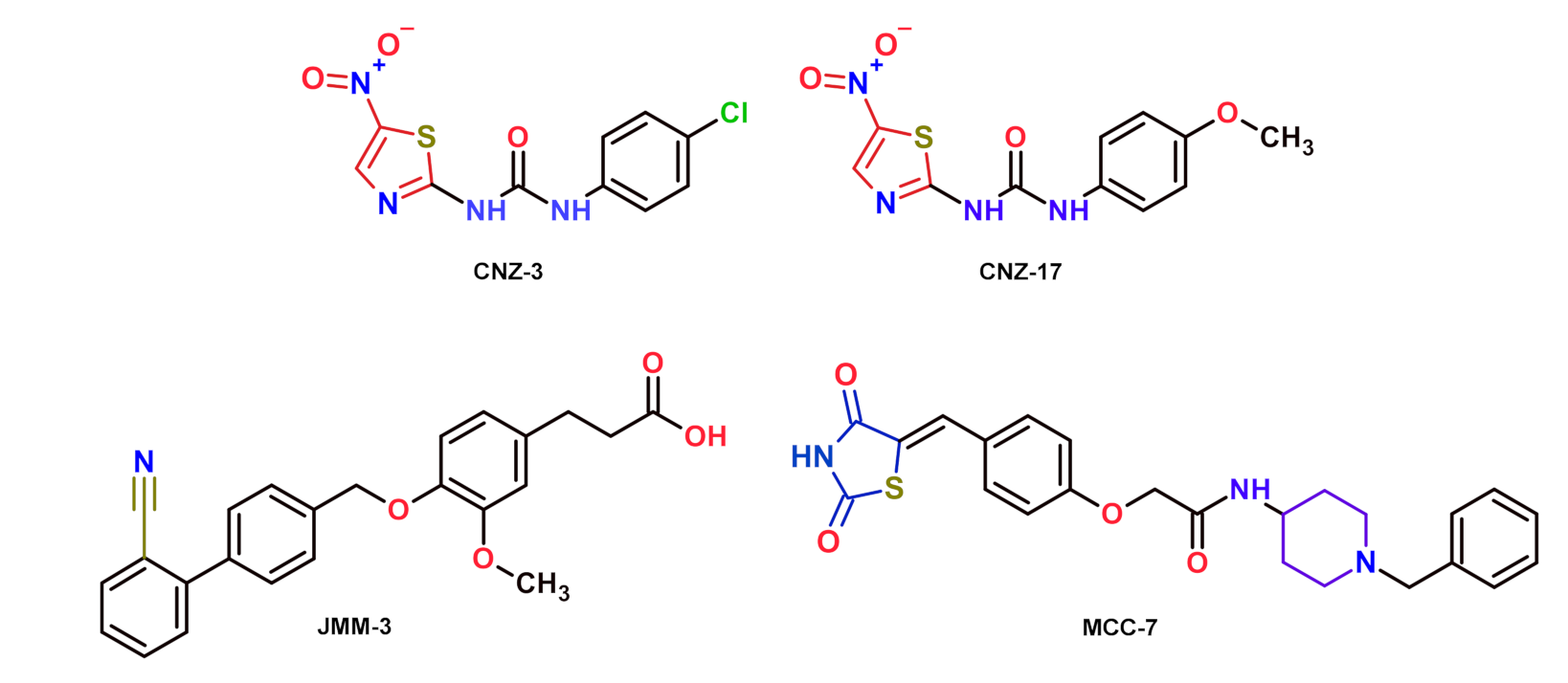
2.2.1. Selection of Compounds That Inhibit the Catalytic Activity of Fused TvG6PD::6PGL Enzyme
2.2.2. Orthogonal Assay
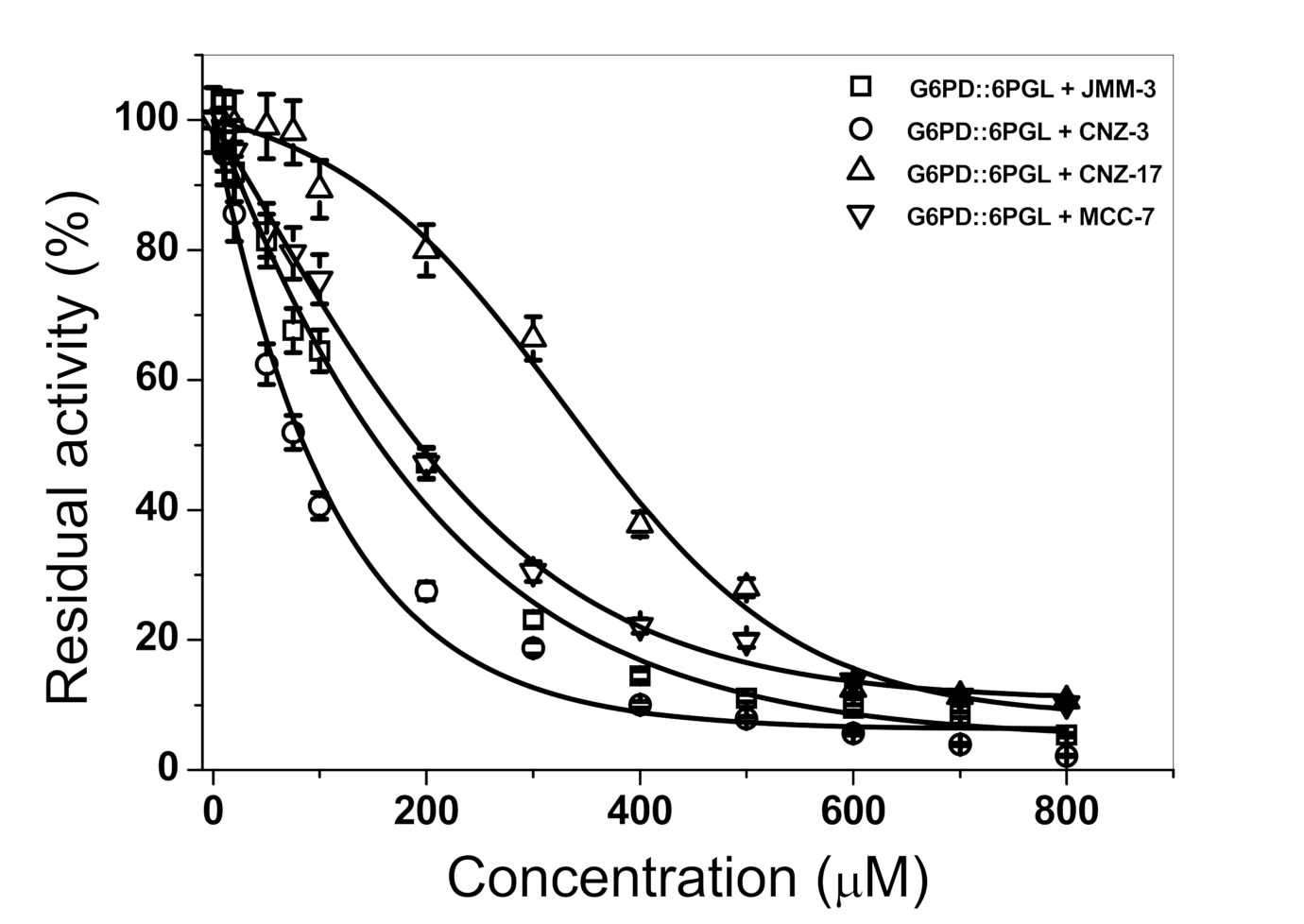
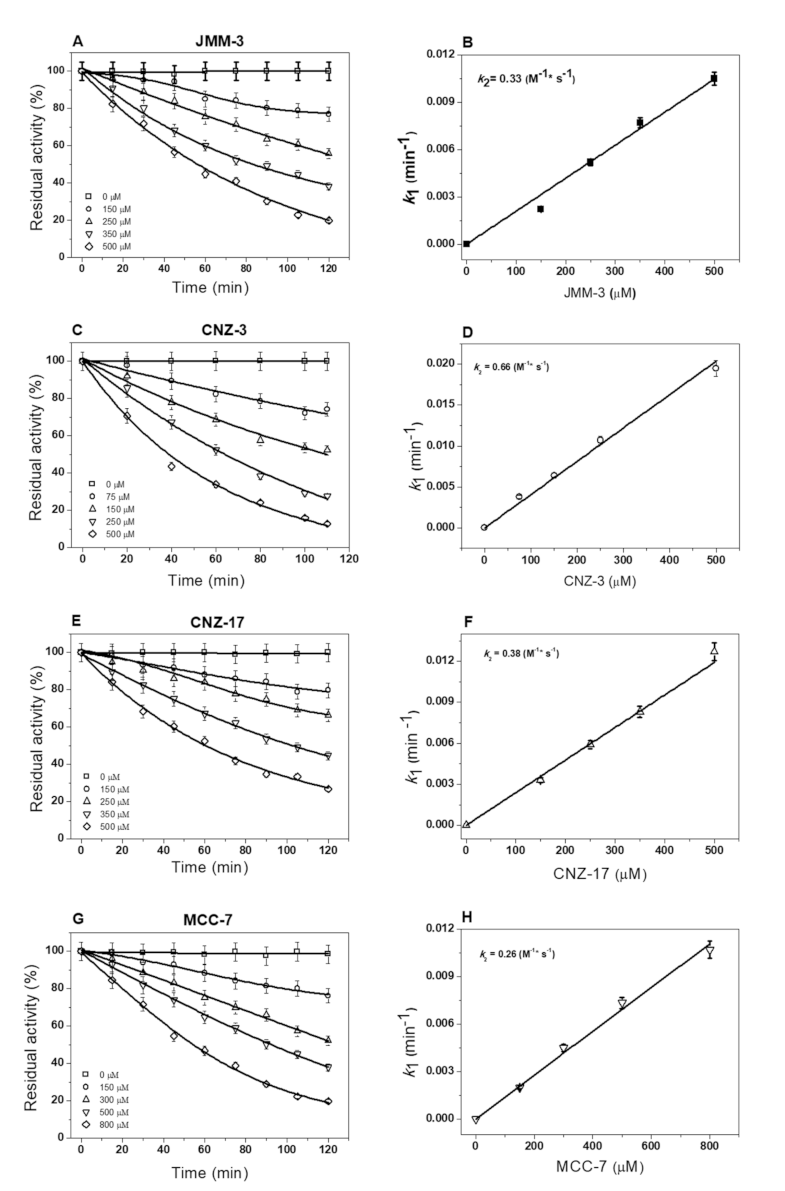
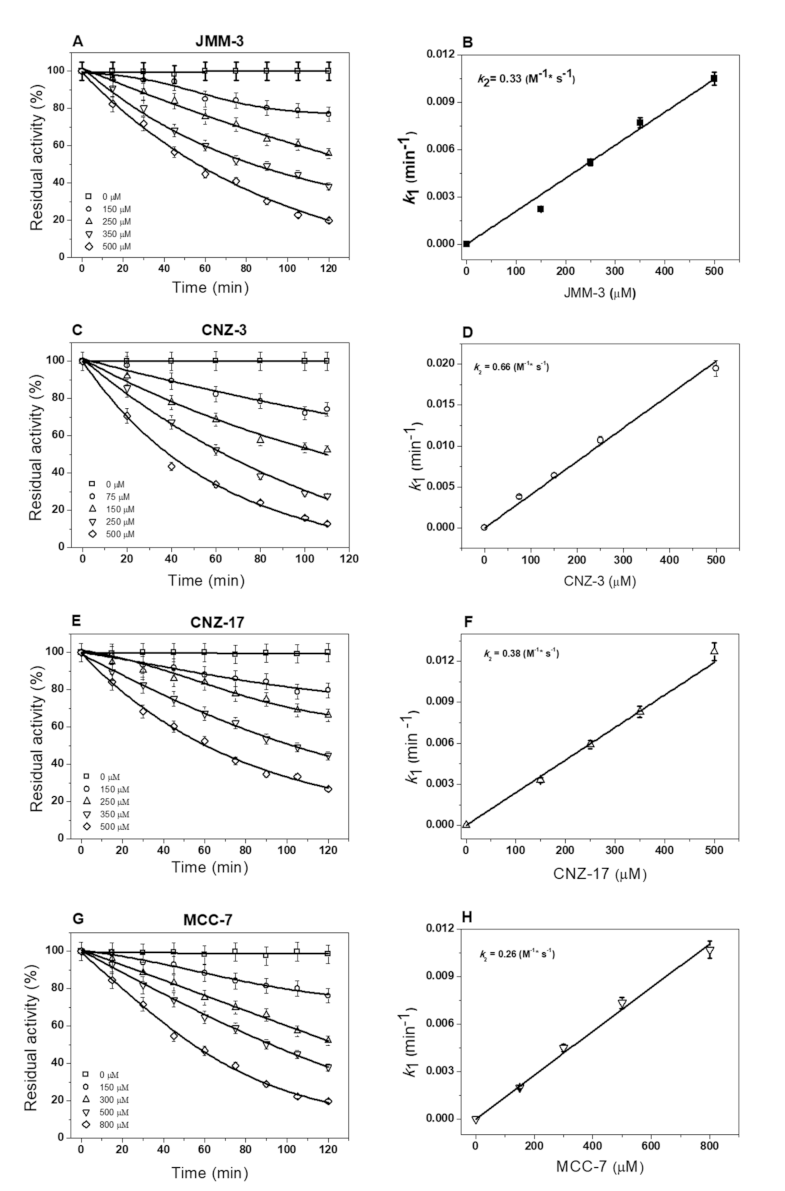
2.2.3. Inactivation of Fused TvG6PD::6PGL Enzyme by Library Compounds
2.2.4. Inhibition Type
2.3. Structural Studies
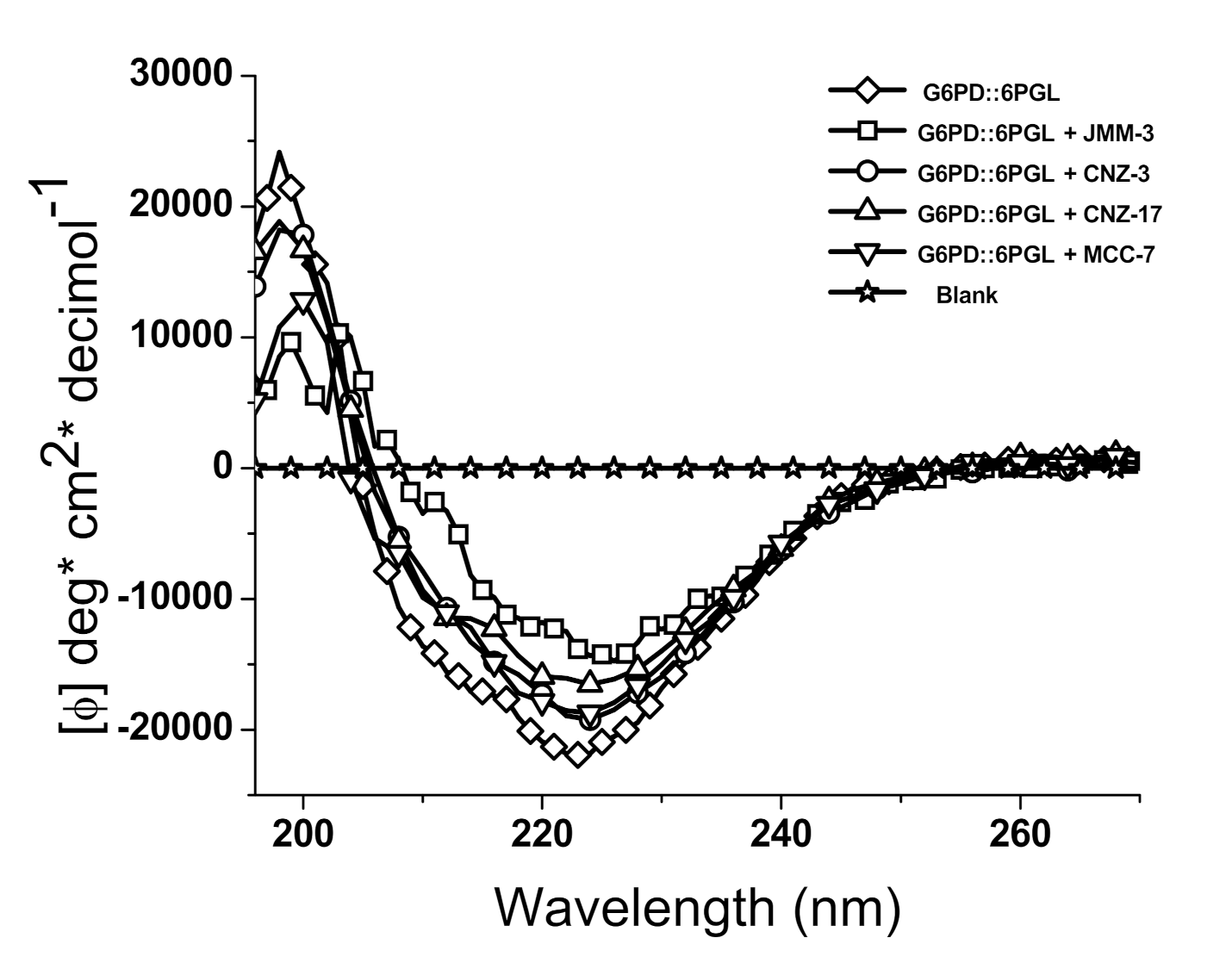
2.3.1. Circular Dichroism
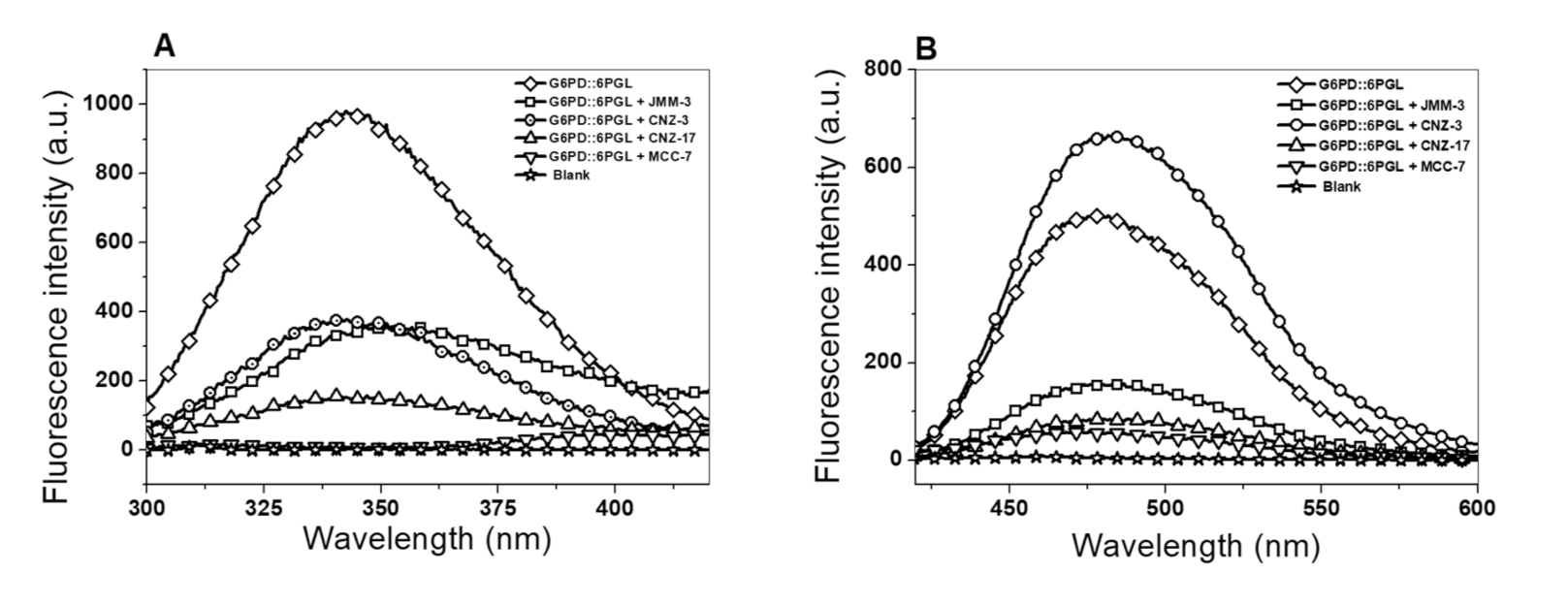
2.3.2. Intrinsic and Extrinsic Fluorescence Assays
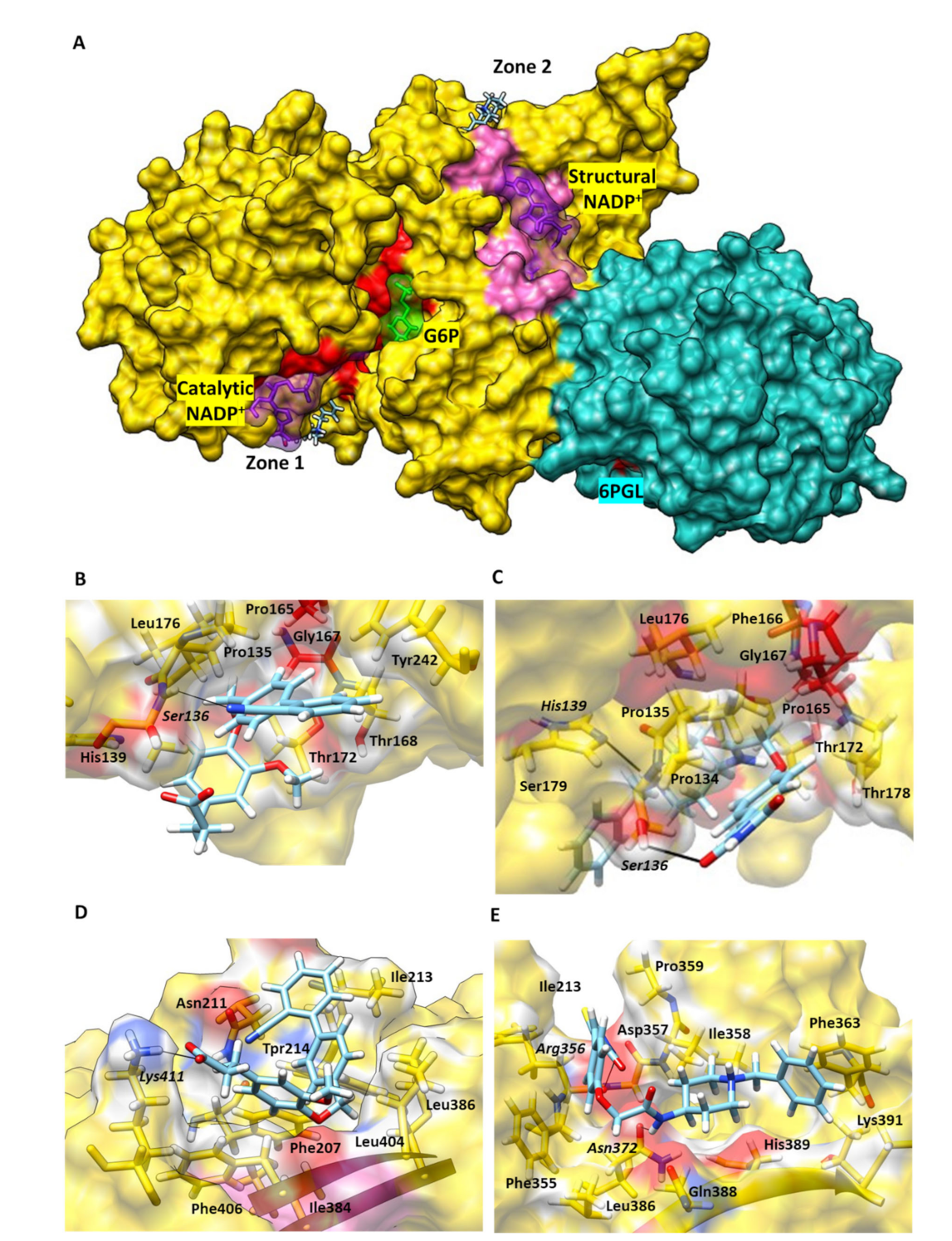
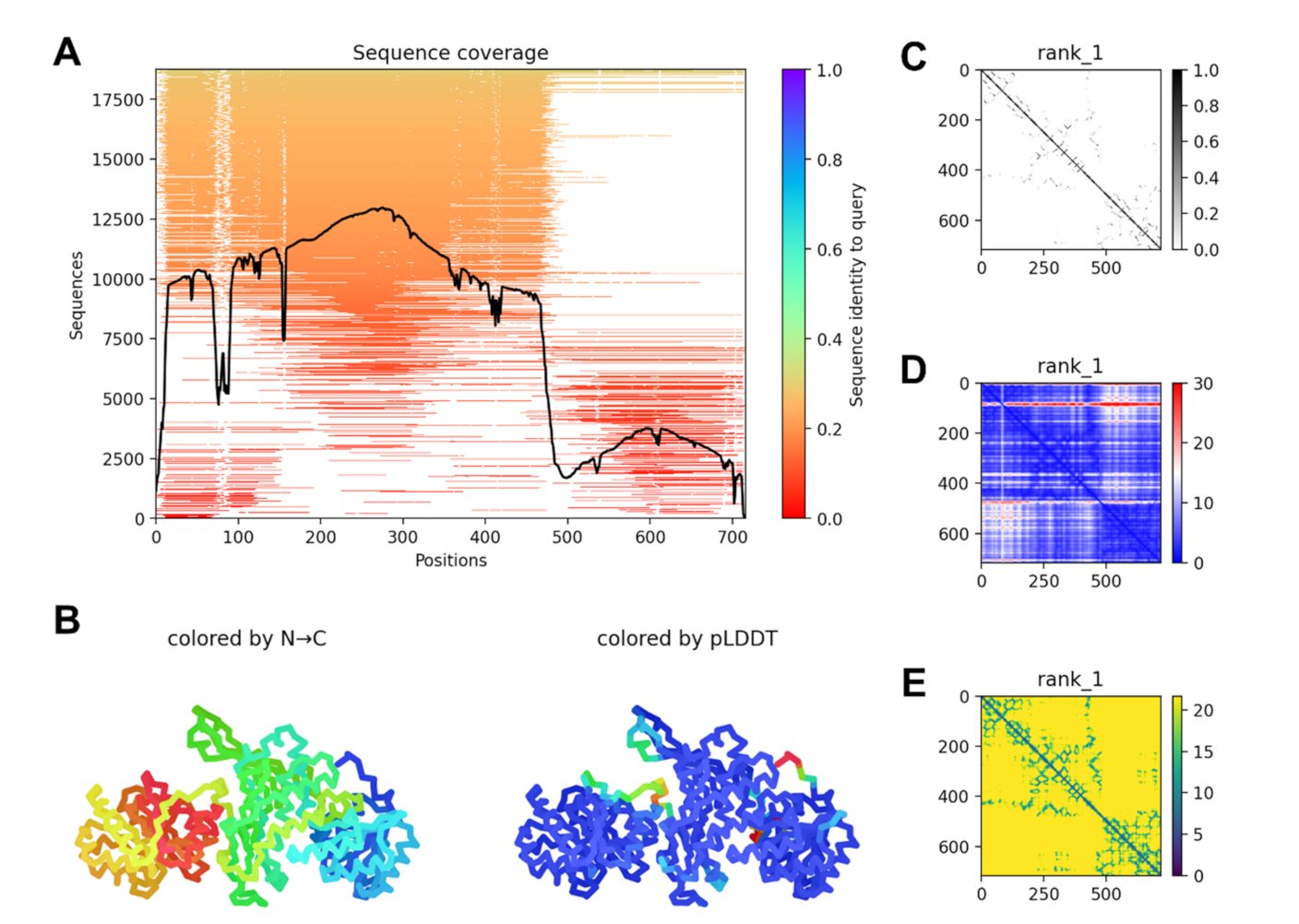
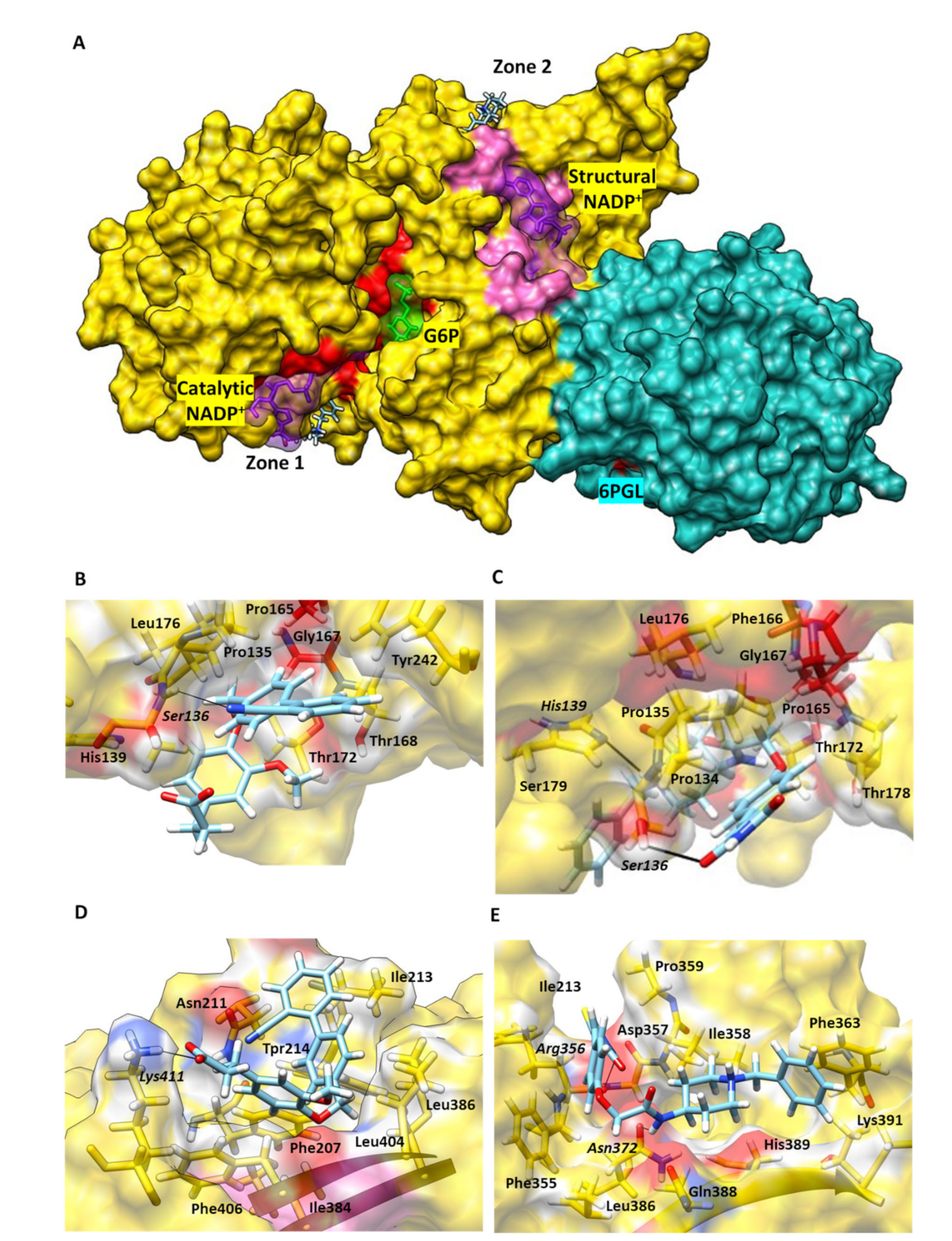
2.4. G6PD::6PGL Model Generation and Selected Hit Inhibitors Molecular Docking
3. Materials and Methods
3.1. Expression and Purification of the Recombinant Fused G6PD:6PGLPprotein
3.2. Functional Assays
3.2.1. Selection of Compounds That Inhibit the Catalytic Activity of Fused TvG6PD::6PGL Enzyme
3.2.2. Orthogonal Assays
3.2.3. Second-Order Rate Constant (k2) of Selected Hit Compounds Showing More Than 50% Inhibition
3.2.4. Determination of Inhibition Type
3.3. Structural Studies
3.3.1. Circular Dichroism Experiments
3.3.2. Intrinsic and Extrinsic Fluorescence Assays
3.4. Blind Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- De Aquino, M.F.K.; Hinderfeld, A.S.; Simoes-Barbosa, A. Trichomonas vaginalis . Trends Parasitol. 2020, 36, 646–647. [Google Scholar] [CrossRef]
- Kissinger, P. Trichomonas vaginalis: A review of epidemiologic, clinical and treatment issues. BMC Infect. Dis. 2015, 15, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global incidence and prevalence of selected curable sexually transmitted infections: 2008. Reprod. Health. Matt 2012.
- Núñez-Troconis, J.T. Diagnóstico de la Tricomonas vaginalis en la mujer. Revista Chilena de Obstetricia y Ginecología 2020, 85, 175–184. [Google Scholar] [CrossRef]
- Allsworth, J.E.; Ratner, J.A.; Peipert, J.F. Trichomoniasis and other sexually transmitted infections: Results from the 2001-2004 National Health and Nutrition Examination Surveys. Sex. Transm. Dis. 2009, 36, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchemal, K.; Bories, C.; Loiseau, P.M. Strategies for Prevention and Treatment of Trichomonas vaginalis Infections. Clin. Microbiol. Rev. 2017, 30, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Gerwen, O.T.V.; Muzny, C.A. Recent advances in the epidemiology, diagnosis, and management of Trichomonas vaginalis infection. F1000 Research 2019, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, C.G.; Karlsson, W.K.; Amin, F.M.; Lindelof, M. Metronidazole-induced encephalopathy: A systematic review. J. Neurol. 2020, 267, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.; Panwar, A.; Pandit, A.; Das, S.K.; Joshi, B. Clinical and neuroradiological spectrum of metronidazole induced encephalopathy: Our experience and the review of literature. Clin. Diagn. Res. 2016, 10, OE01. [Google Scholar] [CrossRef] [PubMed]
- Dunne, R.L.; Dunn, L.A.; Upcroft, P.; O’Donoghue, P.J.; Upcroft, J.A. Drug resistance in the sexually transmitted protozoan Trichomonas vaginalis. Cell Res. 2003, 13, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Upcroft, P.; Upcroft, J.A. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin. Microbiol. Rev. 2001, 14, 150–164. [Google Scholar] [CrossRef] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval1, V.; Grüning, N.M.; Krüger, A.; Alam, M.T.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Morales, Y.Y.; Vanoye-Carlo, A.; Castillo-Rodríguez, R.A.; Serrano-Posada, H.; González-Valdez, A.; Ortega-Cuellar, D.; Hernández-Ochoa, B.; Moreno-Vargas, L.M.; Prada-Gracia, D.; Sierra-Palacios, E.; et al. Cloning and biochemical characterization of three glucose-6-phosphate dehydrogenase mutants presents in the Mexican population. Int. J. Biol. Macromol. 2018, 119, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rosas, V.; Juárez-Cruz, M.V.; Ramírez-Nava, E.J.; Hernández-Ochoa, B.; Morales-Luna, L.; González-Valdez, A.; Serrano-Posada, H.; Cárdenas-Rodríguez, N.; Ortiz-Ramírez, P.; Centeno-Leija, S.; et al. Effects of Single and Double Mutants in Human Glucose-6-Phosphate Dehydrogenase Variants Present in the Mexican Population: Biochemical and Structural Analysis. Int. J. Mol. Sci. 2020, 21, 2732. [Google Scholar] [CrossRef] [Green Version]
- Morales-Luna, L.; Serrano-Posada, H.; González-Valdez, A.; Ortega-Cuellar, D.; Vanoye-Carlo, A.; Hernández-Ochoa, B.; Sierra-Palacios, E.; Rufino-González, Y.; Castillo-Rodríguez, R.A.; Pérez de la cruz, V.; et al. Biochemical Characterization and Structural Modeling of Fused Glucose-6-Phosphate Dehydrogenase-Phosphogluconolactonase from Giardia lamblia. Int. J. Mol. Sci. 2018, 19, 2518. [Google Scholar] [CrossRef] [Green Version]
- Preuss, J.; Hedrick, M.; Sergienko, E.; Pinkerton, A.; Mangravita-Novo, A.; Smith, L.; Marx, C.; Fischer, E.; Jortzik, E.; Rahlfs, S.; et al. High-throughput screening for small-molecule inhibitors of plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase. J. Biomol. Screen. 2012, 17, 738–751. [Google Scholar] [CrossRef] [Green Version]
- Stover, N.A.; Dixon, T.A.; Cavalcanti, A.R. Multiple independent fusions of glucose-6-phosphate dehydrogenase with enzymes in the pentose phosphate pathway. PLoS ONE 2011, 6, e22269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Luna, L.; Hernández-Ochoa, B.; Ramírez-Nava, E.J.; Martínez-Rosas, V.; Ortiz-Ramírez, P.; Fernández-Rosario, F.; González-Valdez, A.; Cárdenas-Rodríguez, N.; Serrano-Posada, H.; Centeno-Leija, S.; et al. Characterizing the Fused TvG6PD::6PGL Protein from the Protozoan Trichomonas vaginalis, and Effects of the NADP+ Molecule on Enzyme Stability. Int. J. Mol. Sci. 2020, 21, 4831. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Nava, E.J.; Hernández-Ochoa, B.; Navarrete-Vázquez, G.; Arreguín-Espinosa, R.; Ortega-Cuellar, D.; González-Valdez, A.; Martínez-Rosas, V.; Morales-Luna, L.; Martínez-Miranda, J.; Sierra-Palacios, E.; et al. Novel inhibitors of human glucose-6-phosphate dehydrogenase (HsG6PD) affect the activity and stability of the protein. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129828. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool1, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making protein folding accesible to all. BioRxiv 2021, 456425. [Google Scholar] [CrossRef]
- Domínguez-Mendoza, E.A.; Galván-Ciprés, Y.; Martínez-Miranda, J.; Miranda-González, C.; Colín-Lozano, B.; Hernández- Núñez, E.; Hernández-Bolio, G.I.; Palomino-Hernández, O.; Navarrete-Vázquez, G. Design, Synthesis, and In Silico Multitarget Pharmacological Simulations of Acid Bioisosteres with a Validated In Vivo Antihyperglycemic Effect. Molecules 2021, 26, 799. [Google Scholar] [CrossRef]
- Nava-Zuazo, C.; Chávez-Silva, F.; Moo-Puc, R.; Chan-Bacab, M.J.; Ortega-Morales, B.O.; Moreno-Díaz, H.; Díaz-Coutiño, D.; Hernández-Núñez, E.; Navarrete-Vázquez, G. 2-acylamino-5-nitro-1,3-thiazoles: Preparation and in vitro bioevaluation against four neglected protozoan parasites. Bioorg. Med. Chem. 2014, 22, 1626–1633. [Google Scholar] [CrossRef]
- Hernández-Ochoa, B.; Navarrete-Vázquez, G.; Aguayo-Ortiz, R.; Ortiz-Ramírez, P.; Morales-Luna, L.; Martínez-Rosas, V.; González-Valdez, A.; Gómez-Chávez, F.; Enríquez-Flores, S.; Wong-Baeza, C.; et al. Identification and In Silico Characterization of Novel Helicobacter pylori Glucose-6-Phosphate Dehydrogenase Inhibitors. Molecules 2021, 26, 4955. [Google Scholar] [CrossRef]
- García-Torres, I.; de la Mora-de la Mora, I.; Marcial-Quino, J.; Gómez-Manzo, S.; Vanoye-Carlo, A.; Navarrete-Vázquez, G.; Colín-Lozano, B.; Gutíerrez-Castrellón, P.; Sierra-Palacios, E.; López-Velázquez, G.; et al. Proton pump inhibitors drastically modify triosephosphate isomerase from Giardia lamblia at functional and structural levels, providing molecular leads in the design of new antigiardiasic drugs. Biochim. Biophys. Acta 2016, 1860, 97–107. [Google Scholar] [CrossRef]
- Gómez-Manzo, S.; Terrón-Hernández, J.; De la Mora-De la Mora, I.; González-Valdez, A.; Marcial-Quino, J.; García-Torres, I.; Vanoye-Carlo, A.; López-Velázquez, G.; Hernández-Alcántara, G.; Oria-Hernández, J.; et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int. J. Mol. Sci. 2014, 15, 21179–21201. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Manzo, S.; Marcial-Quino, J.; Vanoye-Carlo, A.; Serrano-Posada, H.; Ortega-Cuellar, D.; González-Valdez, A.; Castillo-Rodríguez, R.A.; Hernandez-Ochoa, B.; Sierra-Palacios, E.; Rodríguez-Bustamante, E.; et al. Glucose-6-Phosphate Dehydrogenase: Update and Analysis of New Mutations around the World. Int. J. Mol. Sci. 2016, 17, 2069. [Google Scholar] [CrossRef] [Green Version]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic. Acids. Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein. Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- ACD/ChemSketch, version 2020.2.0, Advanced Chemistry Development, Inc., Toronto, ON, Canada. 2020. Available online: www.acdlabs.com (accessed on 26 October 2021).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds (400 μM) | G6PD::6PGL Inhibition (%) | HsG6PD Inhibition (%) |
|---|---|---|
| JMM-2 | 79 | 81 |
| JMM-3 | 56 | 3 |
| CNZ-3 | 93 | 92 |
| CNZ-7 | 52 | 68 |
| CNZ-8 | 51 | 43 |
| CNZ-16 | 61 | 31 |
| CNZ-17 | 62 | 0 |
| CMC-1 | 63 | 34 |
| TDA-5 | 54 | 17 |
| MCC-7 | 80 | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Rosas, V.; Hernández-Ochoa, B.; Navarrete-Vázquez, G.; Martínez-Conde, C.; Gómez-Chávez, F.; Morales-Luna, L.; González-Valdez, A.; Arreguin-Espinosa, R.; Enríquez-Flores, S.; Pérez de la Cruz, V.; et al. Kinetic and Molecular Docking Studies to Determine the Effect of Inhibitors on the Activity and Structure of Fused G6PD::6PGL Protein from Trichomonas vaginalis. Molecules 2022, 27, 1174. https://doi.org/10.3390/molecules27041174
Martínez-Rosas V, Hernández-Ochoa B, Navarrete-Vázquez G, Martínez-Conde C, Gómez-Chávez F, Morales-Luna L, González-Valdez A, Arreguin-Espinosa R, Enríquez-Flores S, Pérez de la Cruz V, et al. Kinetic and Molecular Docking Studies to Determine the Effect of Inhibitors on the Activity and Structure of Fused G6PD::6PGL Protein from Trichomonas vaginalis. Molecules. 2022; 27(4):1174. https://doi.org/10.3390/molecules27041174
Chicago/Turabian StyleMartínez-Rosas, Víctor, Beatriz Hernández-Ochoa, Gabriel Navarrete-Vázquez, Carlos Martínez-Conde, Fernando Gómez-Chávez, Laura Morales-Luna, Abigail González-Valdez, Roberto Arreguin-Espinosa, Sergio Enríquez-Flores, Verónica Pérez de la Cruz, and et al. 2022. "Kinetic and Molecular Docking Studies to Determine the Effect of Inhibitors on the Activity and Structure of Fused G6PD::6PGL Protein from Trichomonas vaginalis" Molecules 27, no. 4: 1174. https://doi.org/10.3390/molecules27041174
APA StyleMartínez-Rosas, V., Hernández-Ochoa, B., Navarrete-Vázquez, G., Martínez-Conde, C., Gómez-Chávez, F., Morales-Luna, L., González-Valdez, A., Arreguin-Espinosa, R., Enríquez-Flores, S., Pérez de la Cruz, V., Aguayo-Ortiz, R., Wong-Baeza, C., Baeza-Ramírez, I., & Gómez-Manzo, S. (2022). Kinetic and Molecular Docking Studies to Determine the Effect of Inhibitors on the Activity and Structure of Fused G6PD::6PGL Protein from Trichomonas vaginalis. Molecules, 27(4), 1174. https://doi.org/10.3390/molecules27041174