
Hydrogen Bonds with Fluorine in Ligand–Protein Complexes-the PDB Analysis and Energy Calculations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
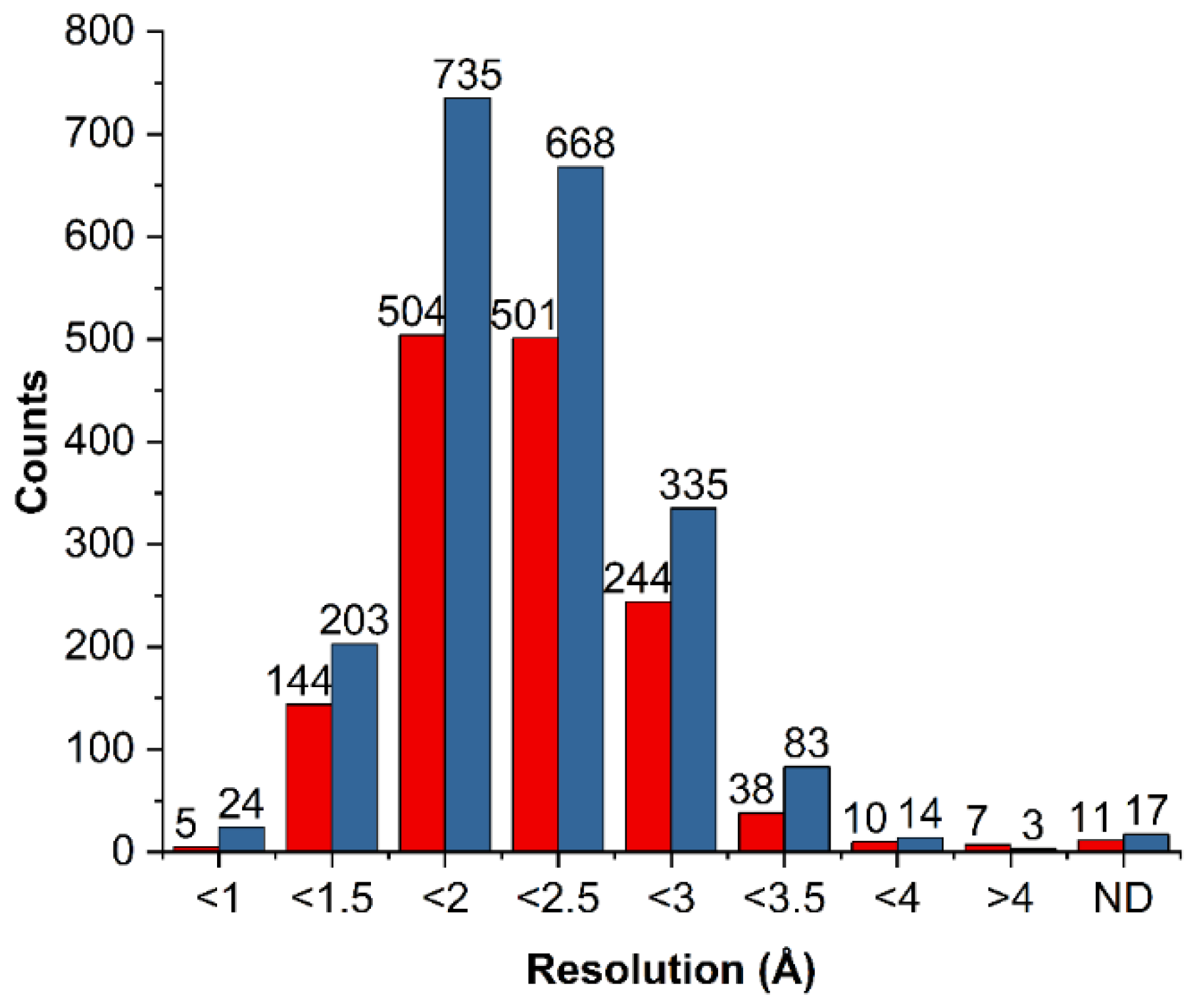
2.1. Choice of a Model System
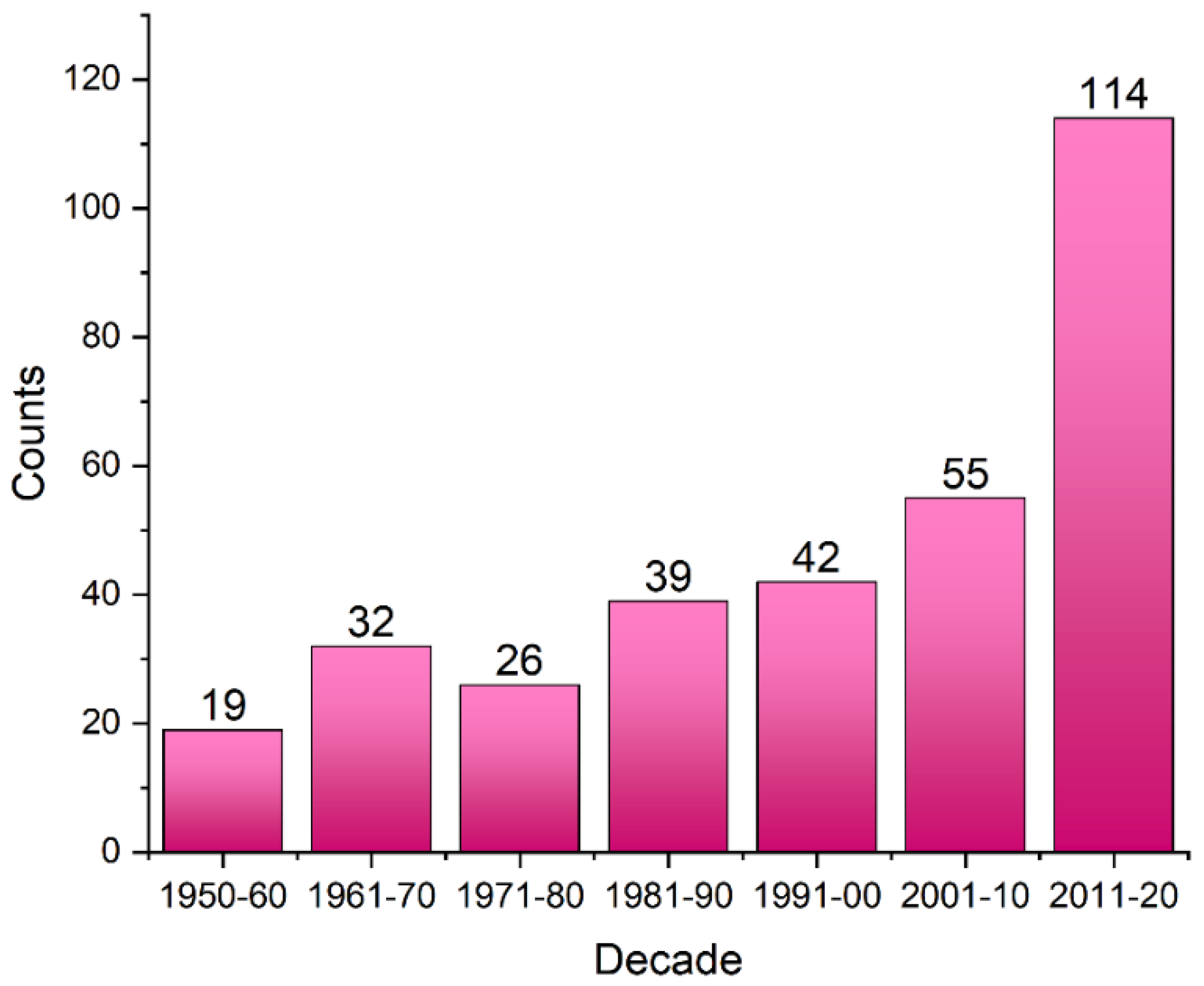
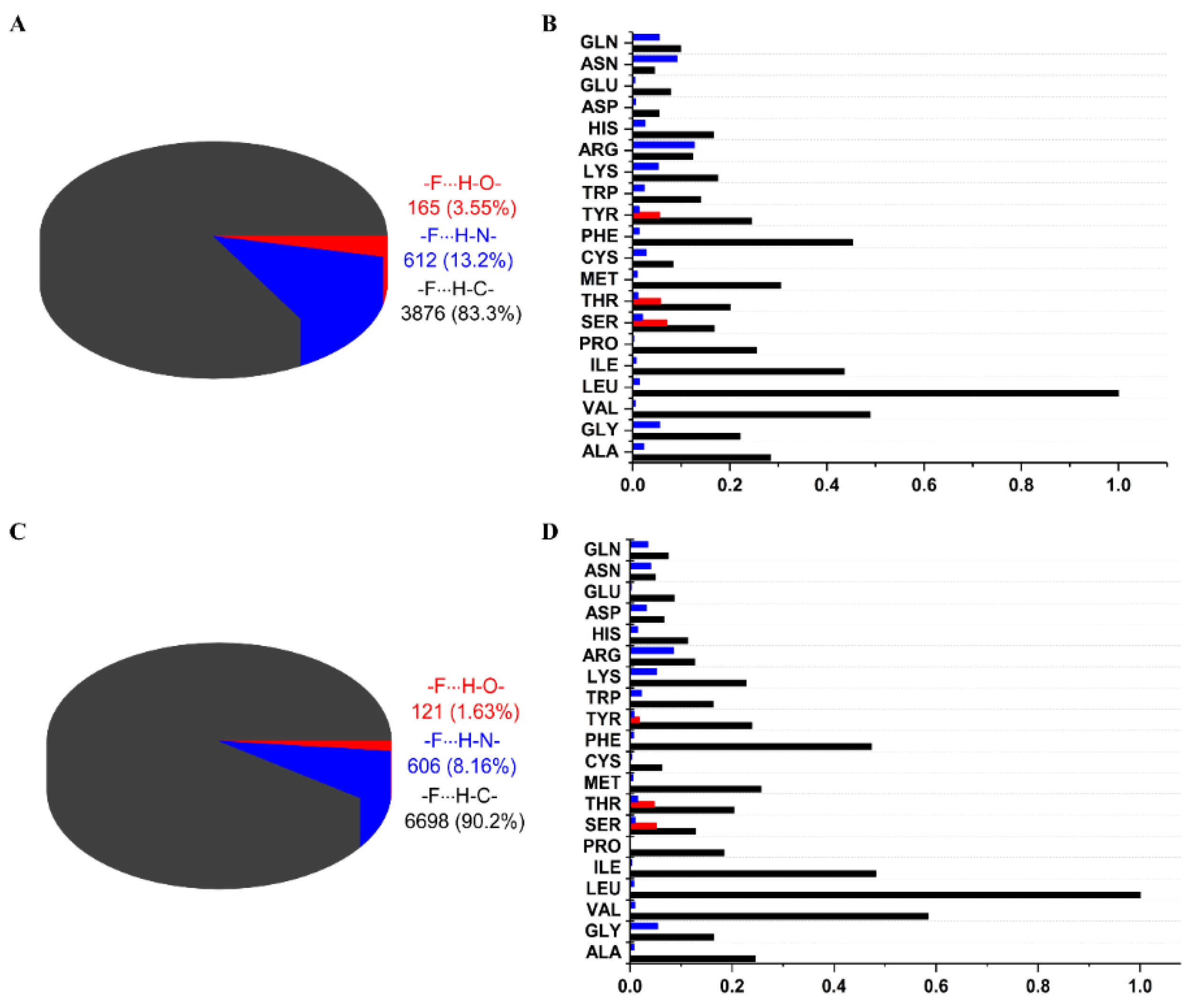
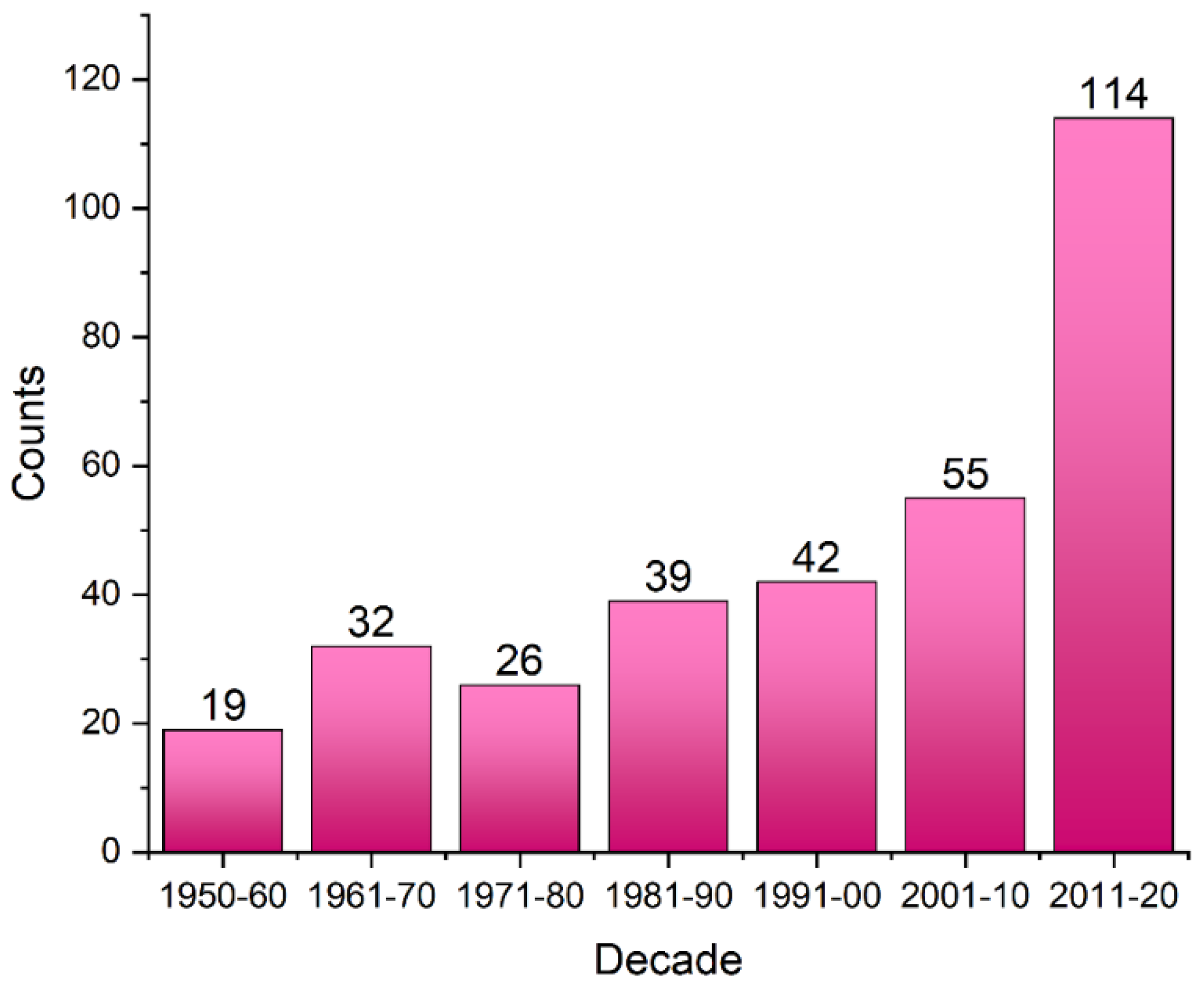
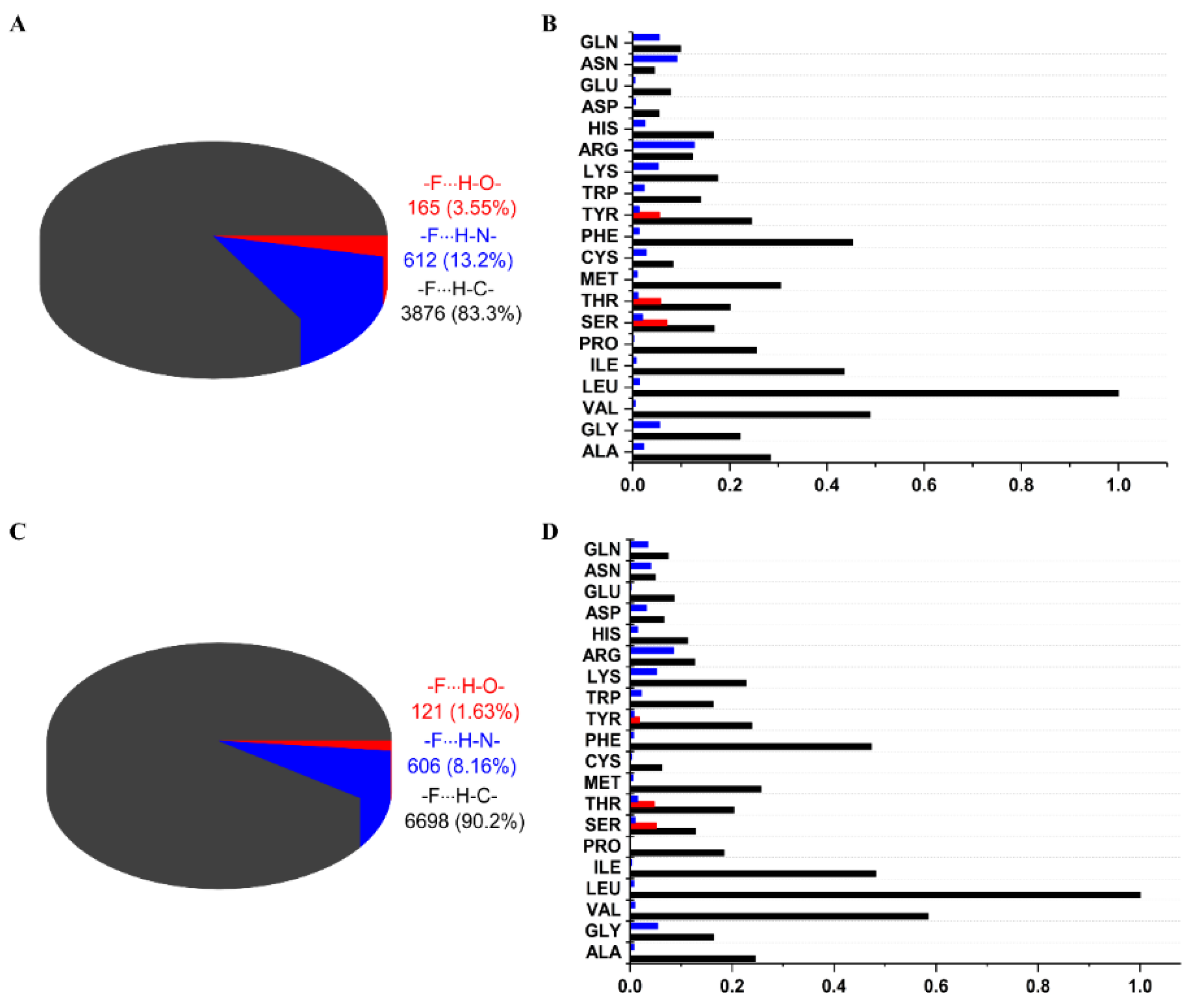
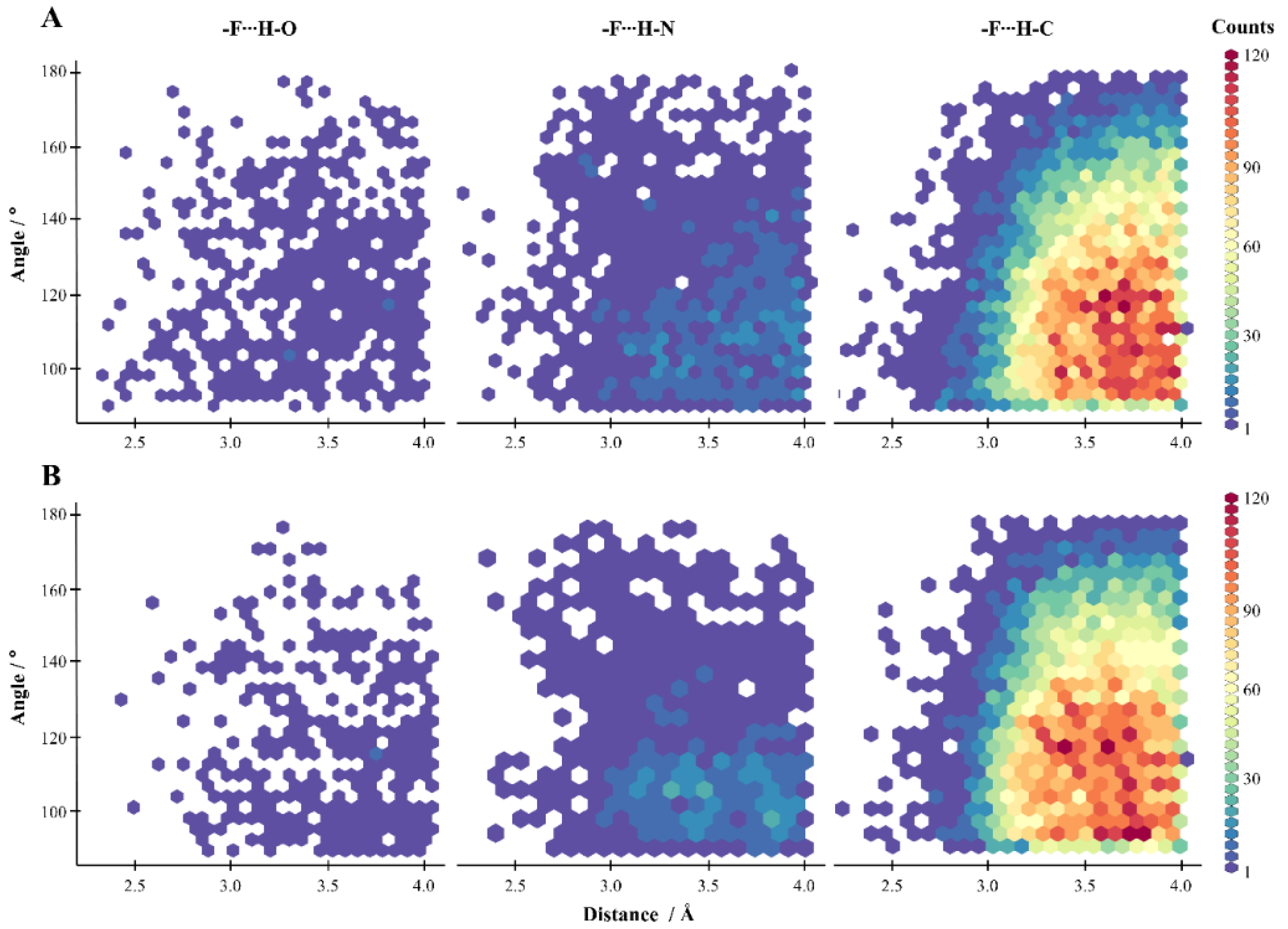
2.2. General Statistics of HBs Containing Fluorine Atoms
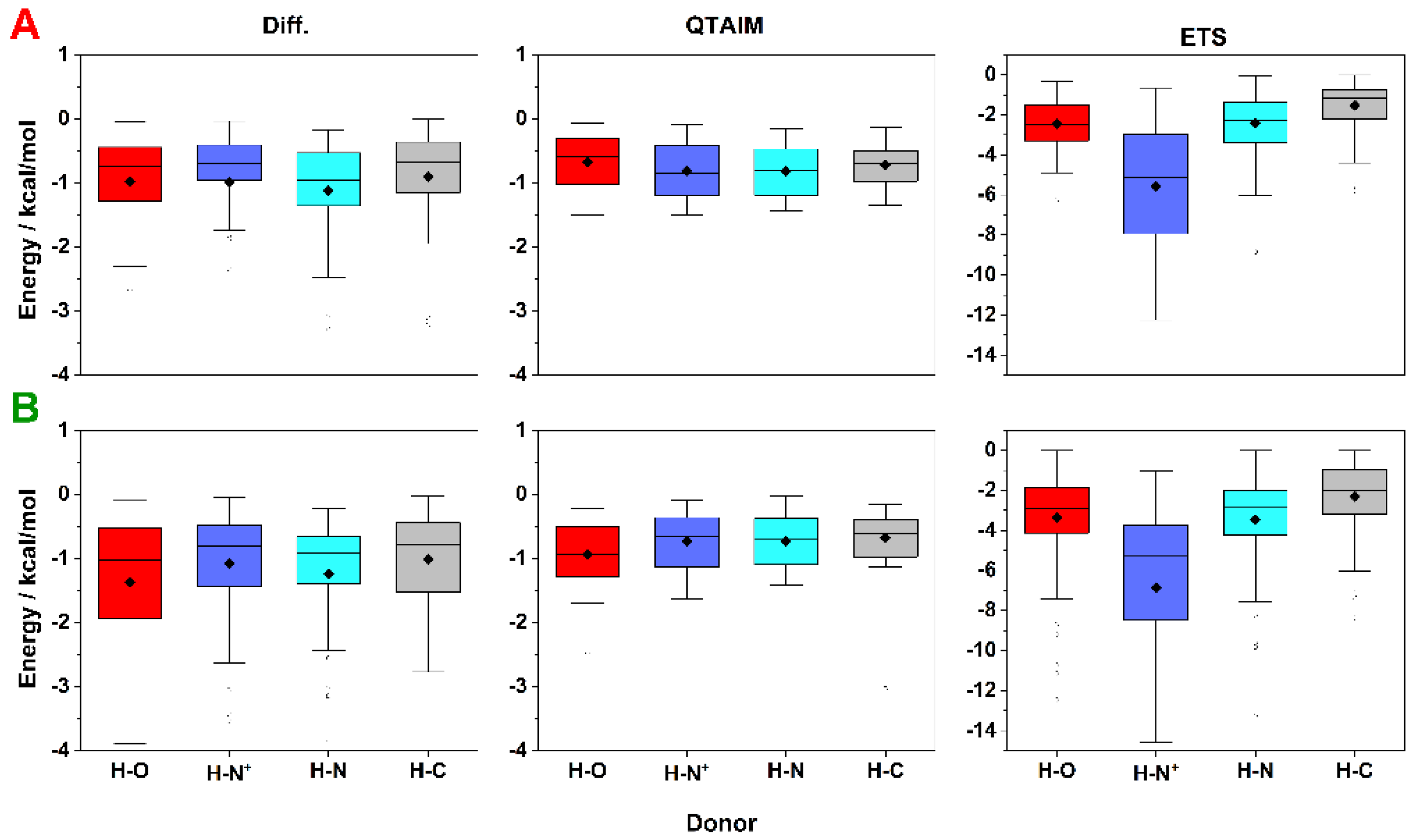
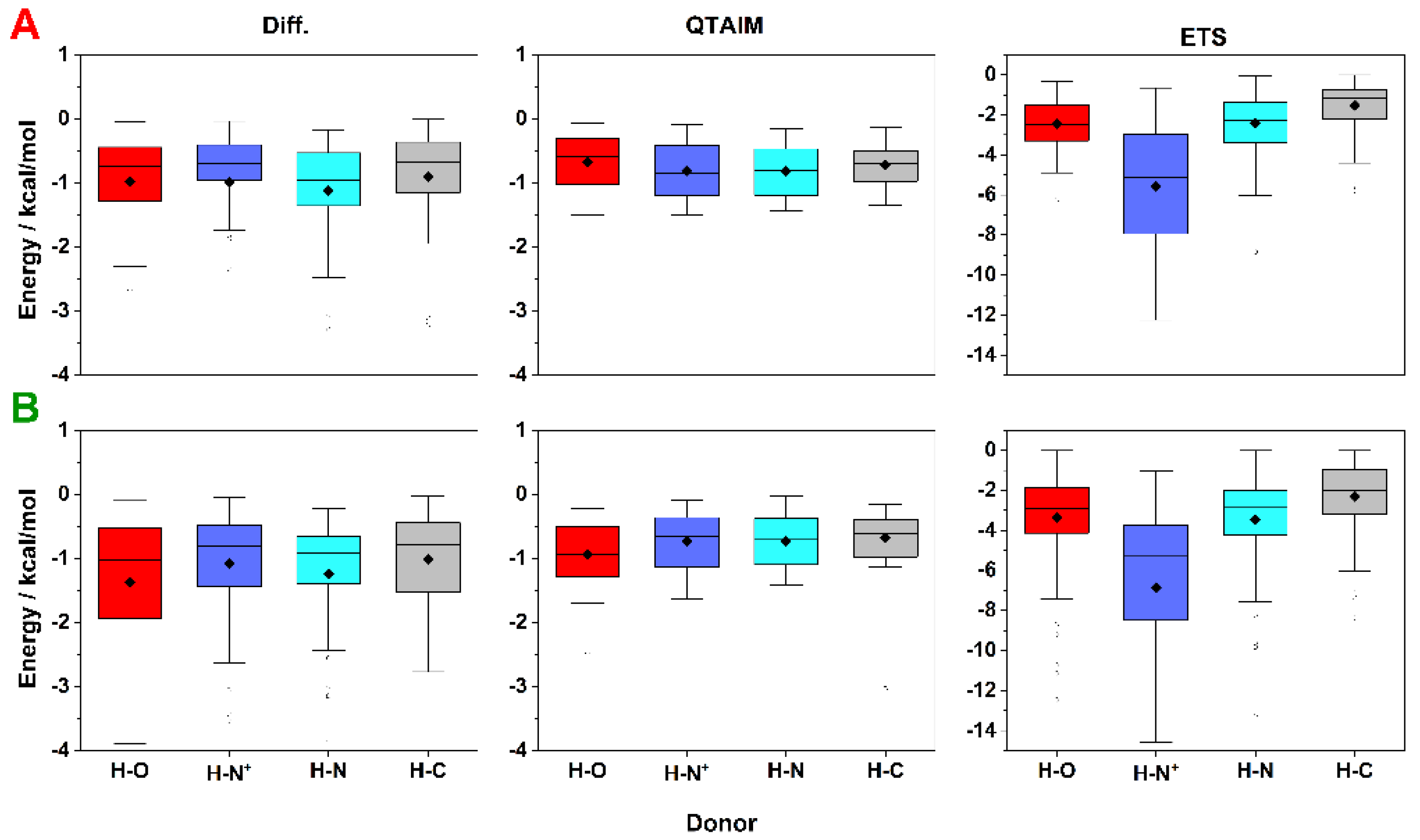
2.3. Energy of HBs with Fluorine
3. Materials and Methods
3.1. PDB Analysis
3.2. Calculation of Interaction Energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Swallow, S. Fluorine in Medicinal Chemistry. In Progress in Medicinal Chemistry; Lawton, G., Witty, D.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 54, pp. 65–133. [Google Scholar]
- Fried, J.; Sabo, E.F. 9α-Fluoro derivatives of cortisone and hydrocortisone. J. Am. Chem. Soc. 1954, 76, 1455–1456. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Tressaud, A. Fluorine, a key element for the 21st century. In Fluorine; Tressaud, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 77–150. [Google Scholar]
- Brown, D.G.; Wobst, H.J. A Decade of FDA-Approved Drugs (2010–2019): Trends and Future Directions. J. Med. Chem. 2021, 64, 2312–2338. [Google Scholar] [CrossRef] [PubMed]
- Ursu, O.; Holmes, J.; Bologa, C.G.; Yang, J.J.; Mathias, S.L.; Stathias, V.; Nguyen, D.T.; Schürer, S.; Oprea, T. DrugCentral 2018: An update. Nucleic Acids Res. 2019, 47, D963–D970. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitencourt-Ferreira, G.; Veit-Acosta, M.; de Azevedo, W.F. Hydrogen bonds in protein-ligand complexes. Methods Mol. Biol. 2019, 2053, 93–107. [Google Scholar] [PubMed]
- Sarkhel, S.; Desiraju, G.R. Hydrogen Bonds in Protein-Ligand Complexes: Strong and Weak Interactions in Molecular Recognition. Proteins Struct. Funct. Genet. 2004, 54, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Hogendorf, A.S.; Hogendorf, A.; Popiołek-Barczyk, K.; Ciechanowska, A.; Mika, J.; Satała, G.; Walczak, M.; Latacz, G.; Handzlik, J.; Kieć-Kononowicz, K.; et al. Fluorinated indole-imidazole conjugates: Selective orally bioavailable 5-HT7 receptor low-basicity agonists, potential neuropathic painkillers. Eur. J. Med. Chem. 2019, 170, 261–275. [Google Scholar] [CrossRef]
- Howard, J.A.K.; Hoy, V.J.; O’Hagan, D.; Smith, G.T. How good is fluorine as a hydrogen bond acceptor? Tetrahedron 1996, 52, 12613–12622. [Google Scholar] [CrossRef]
- Pietruś, W.; Kurczab, R.; Kafel, R.; Machalska, E.; Kalinowska-Tłuścik, J.; Hogendorf, A.; Żylewski, M.; Baranska, M.; Bojarski, A.J. How can fluorine directly and indirectly affect the hydrogen bonding in molecular systems?—A case study for monofluoroanilines. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 252, 119536. [Google Scholar] [CrossRef] [PubMed]
- Pietruś, W.; Kurczab, R.; Kalinowska-Tłuścik, J.; Machalska, E.; Golonka, D.; Barańska, M.; Bojarski, A. Influence of fluorine substitution on nonbonding interactions in selected para-halogeno anilines. ChemPhysChem 2021. [Google Scholar] [CrossRef]
- Zhang, G.; He, W.; Chen, D. On difference of properties between organic fluorine hydrogen bond C-H F-C and conventional hydrogen bond. Mol. Phys. 2014, 112, 1736–1744. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Taylor, R. Organic Fluorine Hardly Ever Accepts Hydrogen Bonds. Chem. A Eur. J. 1997, 3, 89–98. [Google Scholar] [CrossRef]
- Ferreira De Freitas, R.; Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogen bonds from 1H NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef] [PubMed]
- Šolínová, V.; Kašička, V. Determination of acidity constants and ionic mobilities of polyprotic peptide hormones by CZE. Electrophoresis 2013, 34, 2655–2665. [Google Scholar] [CrossRef] [PubMed]
- Graton, J.; Besseau, F.; Brossard, A.M.; Charpentier, E.; Deroche, A.; Le Questel, J.Y. Hydrogen-bond acidity of OH groups in various molecular environments (phenols, alcohols, steroid derivatives, and amino acids structures): Experimental measurements and density functional theory calculations. J. Phys. Chem. A 2013, 117, 13184–13193. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein, and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Team, R.C. Computational Many-Particle Physics. In Lecture Notes in Physics; Fehske, H., Schneider, R., Weiße, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 739. [Google Scholar]
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes; R Package Version 1.1.2; R Package: Vienna, Austria, 2014; Available online: https://CRAN.R-project.org/package=RColorBrewer (accessed on 29 December 2021).
- Lewin-Koh, N. Hexagon Binning: An Overview; R Core Team: Vienna, Austria, 2014. [Google Scholar]
- Bokeh Development Team. Bokeh: Python Library for Interactive Visualization. 2014. Available online: http://www.bokeh.pydata.org (accessed on 29 December 2021).
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis (2nd ed.). Meas. Interdiscip. Res. Perspect. 2019, 17, 160–167. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided. Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Jabłoński, M. A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules 2020, 25, 5512. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functions. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-S.; Li, G.-D.; Mao, S.-P.; Chai, J.-D. Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections. J. Chem. Theory Comput. 2013, 9, 263–272. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Mennucci, B.; Cammi, R.; Tomasi, J. Analytical free energy second derivatives with respect to nuclear coordinates: Complete formulation for electrostatic continuum solvation models. J. Chem. Phys. 1999, 110, 6858–6870. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll; TK Gristmill Software: Overland Park, KS, USA, 2015. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method—I. The transition state method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Michalak, A.; Mitoraj, M.; Ziegler, T. Bond orbitals from chemical valence theory. J. Phys. Chem. A 2008, 112, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.; Michalak, A. Applications of natural orbitals for chemical valence in a description of bonding in conjugated molecules. J. Mol. Model. 2008, 14, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.; Michalak, A. Natural orbitals for chemical valence as descriptors of chemical bonding in transition metal complexes. J. Mol. Model. 2007, 13, 347–355. [Google Scholar] [CrossRef]
- Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ros, P. Self-consistent molecular Hartree-Fock-Slater calculations II. The effect of exchange scaling in some small molecules. Chem. Phys. 1973, 2, 52–59. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ellis, D.E.; Ros, P. Self-consistent molecular Hartree-Fock-Slater calculations I. The computational procedure. Chem. Phys. 1973, 2, 41–51. [Google Scholar] [CrossRef]
- Velde, G.; Baerends, E.J. Numerical integration for polyatomic systems. J. Comput. Phys. 1992, 99, 84–98. [Google Scholar] [CrossRef]
- Etter, M.C. Hydrogen bonds as design elements in organic chemistry. J. Phys. Chem. 1991, 95, 4601–4610. [Google Scholar] [CrossRef]
- Taylor, R. The hydrogen bond between N-H or O-H and organic fluorine: Favourable yes, competitive no. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 474–488. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietruś, W.; Kafel, R.; Bojarski, A.J.; Kurczab, R. Hydrogen Bonds with Fluorine in Ligand–Protein Complexes-the PDB Analysis and Energy Calculations. Molecules 2022, 27, 1005. https://doi.org/10.3390/molecules27031005
Pietruś W, Kafel R, Bojarski AJ, Kurczab R. Hydrogen Bonds with Fluorine in Ligand–Protein Complexes-the PDB Analysis and Energy Calculations. Molecules. 2022; 27(3):1005. https://doi.org/10.3390/molecules27031005
Chicago/Turabian StylePietruś, Wojciech, Rafał Kafel, Andrzej J. Bojarski, and Rafał Kurczab. 2022. "Hydrogen Bonds with Fluorine in Ligand–Protein Complexes-the PDB Analysis and Energy Calculations" Molecules 27, no. 3: 1005. https://doi.org/10.3390/molecules27031005
APA StylePietruś, W., Kafel, R., Bojarski, A. J., & Kurczab, R. (2022). Hydrogen Bonds with Fluorine in Ligand–Protein Complexes-the PDB Analysis and Energy Calculations. Molecules, 27(3), 1005. https://doi.org/10.3390/molecules27031005