
EGFR and COX-2 Dual Inhibitor: The Design, Synthesis, and Biological Evaluation of Novel Chalcones

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
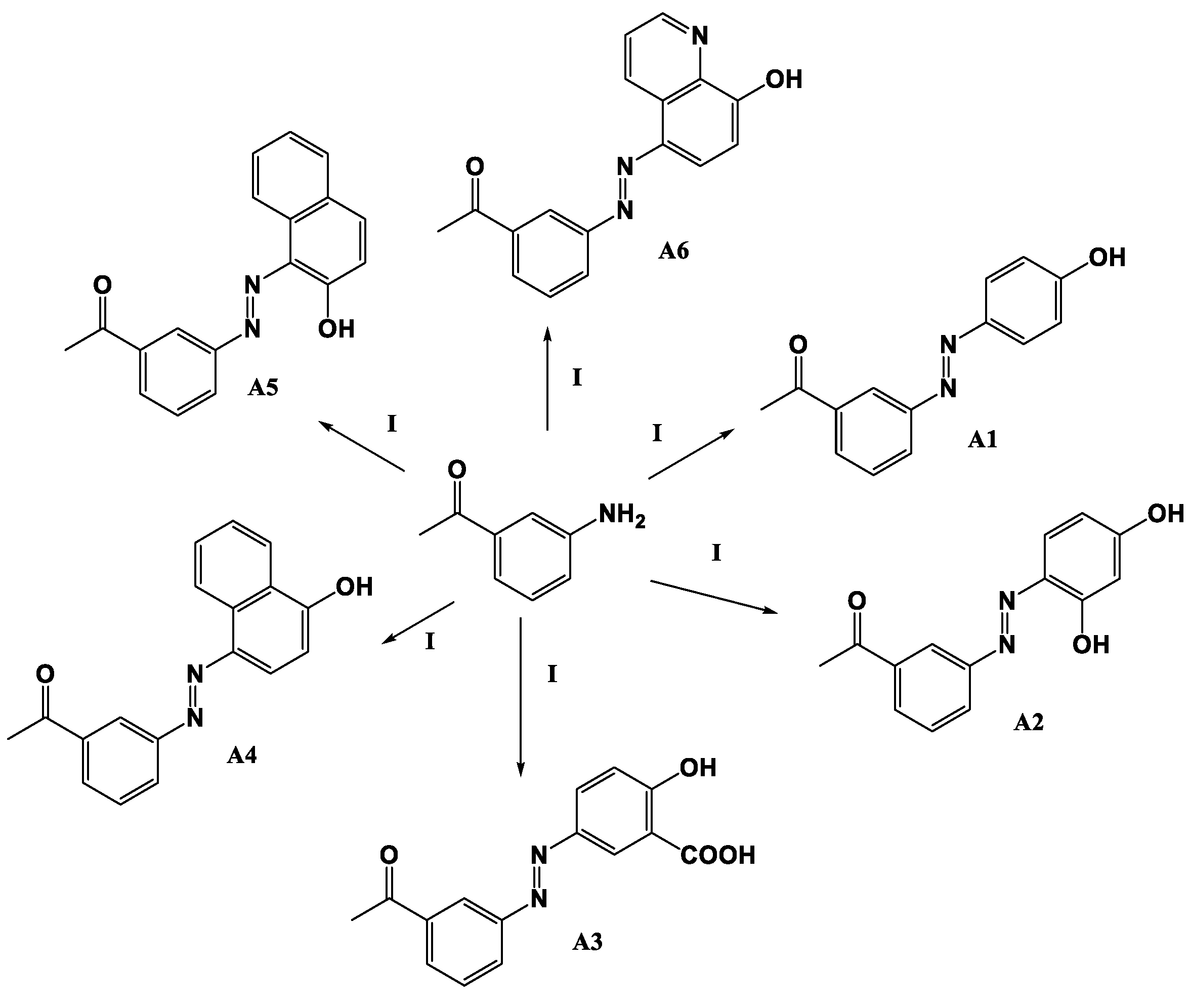
2.1. Chemistry
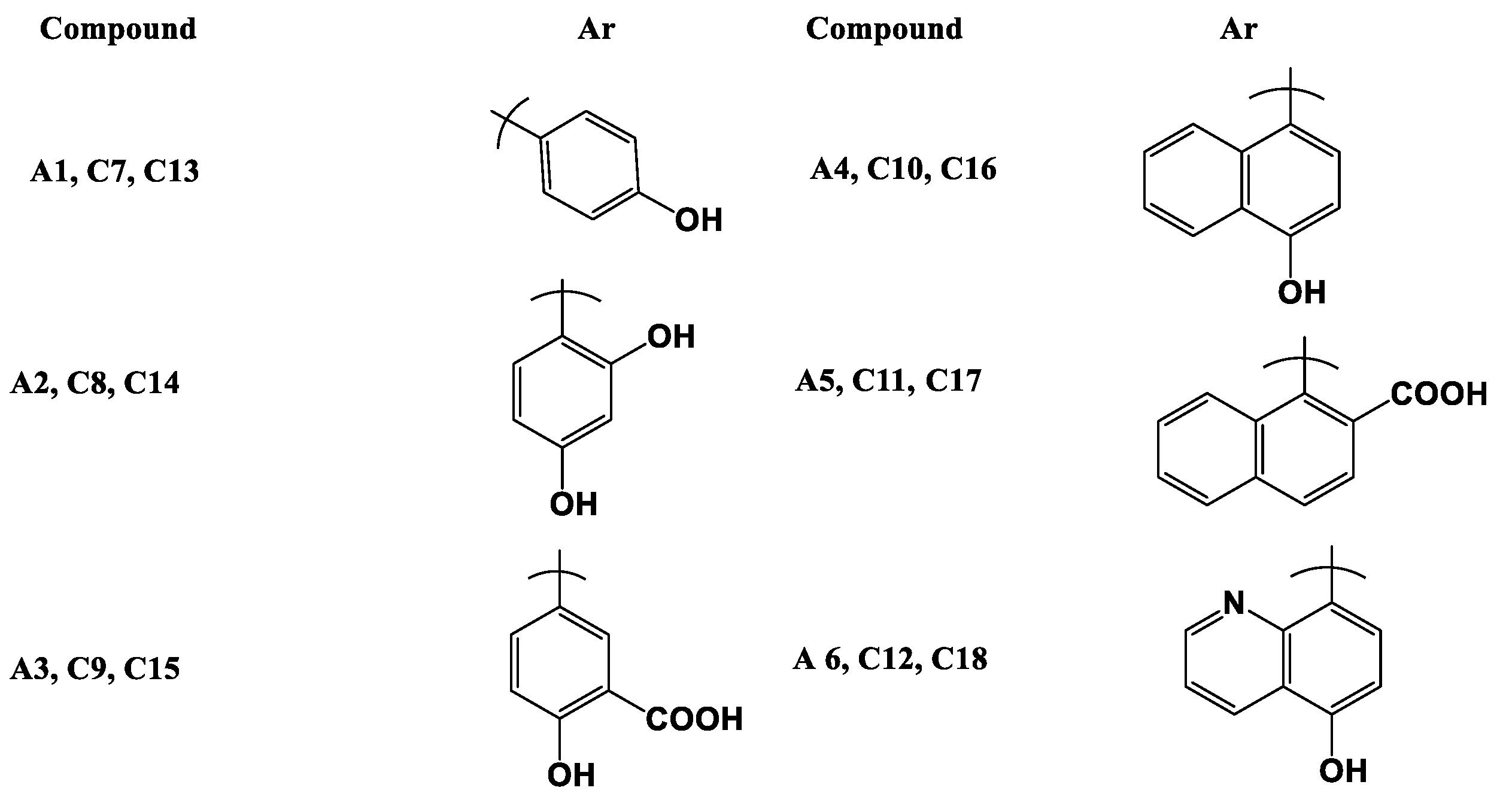
- (E)-1-(3-((2,4-Dihydroxyphenyl)daizenyl)phenyl)ethan-1-one (A2)
- (E)-5-((3-Acetylphenyl)diazenyl)-2-hydroxybenzoic acid (A3)
- (E)-1-(3-((4-Hydroxynaphthalen-1-yl)diazenyl)phenyl)ethan-1-one (A4)
- (E)-1-(3-((2-Hydroxynaphthalen-1-yl)diazenyl)phenyl)ethan-1-one (A5)
- (E)-1-(3-((8-Hydroxyquinolin-5-yl)diazenyl)phenyl)ethan-1-one (A6)
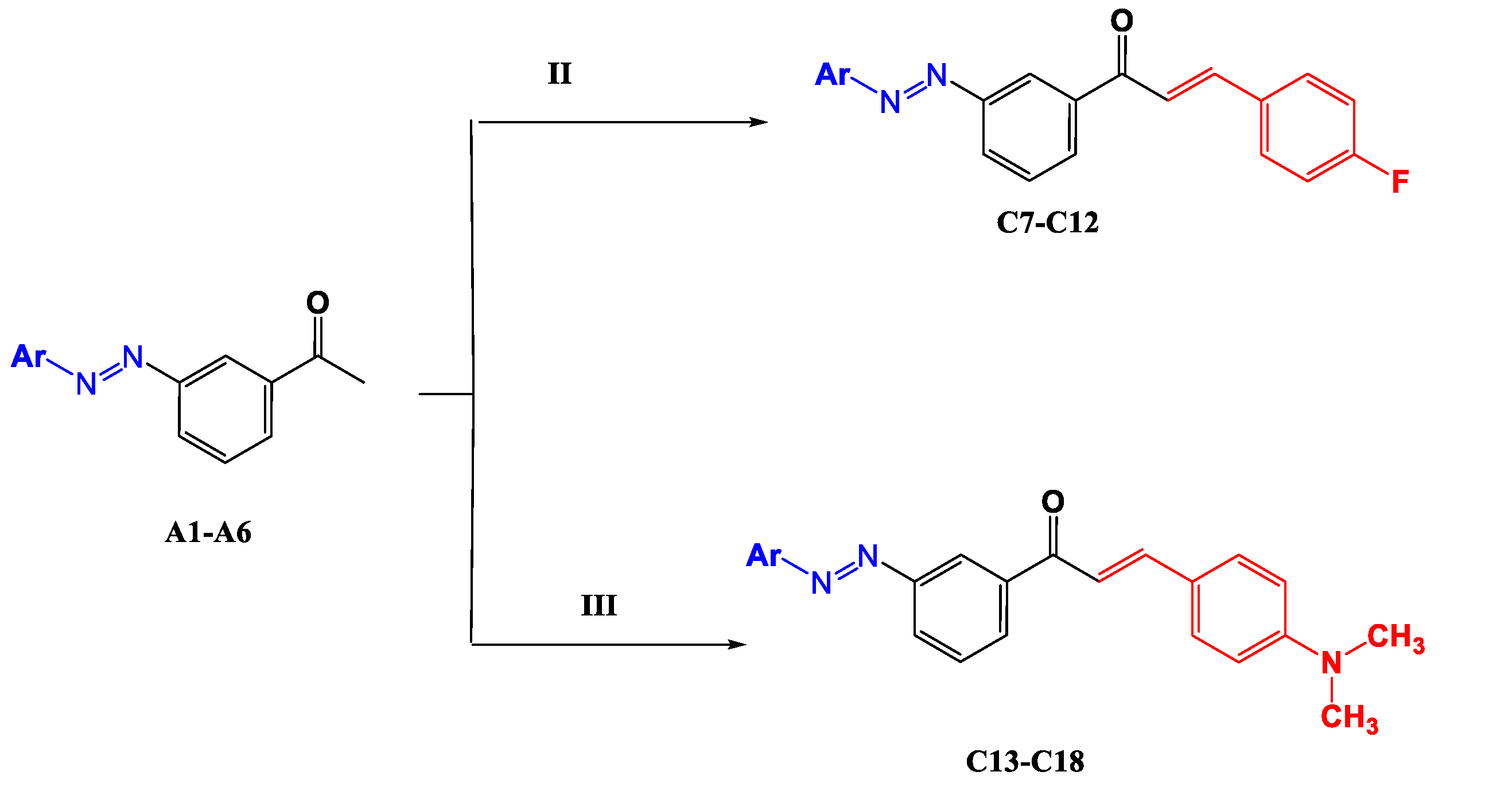
- (E)-3-(4-Fluorophenyl)-1-(3-((E)-(4-hydroxyphenyl) diazenyl)phenyl) prop-2-en-1-one (C7)
- (E)-1-(3-((E)-(2,4-Dihydroxyphenyl) diazenyl) phenyl)-3-(4-fluorophenyl) prop-2-en-1-one (C8)
- 5.
- -((Z)-(3-((E)-3-(4-Fluorophenyl)acryloyl)phenyl)diazenyl)-2-hydroxybenzoic acid (C9)
- (E)-3-(4-Fluorophenyl)-1-(4-((E)-(4-hydroxynaphthalen-1-yl)diazenyl)phenyl) prop-2-en-1-one (C10)
- (E)-3-(4-Fluorophenyl)-1-(4-((E)-(2-hydroxynaphthalen-1-yl)diazenyl)phenyl)prop-2-en-1-one (C11)
- (E)-3-(4-Fluorophenyl)-1-(4-((E)-(8-hydroxyquinolin-5-yl)diazenyl)phenyl)prop-2-en-1-one (C12)
- (E)-3-(4-(Dimethylamino)phenyl)-1-(3-((E)-(4-hydroxyphenyl)diazenyl)phenyl)prop-2-en-1-one (C13)
- (E)-1-(3-((E)-(2,4-Dihydroxyphenyl)diazenyl)phenyl)-3-(4-(dimethylamino) phenyl) prop-2-en-1-one (C14)
- 5.
- -((E)-(4-((E)-3-(4-(Dimethylamino)phenyl)acryloyl)phenyl)diazenyl)-2-hydroxy benzoic acid (C15)
- (E)-3-(4-(Dimethylamino)phenyl)-1-(3-((E)-(4-hydroxynaphthalen-1-yl)diazenyl) phenyl) prop-2-en-1-one (C16)
- (E)-3-(4-(dimethylamino)phenyl)-1-(3-((Z)-(2-hydroxynaphthalen-1-yl)diazenyl) phenyl) prop-2-en-1-one (C17)
- (E)-3-(4-(dimethylamino)phenyl)-1-(3-((Z)-(8-hydroxyquinolin-5-yl)diazenyl) phenyl) prop-2-en-1-one (C18)
2.2. Cytotoxic Activity
2.3. Protein Kinase Inhibition Activity
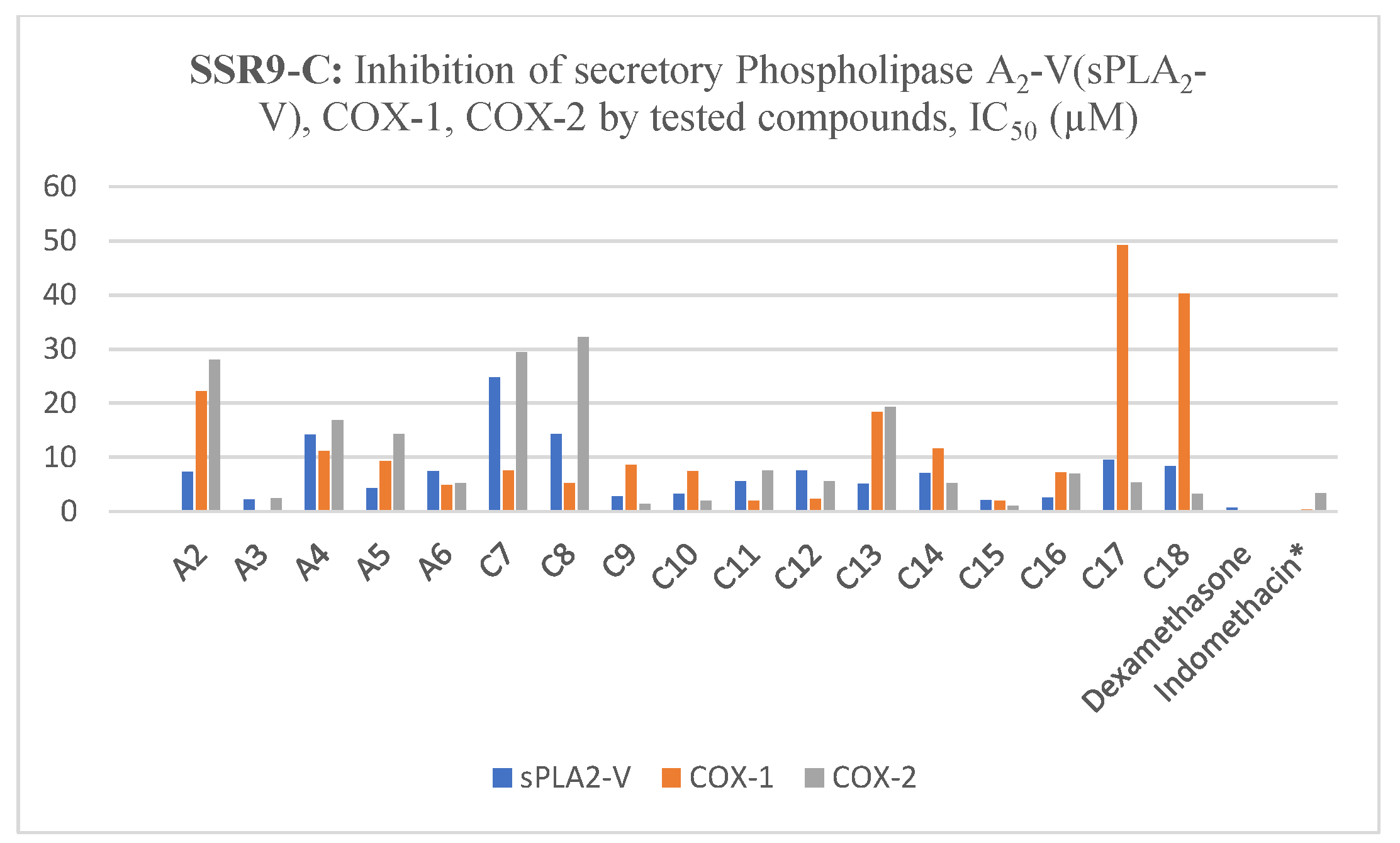
2.4. Inhibition of Secretory PLA2-V, COX-1, COX-2 by Tested Compounds
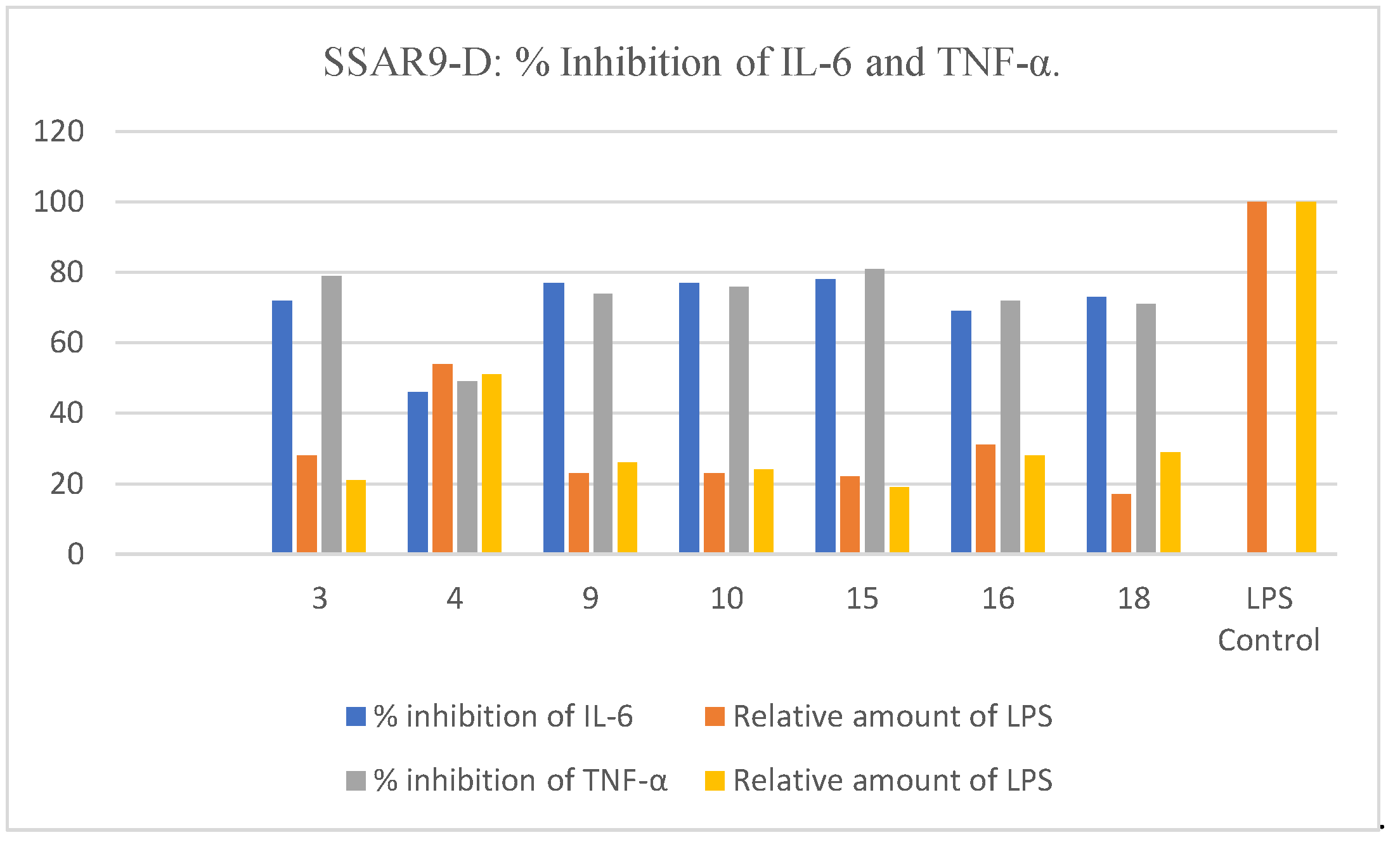
2.5. Inhibition of Release of IL-6 and TNF-α in LPS-Stimulated Macrophages
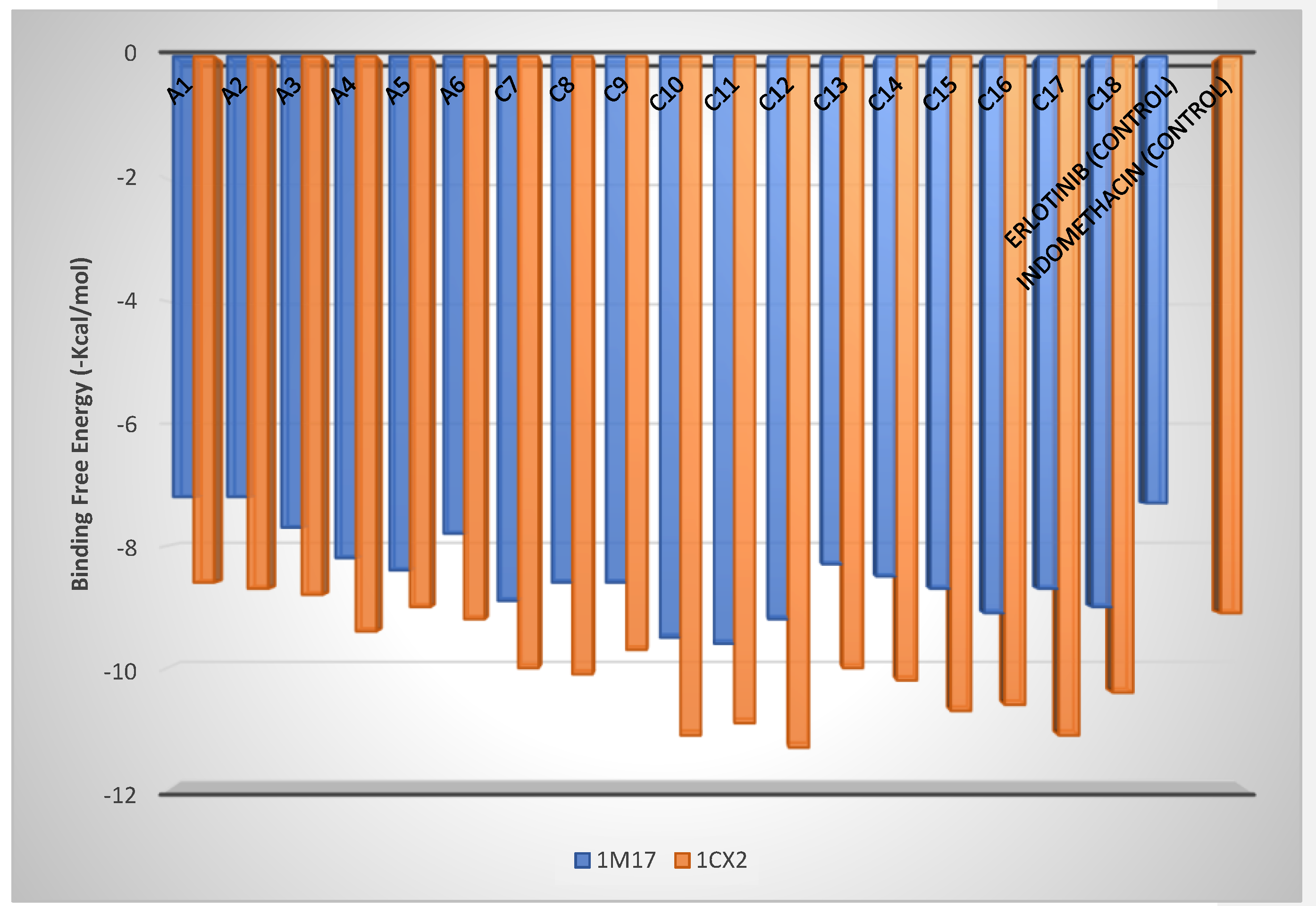
2.6. Molecular Docking
3. Materials and Methods
3.1. Synthesis of Azo-Derivatives (A1–A6)
3.2. Synthesis of Chalcones C7–C18, General Method
3.3. Cytotoxicity Assay
3.4. EGFR Inhibitory Activity
3.5. Assay of Secretory PLA2-V Activity
3.6. Cyclooxygenase Assay
3.7. Cell Treatment and ELISA Assay for IL-6 and TNF-α
3.8. Statistical Analysis
3.9. Virtual Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yabroff, K.R.; Wu, X.-C.; Negoita, S.; Stevens, J.; Coyle, L.; Zhao, J.; Mumphrey, B.J.; Jemal, A.; Ward, K.C. Association of the COVID-19 Pandemic with Patterns of Statewide Cancer Services. J. Nat. Canc. Inst. 2021. [Google Scholar] [CrossRef]
- Phillips, P.; Phillips, J. Hysterectomy with radiotherapy or chemotherapy or both for women with locally advanced cervical cancer. Clin. Nurse Spec. 2017, 31, 189–190. [Google Scholar] [CrossRef]
- Dhama, K.; Chauhan, R.; Singhal, L. Anti-cancer activity of cow urine: Current status and future directions. Int J. Cow Sci. 2005, 1, 1–25. [Google Scholar]
- Yang, C.-C.; Chang, K.-W. Eicosanoids and HB-EGF/EGFR in cancer. Cancer Metastasis Rev. 2018, 37, 385–395. [Google Scholar] [CrossRef]
- Musa, A.; Al-Sanea, M.M.; Alotaibi, N.H.; Alnusaire, T.S.; Ahmed, S.R.; Mostafa, E.M. In silico Study, Protein Kinase Inhibition and Antiproliferative Potential of Flavonoids Isolated from Bassia eriophora (Schrad.) Growing in KSA. Ind. J. Pharmac. Edu. Res. 2021, 55, 483–490. [Google Scholar] [CrossRef]
- Scheff, R.J.; Schneider, B.J. Seminars in interventional radiology. In Non–Small-Cell Lung Cancer: Treatment of Late Stage Disease: Chemotherapeutics and New Frontiers; Thieme Medical Publishers: New York, NY, USA, 2013; pp. 191–198. [Google Scholar]
- Yokouchi, H.; Kanazawa, K.; Ishida, T.; Oizumi, S.; Shinagawa, N.; Sukoh, N.; Harada, M.; Ogura, S.; Munakata, M.; Dosaka-Akita, H. Cyclooxygenase-2 inhibitors for non-small-cell lung cancer: A phase II trial and literature review. Mol. Clinic. Oncol. 2014, 2, 744–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.L.; Langenbach, R. Why there are two cyclooxygenase isozymes. J. Clinic. Investig. 2001, 107, 1491–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandler, A.B.; Dubinett, S.M. COX-2 inhibition and Lung Cancer. Semin. Oncol. 2004, 31, 45–52. [Google Scholar]
- Shtivelman, E.; Hensing, T.; Simon, G.R.; Dennis, P.A.; Otterson, G.A.; Bueno, R.; Salgia, R. Molecular pathways and therapeutic targets in lung cancer. Oncotarget 2014, 5, 1392. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Stiegler, A.L.; Boggon, T.J.; Kobayashi, S.; Halmos, B. EGFR-mutated lung cancer: A paradigm of molecular oncology. Oncotarget 2010, 1, 497. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhou, Z.; Sun, X.; Yang, Z.; Zheng, P.; Xu, S.; Zhu, W. The new opportunities in medicinal chemistry of fourth-generation EGFR inhibitors to overcome C797S mutation. Eur. J. Med. Chem. 2021, 210, 112995. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by design: A medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem. 2018, 62, 420–444. [Google Scholar] [CrossRef]
- Alzarea, S.I.; Elmaidomy, A.H.; Saber, H.; Musa, A.; Al-Sanea, M.M.; Mostafa, E.M.; Hendawy, O.M.; Youssif, K.A.; Alanazi, A.S.; Alharbi, M. Potential anticancer lipoxygenase inhibitors from the red sea-derived brown algae sargassum cinereum: An in-silico-supported In-Vitro Study. Antibiotics 2021, 10, 416. [Google Scholar] [CrossRef]
- Abdellatif, K.R.; Abdelgawad, M.A.; Elshemy, H.A.; Alsayed, S.S.; Kamel, G. Synthesis and anti-inflammatory evaluation of new 1, 3, 5-triaryl-4, 5-dihydro-1 H-pyrazole derivatives possessing an aminosulphonyl pharmacophore. Arch. Pharm. Res. 2015, 38, 1932–1942. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Ahmad, W.; Al-Sanea, M.M.; Elshemy, H.A. New pyrimidine-benzoxazole/benzimidazole hybrids: Synthesis, antioxidant, cytotoxic activity, in vitro cyclooxygenase and phospholipase A2-V inhibition. Bioorg. Chem. 2019, 92, 103218. [Google Scholar] [CrossRef]
- Abdellatif, K.R.; Abdelgawad, M.A.; Labib, M.B.; Zidan, T.H. Synthesis and biological evaluation of new diarylpyrazole and triarylimidazoline derivatives as selective COX-2 inhibitors. Arch. Der Pharm. 2017, 350, 1600386. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; El-Gendy, A.O.; Kamel, G.M.; Azouz, A.A.; Bukhari, S.N.A. Discovery of a COX-2 selective inhibitor hit with anti-inflammatory activity and gastric ulcer protective effect. Future Med. Chem. 2017, 9, 1899–1912. [Google Scholar] [CrossRef]
- Musa, A. Chemical constituents, antimicrobial and antiinflammatory evaluations of various extracts ofsuaeda vera forssk. Growing in Saudi Arabia. Int. J. Pharm. Res. 2019, 11, 962–967. [Google Scholar]
- Cao, Z.; Yang, J.; Xu, R.; Song, Q.; Zhang, X.; Liu, H.; Qiang, X.; Li, Y.; Tan, Z.; Deng, Y. Design, synthesis and evaluation of 4′-OH-flurbiprofen-chalcone hybrids as potential multifunctional agents for Alzheimer’s disease treatment. Bioorg. Med. Chem. 2018, 26, 1102–1115. [Google Scholar]
- Gaber, M.; El-Ghamry, H.A.; Mansour, M.A. Pd (II) and Pt (II) chalcone complexes. Synthesis, spectral characterization, molecular modeling, biomolecular docking, antimicrobial and antitumor activities. J. Photochem. Photobiol. A Chem. 2018, 354, 163–174. [Google Scholar] [CrossRef]
- Abou-Zied, H.A.; Youssif, B.G.; Mohamed, M.F.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef]
- Sasidharan, R.; Eom, B.H.; Heo, J.H.; Park, J.E.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Manju, S.L.; Mathew, B. Morpholine-based chalcones as dual-acting monoamine oxidase-B and acetylcholinesterase inhibitors: Synthesis and biochemical investigations. J. Enzym. Inhib. Med. Chem. 2021, 36, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, M.A.; Mohamed, A.M.; Musa, A.; Mostafa, E.M.; Awad, H.M. Synthesis, chromatographic separation and antimicrobial evolution of new azoquinoline-8-ol. J. Pharm. Sci. Res. 2018, 10, 1314–1318. [Google Scholar]
- Kik, K.; Bukowska, B.; Krokosz, A.; Sicińska, P. Oxidative Properties of Polystyrene Nanoparticles with Different Diameters in Human Peripheral Blood Mononuclear Cells (In Vitro Study). Int. J. Mol. Sci. 2021, 22, 4406. [Google Scholar] [CrossRef]
- Bougueria, H.; Benosmane, A.; Benaouida, M.A.; Bouchoul, A.E.K.; Bouaoud, S.E. 1-(3-Acetylphenyl)-2-(2-oxidonaphthalen-1-yl) diazen-1-ium. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, o1052. [Google Scholar] [CrossRef] [Green Version]
- Wu, J. Synthetic Studies in Nitrogen Chemistry. Ph.D. Dissertation, University of Florida, Gainesville, FL, USA, 1992. [Google Scholar]
- Gao, X.; Xu, Y.X.; Janakiraman, N.; Chapman, R.A.; Gautam, S.C. Immunomodulatory activity of resveratrol: Suppression of lymphocyte proliferation, development of cell-mediated cytotoxicity, and cytokine production. Biochem. Pharmacol. 2001, 62, 1299–1308. [Google Scholar] [CrossRef]
- Gao, X.; Kuo, J.; Jiang, H.; Deeb, D.; Liu, Y.; Divine, G.; Chapman, R.A.; Dulchavsky, S.A.; Gautam, S.C. Immunomodulatory activity of curcumin: Suppression of lymphocyte proliferation, development of cell-mediated cytotoxicity, and cytokine production in vitro. Biochem. Pharmacol. 2004, 68, 51–61. [Google Scholar] [CrossRef]
- Musa, A.; Elmaidomy, A.H.; Sayed, A.M.; Alzarea, S.I.; Al-Sanea, M.M.; Mostafa, E.M.; Hendawy, O.M.; Abdelgawad, M.A.; Youssif, K.A.; Refaat, H. Cytotoxic Potential, Metabolic Profiling, and Liposomes of Coscinoderma sp. Crude Extract Supported by in silico Analysis. Int. J. Nanomed. 2021, 16, 3861. [Google Scholar] [CrossRef]
- Al-Sanea, M.M. Synthesis and biological evaluation of small molecule modulators of cdk8/cyclin c complex with phenylaminoquinoline scaffold. PeerJ 2020, 8, e8649. [Google Scholar] [CrossRef]
- Al-Sanea, M.; Abdelazem, A.; Park, B.; Yoo, K.; Sim, T.; Kwon, Y.; Lee, S. ROS1 kinase inhibitors for molecular-targeted therapies. Curr. Med. Chem. 2016, 23, 142–160. [Google Scholar] [CrossRef]
- Pohanka, M.; Hrabinova, M.; Kuca, K. Diagnosis of intoxication by the organophosphate vx: Comparison between an electrochemical sensor and ellman s photometric method. Sensors 2008, 8, 5229–5237. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, W.; Kumolosasi, E.; Jantan, I.; Bukhari, S.N.; Jasamai, M. Effects of novel diarylpentanoid analogues of curcumin on secretory phospholipase A2, cyclooxygenases, lipo-oxygenase, and microsomal prostaglandin E synthase-1. Chem. Biol. Drug Des. 2014, 83, 670–681. [Google Scholar]
- Abdelgawad, M.A.; Musa, A.; Almalki, A.H.; Alzarea, S.I.; Mostafa, E.M.; Hegazy, M.M.; Mostafa-Hedeab, G.; Ghoneim, M.M.; Parambi, D.G.; Bakr, R.B. Novel Phenolic Compounds as Potential Dual EGFR and COX-2 Inhibitors: Design, Semisynthesis, in vitro Biological Evaluation and in silico Insights. Drug Des. Dev. Ther. 2021, 15, 2325. [Google Scholar] [CrossRef] [PubMed]
- Gogos, C.A.; Drosou, E.; Bassaris, H.P.; Skoutelis, A. Pro-versus anti-inflammatory cytokine profile in patients with severe sepsis: A marker for prognosis and future therapeutic options. J. Infect. Dis. 2000, 181, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Maione, F.; Uddin, G.; Raza, M.; Siddiqui, B.S.; Muhammad, N.; Shah, S.U.A.; Khan, H.; De Feo, V.; Mascolo, N. Biological evaluation and docking analysis of daturaolone as potential cyclooxygenase inhibitor. Evid.-Based Complement. Altern. Med. 2016, 2016, 4098686. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Grimm, M.; Dai, W.-t.; Hou, M.-c.; Xiao, Z.-X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef]
- Farouk, A.; Mohsen, M.; Ali, H.; Shaaban, H.; Albaridi, N. Antioxidant Activity and Molecular Docking Study of Volatile Constituents from Different Aromatic Lamiaceous Plants Cultivated in Madinah Monawara, Saudi Arabia. Molecules 2021, 26, 4145. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N. | Code | Cell Viability % | Antiproliferative Activity IC50 ± SEM (µM) | ||||
|---|---|---|---|---|---|---|---|
| HT-29 | PaCa-2 | A375 | H-460 | Panc-1 | |||
| 1. | A1 | 80.3 ± 1.6 | 8.3 ± 2.1 | 8.6 ± 1.2 | 9.3 ± 1.9 | 7.4 ± 2.2 | 9.1 ± 1.8 |
| 2. | A2 | 90.4 ± 1.9 | 9.2 ± 2.5 | 9.6 ± 1.5 | 8.4 ± 2.1 | 5.9 ± 2.4 | 9.6 ± 2.0 |
| 3. | A3 | 79.6 ± 4.5 | 1.8 ± 0.5 | 2.4 ± 1.5 | 4.0 ± 1.5 | 1.8 ± 0.4 | 2.2 ± 0.9 |
| 4. | A4 | 87.4 ± 1.7 | 2.7 ± 0.6 | 2.0 ± 0.6 | 4.4 ± 1.9 | 3.4 ± 1.0 | 2.0 ± 0.4 |
| 5. | A5 | 88.2 ± 1.2 | 11.9 ± 2.5 | 10.5 ± 1.9 | 9.6 ± 2.4 | 9.2 ± 1.2 | 9.6 ± 1.8 |
| 6. | A6 | 82.5 ± 1.7 | 7.6 ± 1.4 | 9.5 ± 1.9 | 9.5 ± 1.5 | 8.8 ± 2.3 | 6.9 ± 2.1 |
| 7. | C7 | 82.6 ± 1.2 | 4.5 ± 1.7 | 2.9 ± 1.7 | 5.3 ± 1.5 | 5.0 ± 1.4 | 4.8 ± 1.6 |
| 8. | C8 | 83.5 ± 1.2 | 2.5 ± 07 | 2.8 ± 0.6 | 2.6 ± 0.5 | 2.2 ± 0.5 | 1.9 ± 0.7 |
| 9. | C9 | 90.2 ± 4.1 | 1.2 ± 0.3 | 2.1 ± 0.3 | 3.2 ± 1.1 | 1.2 ± 0.2 | 1.8 ± 0.4 |
| 10. | C10 | 91.2 ± 2.9 | 1.1 ± 0.2 | 0.9 ± 0.4 | 2.6 ± 1.0 | 1.2 ± 0.1 | 1.6 ± 0.5 |
| 11. | C11 | 82.2 ± 2.4 | 2.9 ± 1.0 | 1.6 ± 0.5 | 3.5 ± 1.2 | 2.8 ± 0.7 | 1.6 ± 0.5 |
| 12. | C12 | 92.0 ± 2.0 | 1.4 ± 0.5 | 0.8 ± 0.4 | 2.4 ± 1.1 | 1.9 ± 0.3 | 1.2 ± 0.7 |
| 13. | C13 | 90.6 ± 1.7 | 7.8 ± 1.8 | 7.4 ± 1.7 | 8.4 ± 1.5 | 6.6 ± 1.2 | 8.7 ± 1.4 |
| 14. | C14 | 89.4 ± 2.1 | 6.5 ± 1.4 | 5.9 ± 1.9 | 4.9 ± 1.6 | 5.2 ± 1.3 | 4.6 ± 1.7 |
| 15. | C15 | 87.3 ± 2.1 | 1.6 ± 0.2 | 2.3 ± 0.4 | 3.4 ± 1.8 | 1.7 ± 0.2 | 1.9 ± 0.1 |
| 16. | C16 | 92.5 ± 2.0 | 1.8 ± 0.4 | 1.9 ± 0.5 | 2.8 ± 1.0 | 2.9 ± 1.0 | 1.8 ± 0.5 |
| 17. | C17 | 87.2 ± 1.4 | 9.2 ± 1.6 | 7.5 ± 1.9 | 8.8 ± 2.5 | 5.6 ± 2.1 | 2.5 ± 1.2 |
| 18. | C18 | 88.4 ± 2.9 | 6.7 ± 0.6 | 6.4 ± 1.5 | 8.8 ± 2.5 | 6.3 ± 1.4 | 6.4 ± 0.4 |
| 19. | Erlotinib | - | 0.07 ± 0.04 | 0.06 ± 0.04 | 4.14 ± 1.2 | 0.04 ± 0.02 | 0.05 ± 0.02 |
| Compound | EGFR Inhibition IC50 ± SEM (µM) |
|---|---|
| A3 | 4.3 ± 0.7 |
| A4 | 3.2 ± 0.8 |
| C7 | 5.8 ± 1.1 |
| C8 | 4.9 ± 0.9 |
| C9 | 0.8 ± 0.3 |
| C10 | 1.1 ± 0.0.2 |
| C11 | 7.2 ± 1.4 |
| C12 | 6.3 ± 1.7 |
| C15 | 2.8 ± 0.5 |
| C16 | 2.9 ± 0.4 |
| Erlotinib | 0.05 ± 0.02 |
| Compound | sPLA2-V IC50 (µM) | COX-1 IC50 (µM) | COX-2 IC50 (µM) |
|---|---|---|---|
| A2 | 7.25 ± 1.24 | 22.2 ± 2.45 | 27.92 ± 1.65 |
| A3 | 2.14 ± 0.94 | 0.21 ± 0.07 | 2.40 ± 0.8 |
| A4 | 14.14 ± 1.52 | 11.12 ± 1.05 | 16.78 ± 2.43 |
| A5 | 4.24 ± 1.16 | 9.23 ± 1.64 | 14.25 ± 1.79 |
| A6 | 7.42 ± 1.41 | 4.85 ± 1.20 | 5.21 ± 1.2 |
| C7 | 24.72 ± 1.59 | 7.52 ± 1.42 | 29.42 ± 1.73 |
| C8 | 14.21 ± 1.32 | 5.14 ± 1.27 | 32.15 ± 1.40 |
| C9 | 2.74 ± 1.24 | 8.57 ± 1.89 | 1.27 ± 0.3 |
| C10 | 3.14 ± 0.64 | 7.37 ± 1.44 | 1.88 ± 0.4 |
| C11 | 5.57 ± 1.19 | 1.95 ± 1.07 | 7.53 ± 0.9 |
| C12 | 7.45 ± 1.23 | 2.24 ± 1.06 | 5.47 ± 0.8 |
| C13 | 5.06 ± 1.20 | 18.29 ± 1.27 | 19.22 ± 1.52 |
| C14 | 7.04 ± 1.29 | 11.55 ± 1.20 | 5.14 ± 1.1 |
| C15 | 2.01 ± 1.11 | 1.9 ± 0.23 | 0.95 ± 0.2 |
| C16 | 2.51 ± 1.05 | 7.10 ± 1.29 | 6.89 ± 0.8 |
| C17 | 9.45 ± 1.67 | 49.11 ± 2.69 | 5.29 ± 1.1 |
| C18 | 8.29 ± 0.88 | 40.17 ± 3.42 | 3.20 ± 0.6 |
| Dexamethasone | 0.57 ± 0.06 | - | - |
| Indomethacin * | - | 0.27 ± 0.04 | 3.29 ± 0.5 |
| Compound | % Inhibition of IL-6 | Relative Amount of LPS | % Inhibition of TNF-α | Relative Amount of LPS |
|---|---|---|---|---|
| 3 | 72 | 28 | 79 | 21 |
| 4 | 46 | 54 | 49 | 51 |
| 9 | 77 | 23 | 74 | 26 |
| 10 | 77 | 23 | 76 | 24 |
| 15 | 78 | 22 | 81 | 19 |
| 16 | 69 | 31 | 72 | 28 |
| 18 | 73 | 17 | 71 | 29 |
| LPS Control | 0 | 100 | 0 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musa, A.; Mostafa, E.M.; Bukhari, S.N.A.; Alotaibi, N.H.; El-Ghorab, A.H.; Farouk, A.; Nayl, A.A.; Ghoneim, M.M.; Abdelgawad, M.A. EGFR and COX-2 Dual Inhibitor: The Design, Synthesis, and Biological Evaluation of Novel Chalcones. Molecules 2022, 27, 1158. https://doi.org/10.3390/molecules27041158
Musa A, Mostafa EM, Bukhari SNA, Alotaibi NH, El-Ghorab AH, Farouk A, Nayl AA, Ghoneim MM, Abdelgawad MA. EGFR and COX-2 Dual Inhibitor: The Design, Synthesis, and Biological Evaluation of Novel Chalcones. Molecules. 2022; 27(4):1158. https://doi.org/10.3390/molecules27041158
Chicago/Turabian StyleMusa, Arafa, Ehab M. Mostafa, Syed Nasir Abbas Bukhari, Nasser Hadal Alotaibi, Ahmed H. El-Ghorab, Amr Farouk, AbdElAziz A. Nayl, Mohammed M. Ghoneim, and Mohamed A. Abdelgawad. 2022. "EGFR and COX-2 Dual Inhibitor: The Design, Synthesis, and Biological Evaluation of Novel Chalcones" Molecules 27, no. 4: 1158. https://doi.org/10.3390/molecules27041158
APA StyleMusa, A., Mostafa, E. M., Bukhari, S. N. A., Alotaibi, N. H., El-Ghorab, A. H., Farouk, A., Nayl, A. A., Ghoneim, M. M., & Abdelgawad, M. A. (2022). EGFR and COX-2 Dual Inhibitor: The Design, Synthesis, and Biological Evaluation of Novel Chalcones. Molecules, 27(4), 1158. https://doi.org/10.3390/molecules27041158