
Understanding Parahydrogen Hyperpolarized Urine Spectra: The Case of Adenosine Derivatives


Abstract
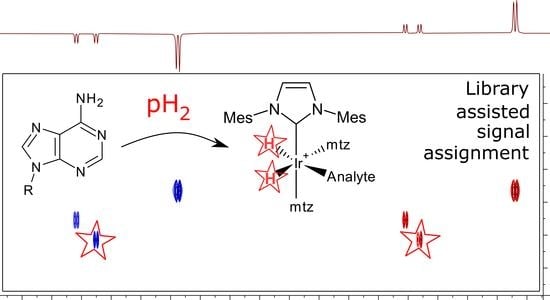
:
1. Introduction
1.1. Parahydrogen Hyperpolarized Chemosensing
1.2. Analytical Value of Adenosine Derivatives
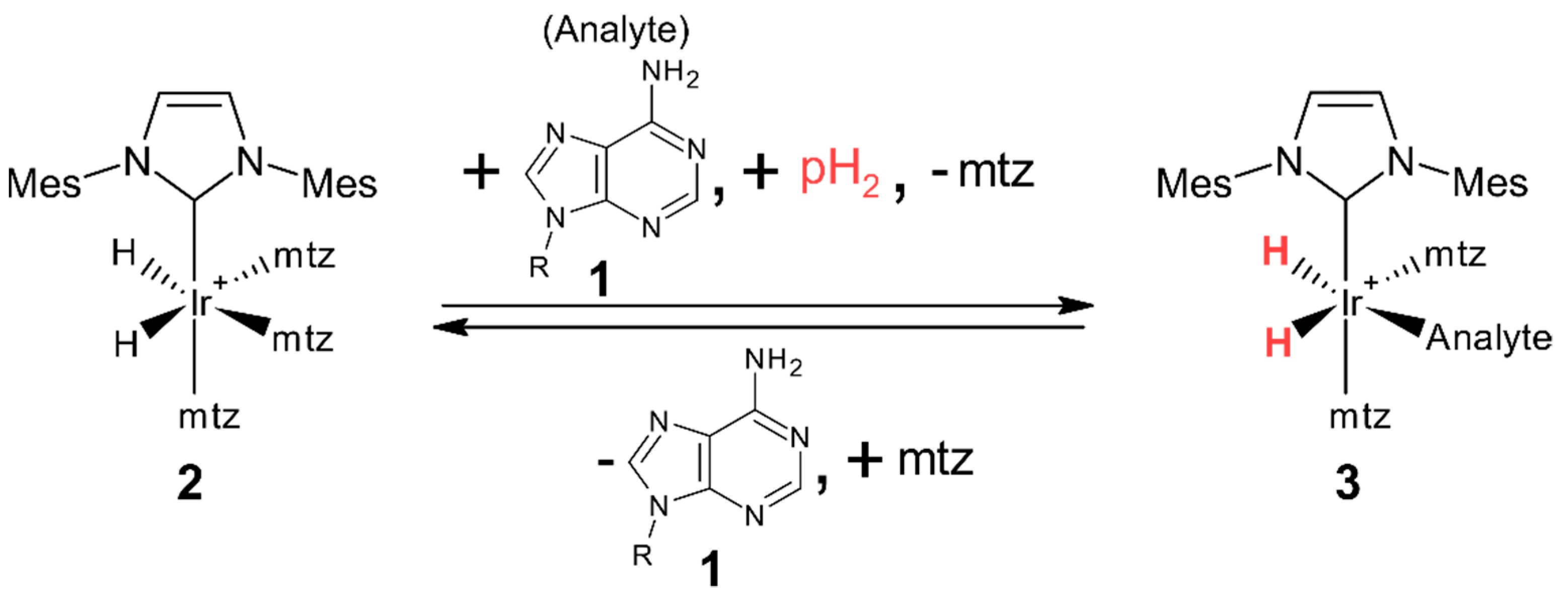
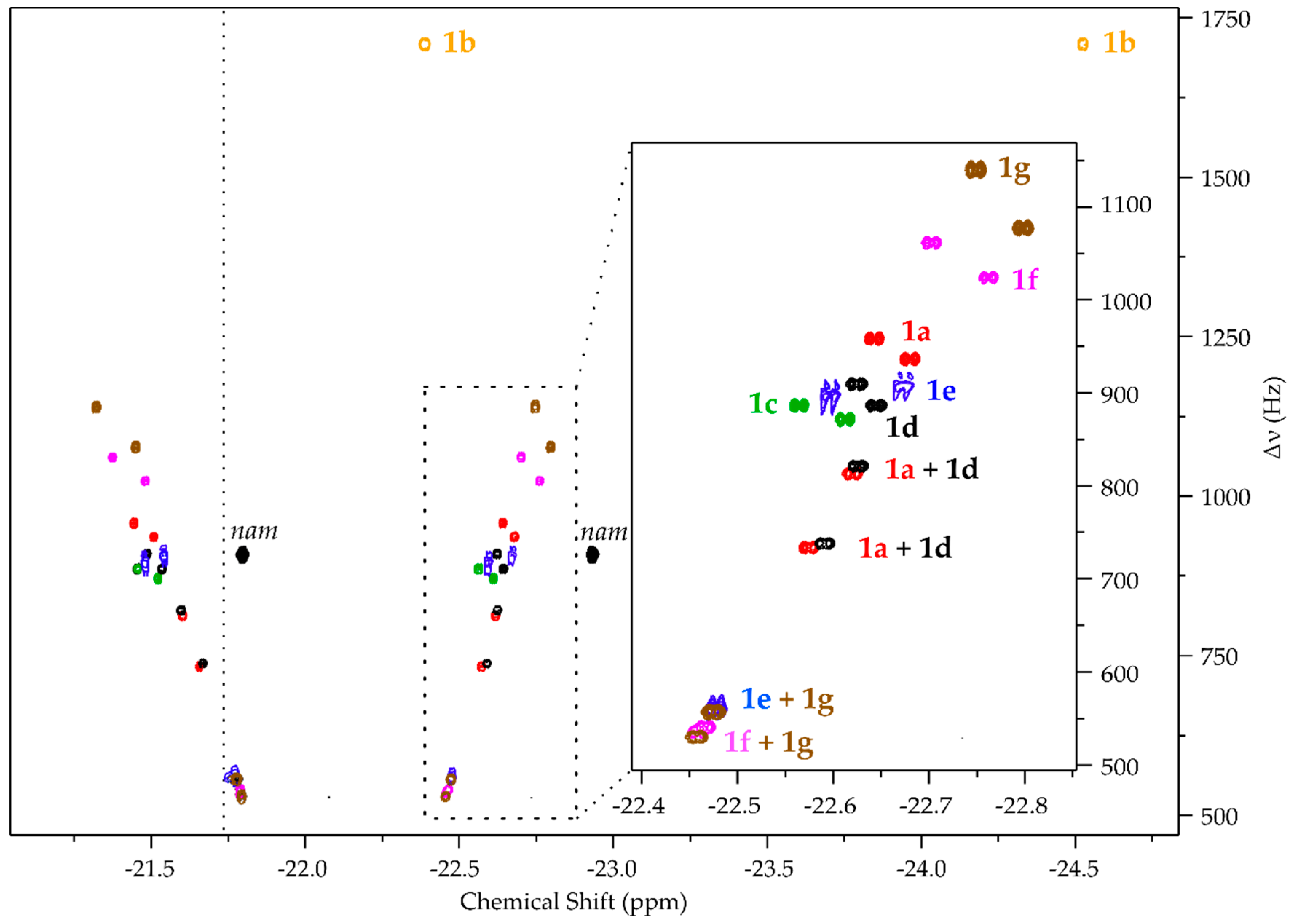
2. Results
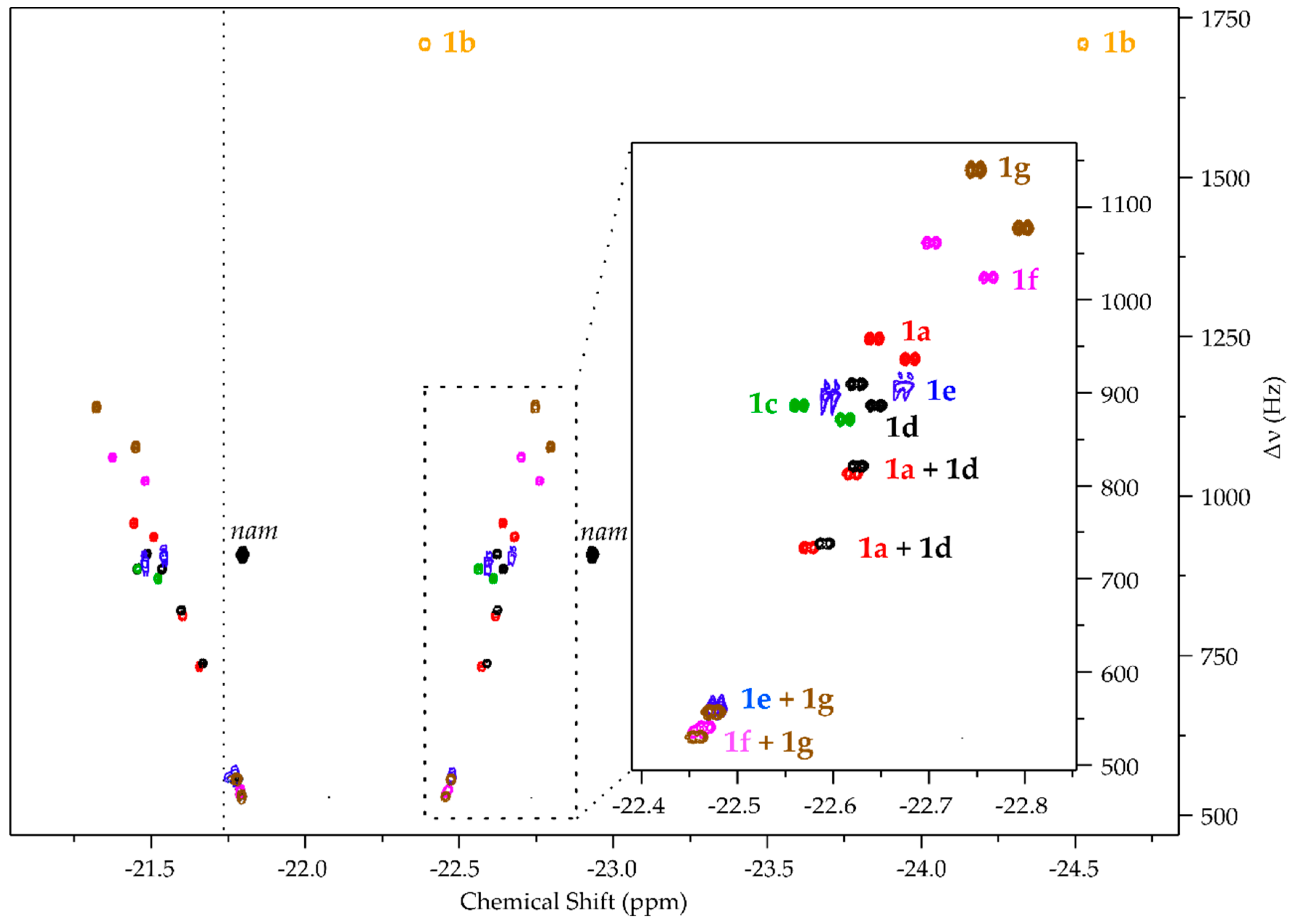
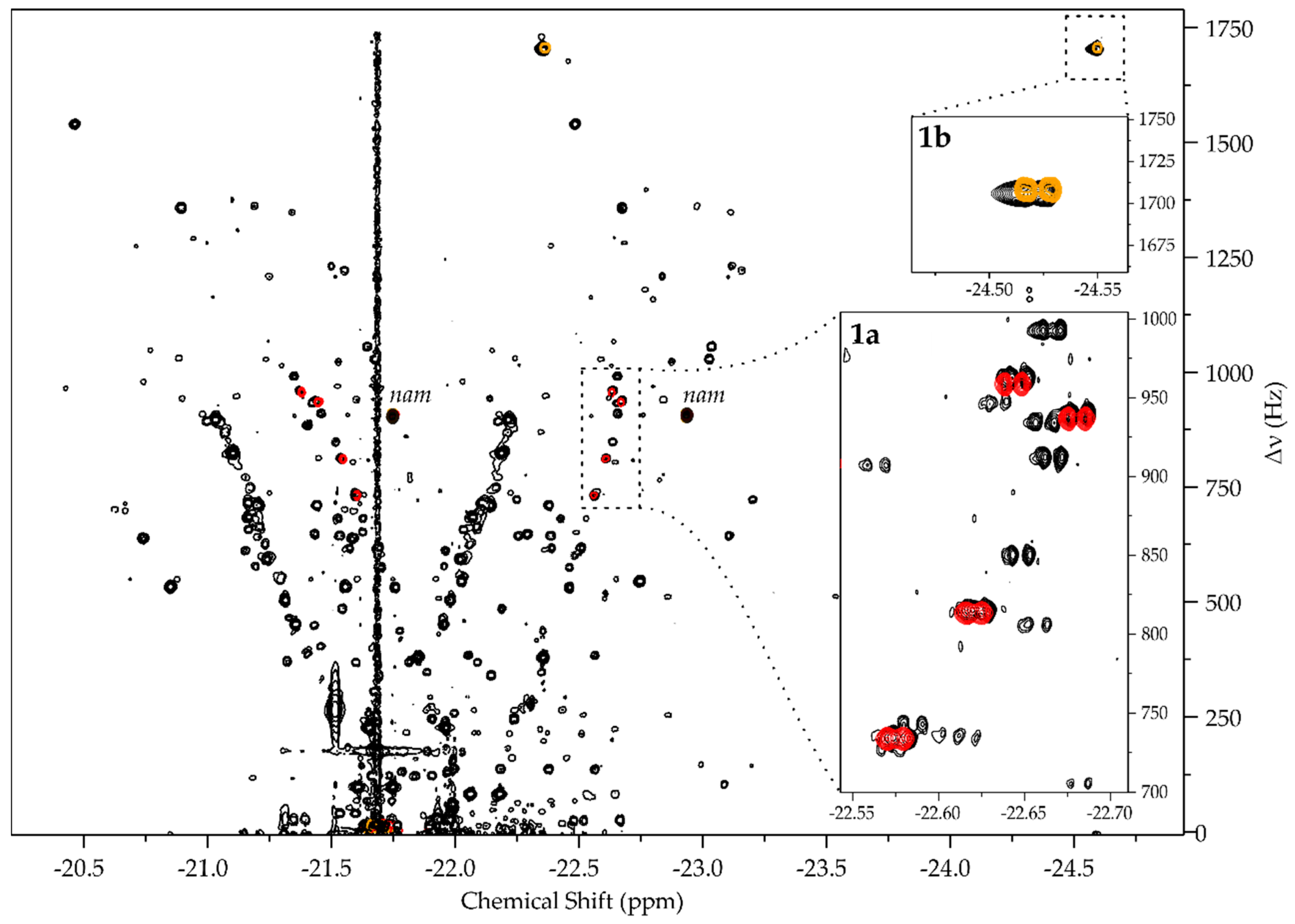
Building the Library
3. Discussion
- pH2 hyperpolarized chemosensing is remarkably sensitive to relatively small changes in analyte structure, allowing for the resolution of series of structurally highly similar metabolites, including closely related isomers (i.e., 1b, 1c, 1d).
- Libraries can be applied by a straightforward superposition of experimental and database spectra.
- The databases would have universality across different instruments, laboratories, and variations in sample preparation procedures.
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Emwas, A.-H.M.; Salek, R.M.; Griffin, J.L.; Merzaban, J. NMR-based metabolomics in human disease diagnosis: Applications, limitations, and recommendations. Metabolomics 2013, 9, 1048–1072. [Google Scholar] [CrossRef]
- Markley, J.L.; Brüschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtunov, K.V.; Pokochueva, E.V.; Salnikov, O.G.; Cousin, S.F.; Kurzbach, D.; Vuichoud, B.; Jannin, S.; Chekmenev, E.Y.; Goodson, B.M.; Barskiy, D.A.; et al. Hyperpolarized NMR Spectroscopy: D-DNP, PHIP, and SABRE Techniques. Chem.-Asian J. 2018, 13, 1857–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermkens, N.K.J.; Eshuis, N.; van Weerdenburg, B.J.A.; Feiters, M.C.; Rutjes, F.P.J.T.; Wijmenga, S.S.; Tessari, M. NMR-Based Chemosensing via p-H2 Hyperpolarization: Application to Natural Extracts. Anal. Chem. 2016, 88, 3406–3412. [Google Scholar] [CrossRef] [Green Version]
- Hermkens, N.K.J.; Aspers, R.L.E.G.; Feiters, M.C.; Rutjes, F.P.J.T.; Tessari, M. Trace analysis in water-alcohol mixtures by continuous p-H2 hyperpolarization at high magnetic field. Magn. Reson. Chem. 2018, 56, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Sellies, L.; Reile, I.; Aspers, R.L.E.G.; Feiters, M.C.; Rutjes, F.P.J.T.; Tessari, M. Parahydrogen induced hyperpolarization provides a tool for NMR metabolomics at nanomolar concentrations. Chem. Commun. 2019, 55, 7235–7238. [Google Scholar] [CrossRef]
- Ausmees, K.; Reimets, N.; Reile, I. Parahydrogen hyperpolarization of minimally altered urine samples for sensitivity enhanced NMR metabolomics. Chem. Commun. 2022, 58, 463–466. [Google Scholar] [CrossRef]
- Reile, I.; Eshuis, N.; Hermkens, N.K.J.; van Weerdenburg, B.J.A.; Feiters, M.C.; Rutjes, F.P.J.T.; Tessari, M. NMR detection in biofluid extracts at sub-uM concentrations via para-H2 induced hyperpolarization. Analyst 2016, 141, 4001–4005. [Google Scholar] [CrossRef] [Green Version]
- Reimets, N.; Ausmees, K.; Vija, S.; Reile, I. Developing Analytical Applications for Parahydrogen Hyperpolarization: Urinary Elimination Pharmacokinetics of Nicotine. Anal. Chem. 2021, 93, 9480–9485. [Google Scholar] [CrossRef]
- Adelabu, I.; TomHon, P.; Kabir, M.S.H.; Nantogma, S.; Abdulmojeed, M.; Mandzhieva, I.; Ettedgui, J.; Swenson, R.E.; Krishna, M.C.; Goodson, B.M.; et al. Order-Unity 13C Nuclear Polarization of [1-13C]Pyruvate in Seconds and the Interplay of Water and SABRE Enhancement. ChemPhysChem 2022, 23, e202100839. [Google Scholar] [CrossRef]
- Ratajczyk, T.; Gutmann, T.; Bernatowicz, P.; Buntkowsky, G.; Frydel, J.; Fedorczyk, B. NMR Signal Enhancement by Effective SABRE Labeling of Oligopeptides. Chem.-Eur. J. 2015, 21, 12616–12619. [Google Scholar] [CrossRef] [PubMed]
- Ratajczyk, T.; Buntkowsky, G.; Gutmann, T.; Fedorczyk, B.; Mames, A.; Pietrzak, M.; Puzio, Z.; Szkudlarek, P.G. Magnetic Resonance Signal Amplification by Reversible Exchange of Selective PyFALGEA Oligopeptide Ligands Towards Epidermal Growth Factor Receptors. ChemBioChem 2021, 22, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Sellies, L.; Aspers, R.L.E.G.; Feiters, M.C.; Rutjes, F.P.J.T.; Tessari, M. Parahydrogen Hyperpolarization Allows Direct NMR Detection of α-Amino Acids in Complex (Bio)mixtures. Angew. Chem. Int. Ed. 2021, 60, 26954–26959. [Google Scholar] [CrossRef] [PubMed]
- Pravdivtsev, A.N.; Buntkowsky, G.; Duckett, S.B.; Koptyug, I.V.; Hövener, J.-B. Parahydrogen-Induced Polarization of Amino Acids. Angew. Chem. Int. Ed. 2021, 60, 23496–23507. [Google Scholar] [CrossRef]
- Kim, S.; Min, S.; Chae, H.; Jeong, H.J.; Namgoong, S.K.; Oh, S.; Jeong, K. Hyperpolarization of Nitrile Compounds Using Signal Amplification by Reversible Exchange. Molecules 2020, 25, 3347. [Google Scholar] [CrossRef]
- Shchepin, R.V.; Barskiy, D.A.; Coffey, A.M.; Goodson, B.M.; Chekmenev, E.Y. NMR Signal Amplification by Reversible Exchange of Sulfur-Heterocyclic Compounds Found In Petroleum. ChemistrySelect 2016, 1, 2552–2555. [Google Scholar] [CrossRef] [Green Version]
- Hermkens, N.K.J.; Feiters, M.C.; Rutjes, F.P.J.T.; Wijmenga, S.S.; Tessari, M. High field hyperpolarization-EXSY experiment for fast determination of dissociation rates in SABRE complexes. J. Magn. Reson. 2017, 276, 122–127. [Google Scholar] [CrossRef]
- Sellies, L.; Aspers, R.L.E.G.; Tessari, M. Determination of hydrogen exchange and relaxation parameters in PHIP complexes at micromolar concentrations. Magn. Reson. 2021, 2, 331–340. [Google Scholar] [CrossRef]
- Giraudeau, P. Quantitative 2D liquid-state NMR. Magn. Reson. Chem. 2014, 52, 259–272. [Google Scholar] [CrossRef]
- Aspers, R.L.E.G.; Tessari, M. An approach to fast 2D nuclear magnetic resonance at low concentration based on p-H2-induced polarization and nonuniform sampling. Magn. Reson. Chem. 2021, 59, 1236–1243. [Google Scholar] [CrossRef]
- Wood, N.J.; Brannigan, J.A.; Duckett, S.B.; Heath, S.L.; Wagstaff, J. Detection of Picomole Amounts of Biological Substrates by para -Hydrogen-Enhanced NMR Methods in Conjunction with a Suitable Receptor Complex. J. Am. Chem. Soc. 2007, 129, 11012–11013. [Google Scholar] [CrossRef] [PubMed]
- Hövener, J.-B.; Schwaderlapp, N.; Lickert, T.; Duckett, S.B.; Mewis, R.E.; Highton, L.A.R.; Kenny, S.M.; Green, G.G.R.; Leibfritz, D.; Korvink, J.G.; et al. A hyperpolarized equilibrium for magnetic resonance. Nat. Commun. 2013, 4, 2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, A.; Brunner, S.; Seidel, P.; Fritz, G.I.; Herbarth, O. Modified nucleosides: An accurate tumour marker for clinical diagnosis of cancer, early detection and therapy control. Br. J. Cancer 2006, 94, 1726–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patejko, M.; Struck-Lewicka, W.; Siluk, D.; Waszczuk-Jankowska, M.; Markuszewski, M.J. Urinary Nucleosides and Deoxynucleosides. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 83, pp. 1–51. ISBN 9780128152072. [Google Scholar]
- Struck, W.; Siluk, D.; Yumba-Mpanga, A.; Markuszewski, M.; Kaliszan, R.; Markuszewski, M.J. Liquid chromatography tandem mass spectrometry study of urinary nucleosides as potential cancer markers. J. Chromatogr. A 2013, 1283, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-Y.; Chen, C.-J.; Huang, Y.-C.; Tsai, F.-J.; Jeng, L.-B.; Lai, C.-C. Urinary Nucleosides as Biomarkers of Breast, Colon, Lung, and Gastric Cancer in Taiwanese. PLoS ONE 2013, 8, e81701. [Google Scholar] [CrossRef]
- Seidel, A.; Seidel, P.; Manuwald, O.; Herbarth, O. Modified nucleosides as biomarkers for early cancer diagnose in exposed populations. Environ. Toxicol. 2015, 30, 956–967. [Google Scholar] [CrossRef]
- Altobelli, E.; Angeletti, P.M.; Latella, G. Role of Urinary Biomarkers in the Diagnosis of Adenoma and Colorectal Cancer: A Systematic Review and Meta-Analysis. J. Cancer 2016, 7, 1984–2004. [Google Scholar] [CrossRef] [Green Version]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The human urine metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Pucar, D.; Dzeja, P.P.; Bast, P.; Gumina, R.J.; Drahl, C.; Lim, L.; Juranic, N.; Macura, S.; Terzic, A. Mapping hypoxia-induced bioenergetic rearrangements and metabolic signaling by 18O-assisted 31P NMR and 1H NMR spectroscopy. Mol. Cell. Biochem. 2004, 256, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Dreyfus, M.; Dodin, G.; Bensaude, O.; Dubois, J.E. Tautomerism of purines. 2. Amino-imino tautomerism in 1-alkyladenines. J. Am. Chem. Soc. 1977, 99, 7027–7037. [Google Scholar] [CrossRef]
- Lippert, B.; Schöllhorn, H.; Thewalt, U. Metal-stabilized rare tautomers of nucleobases. 4. on the question of adenine tautomerization by a coordinated platinum(II). Inorg. Chim. Acta 1992, 198–200, 723–732. [Google Scholar] [CrossRef]
- Kapinos, L.E.; Operschall, B.P.; Larsen, E.; Sigel, H. Understanding the Acid-Base Properties of Adenosine: The Intrinsic Basicities of N1, N3 and N7. Chem.-Eur. J. 2011, 17, 8156–8164. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, Q.; Wang, M.; Fu, S.; Zhang, Q.; Zhang, Z.; Zhao, H.; Liu, Y.; Huang, Z.; Xie, Z.; et al. Using UHPLC Q-Trap/MS as a complementary technique to in-depth mine UPLC Q-TOF/MS data for identifying modified nucleosides in urine. J. Chromatogr. B 2017, 1051, 108–117. [Google Scholar] [CrossRef]
- Kovacs, H.; Moskau, D.; Spraul, M. Cryogenically cooled probes—A leap in NMR technology. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 46, 131–155. [Google Scholar] [CrossRef]
- Natterer, J.; Bargon, J. Parahydrogen induced polarization. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293–315. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Analyte | No | R @ 1 | R @ 6 | R @ 2′ | R @ 5′ |
|---|---|---|---|---|---|
| Adenosine | 1a | H | H | H | H |
| 1-Methyladenosine * | 1b | CH3 | H | H | H |
| 6N-Methyladenosine | 1c | H | CH3 | H | H |
| 2′-O-Methyladenosine | 1d | H | H | CH3 | H |
| AMP | 1e | H | H | H | PO3H |
| ADP | 1f | H | H | H | PO3-PO3H |
| ATP | 1g | H | H | H | PO3-PO3-PO3H |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ausmees, K.; Reimets, N.; Reile, I. Understanding Parahydrogen Hyperpolarized Urine Spectra: The Case of Adenosine Derivatives. Molecules 2022, 27, 802. https://doi.org/10.3390/molecules27030802
Ausmees K, Reimets N, Reile I. Understanding Parahydrogen Hyperpolarized Urine Spectra: The Case of Adenosine Derivatives. Molecules. 2022; 27(3):802. https://doi.org/10.3390/molecules27030802
Chicago/Turabian StyleAusmees, Kerti, Nele Reimets, and Indrek Reile. 2022. "Understanding Parahydrogen Hyperpolarized Urine Spectra: The Case of Adenosine Derivatives" Molecules 27, no. 3: 802. https://doi.org/10.3390/molecules27030802
APA StyleAusmees, K., Reimets, N., & Reile, I. (2022). Understanding Parahydrogen Hyperpolarized Urine Spectra: The Case of Adenosine Derivatives. Molecules, 27(3), 802. https://doi.org/10.3390/molecules27030802