
ML216-Induced BLM Helicase Inhibition Sensitizes PCa Cells to the DNA-Crosslinking Agent Cisplatin

 , ,
, ,
Abstract
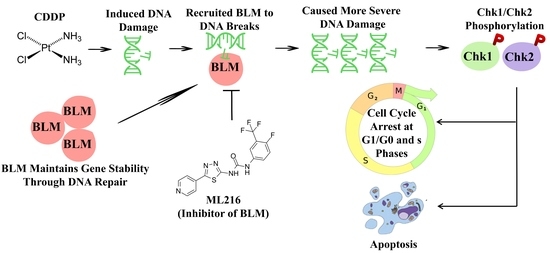
1. Introduction
2. Results
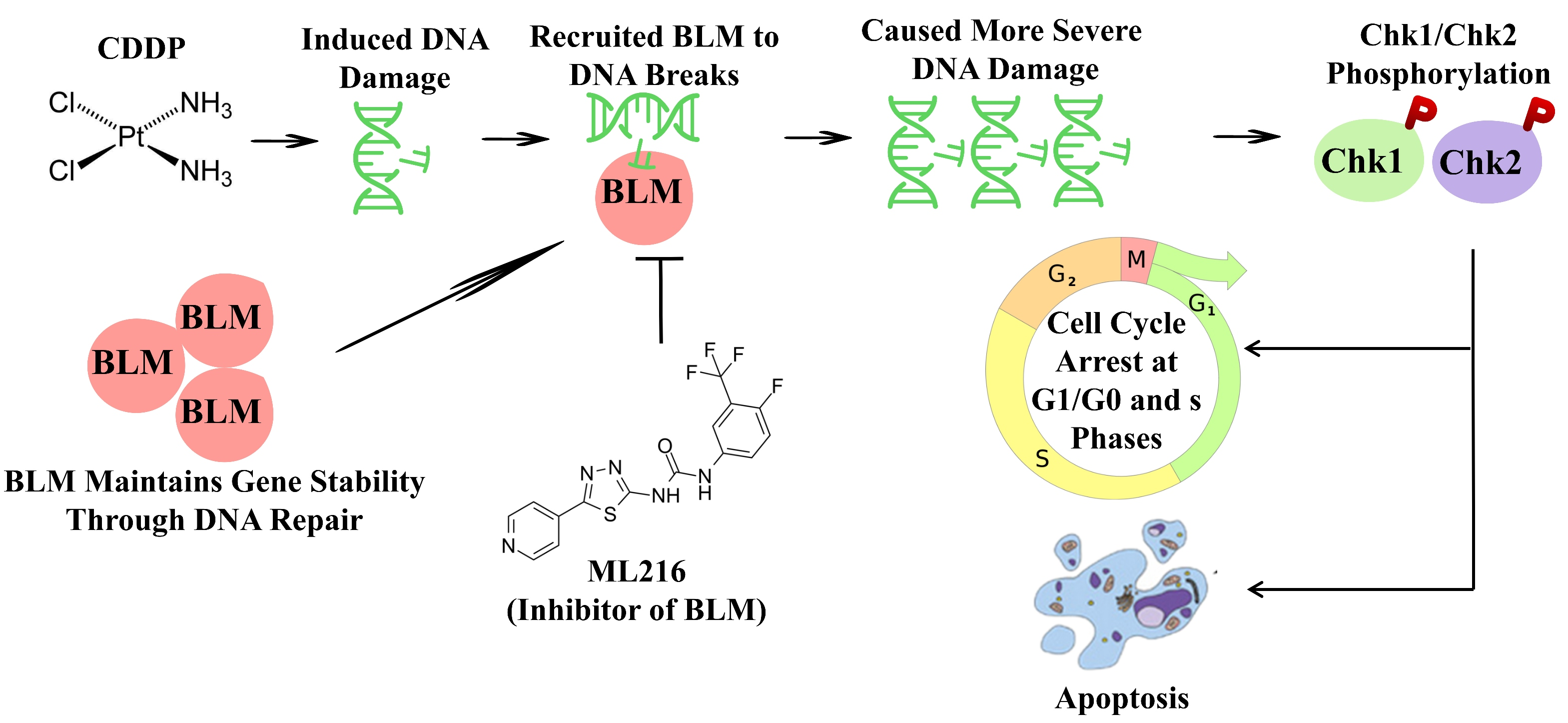
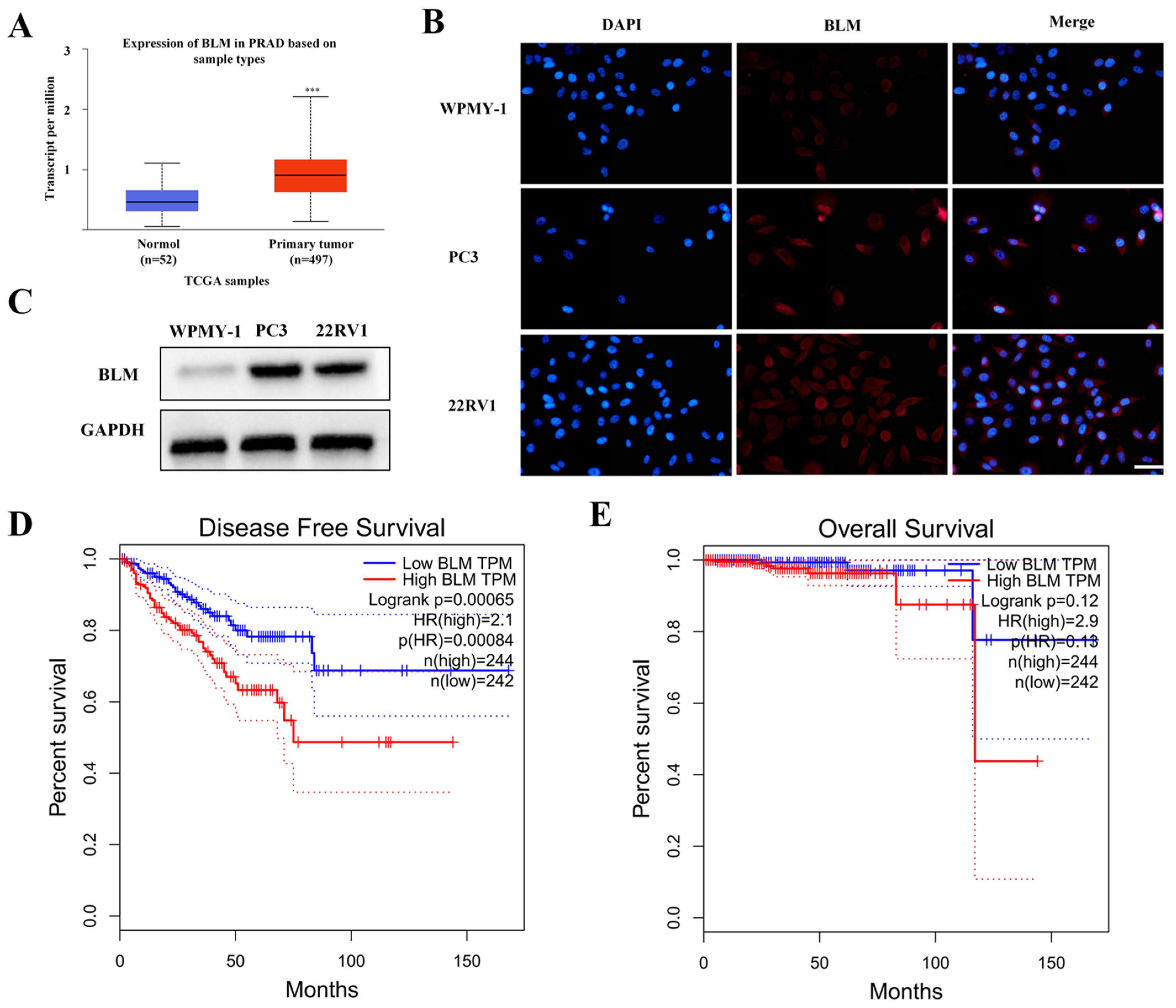
2.1. BLM Could Act as a Therapeutic Drug Target for PCa
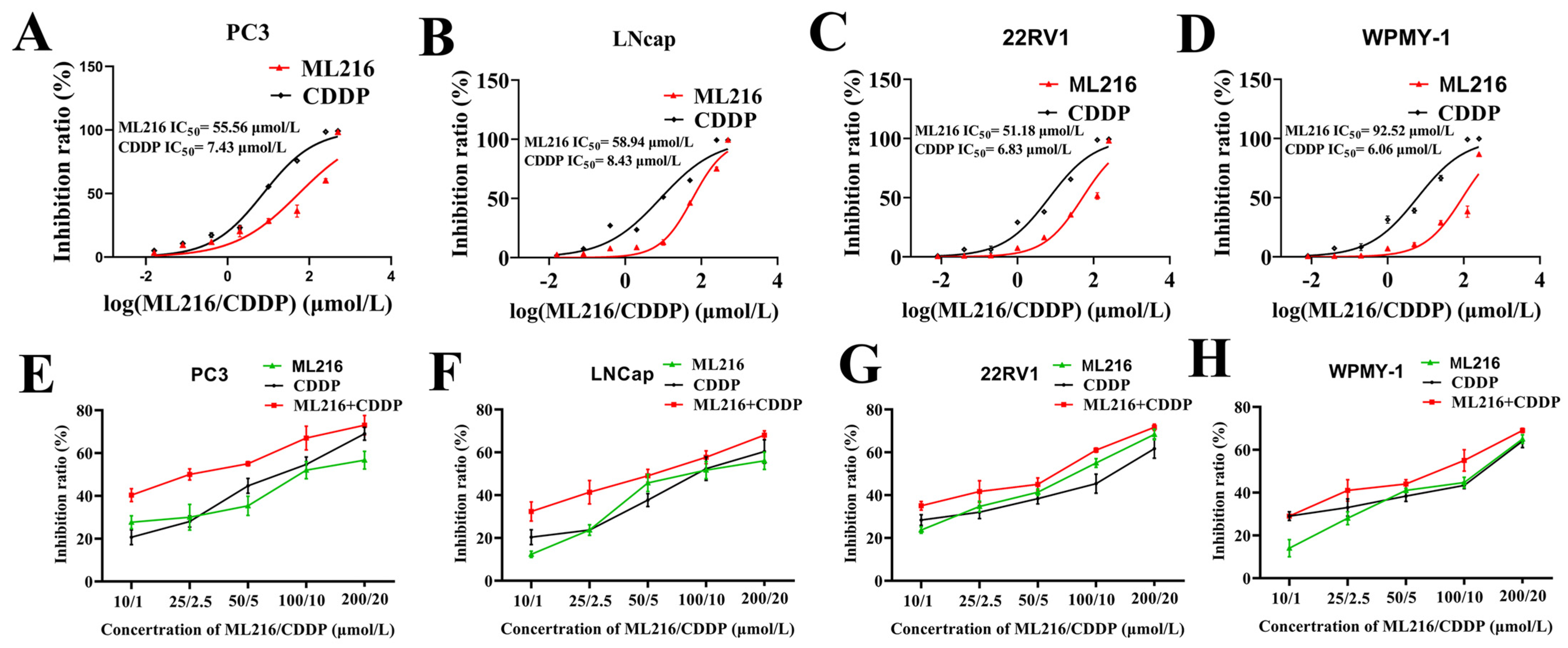
2.2. ML216 Sensitizes CDDP-Induced Cytotoxicity in PCa Cells
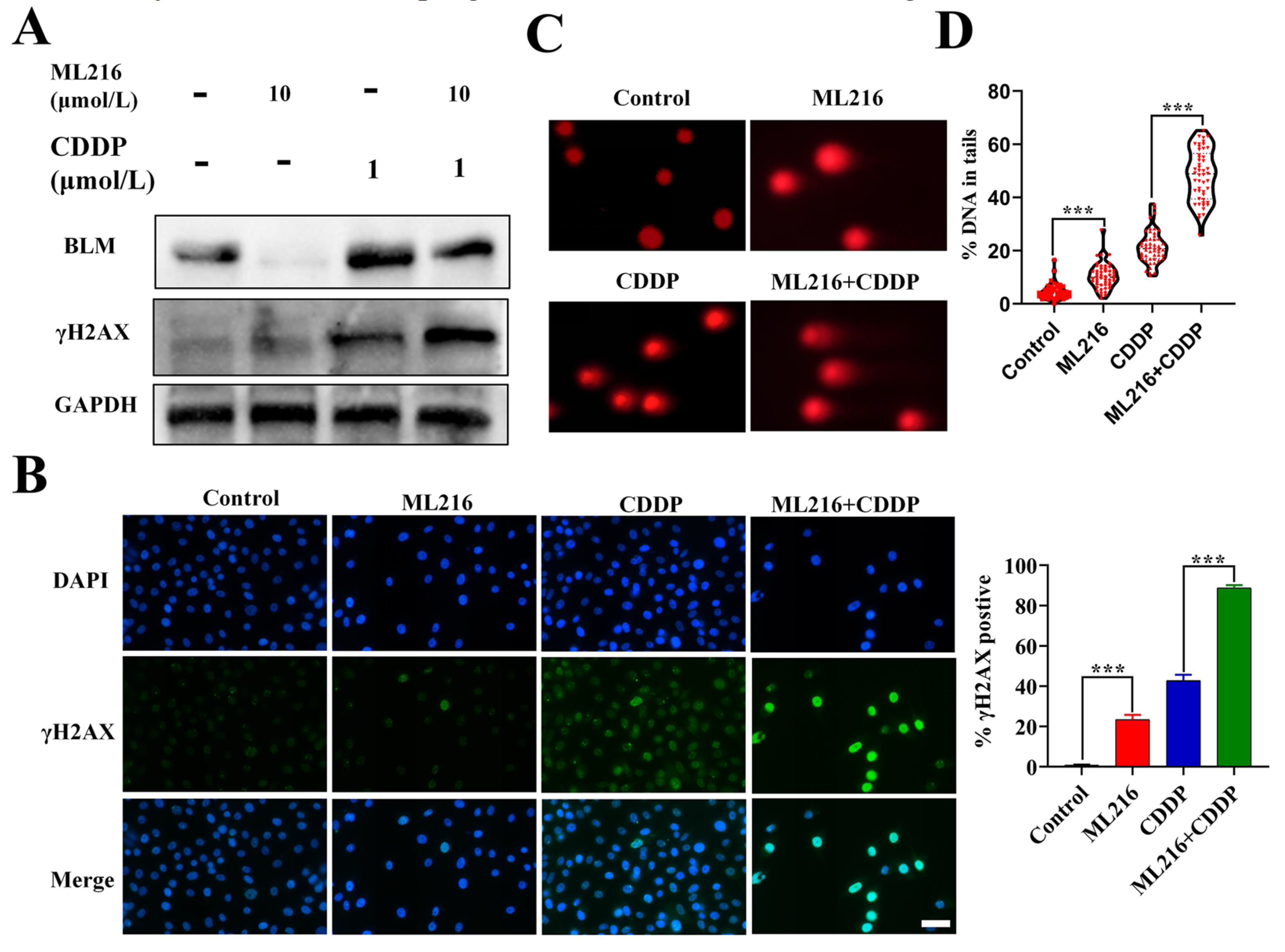
2.3. ML216 Enhances CDDP-Induced DSBs by Inhibiting BLM in PC3 Cells
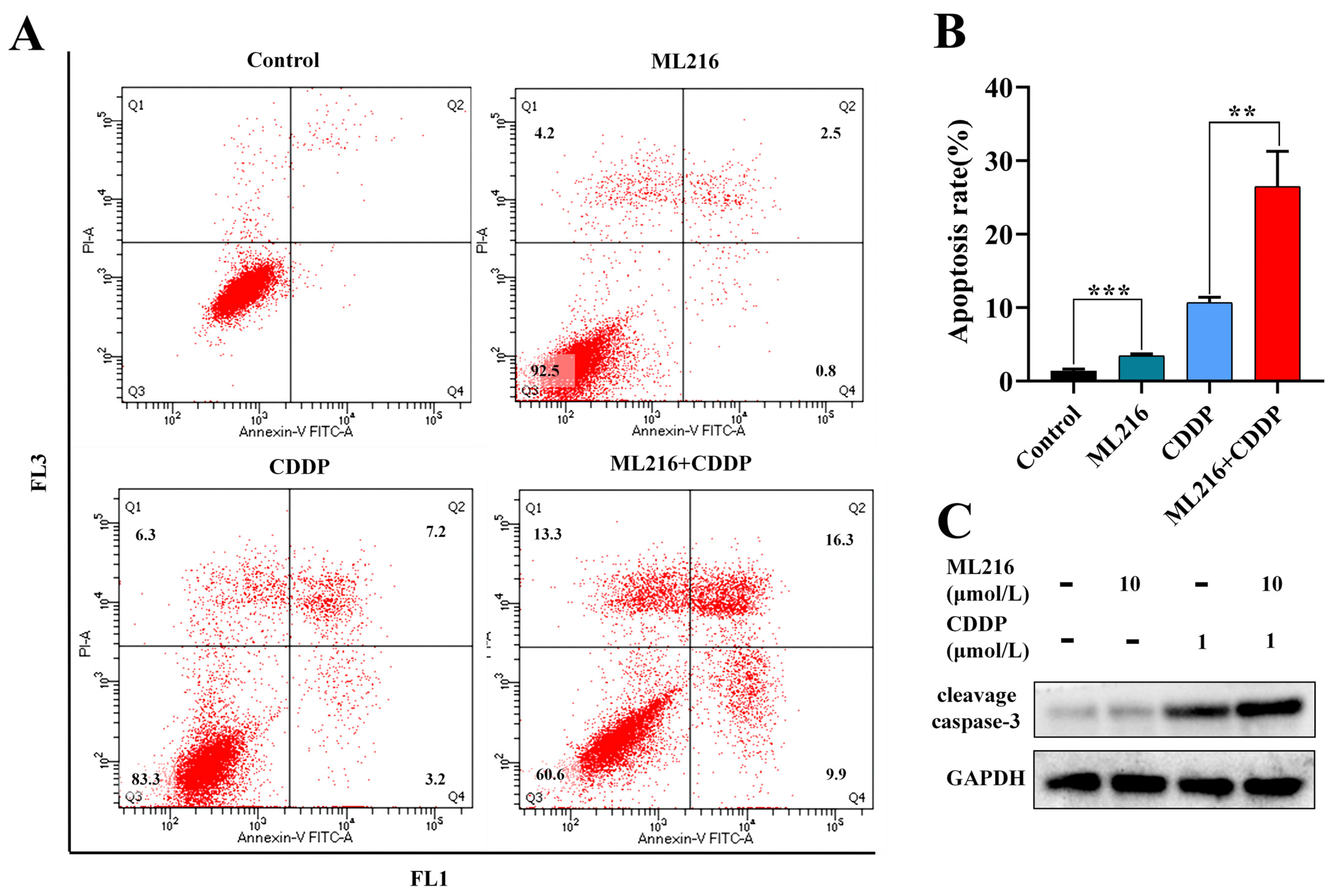
2.4. ML216 Hypersensitizes CDDP-Induced Apoptosis in PC3 Cells
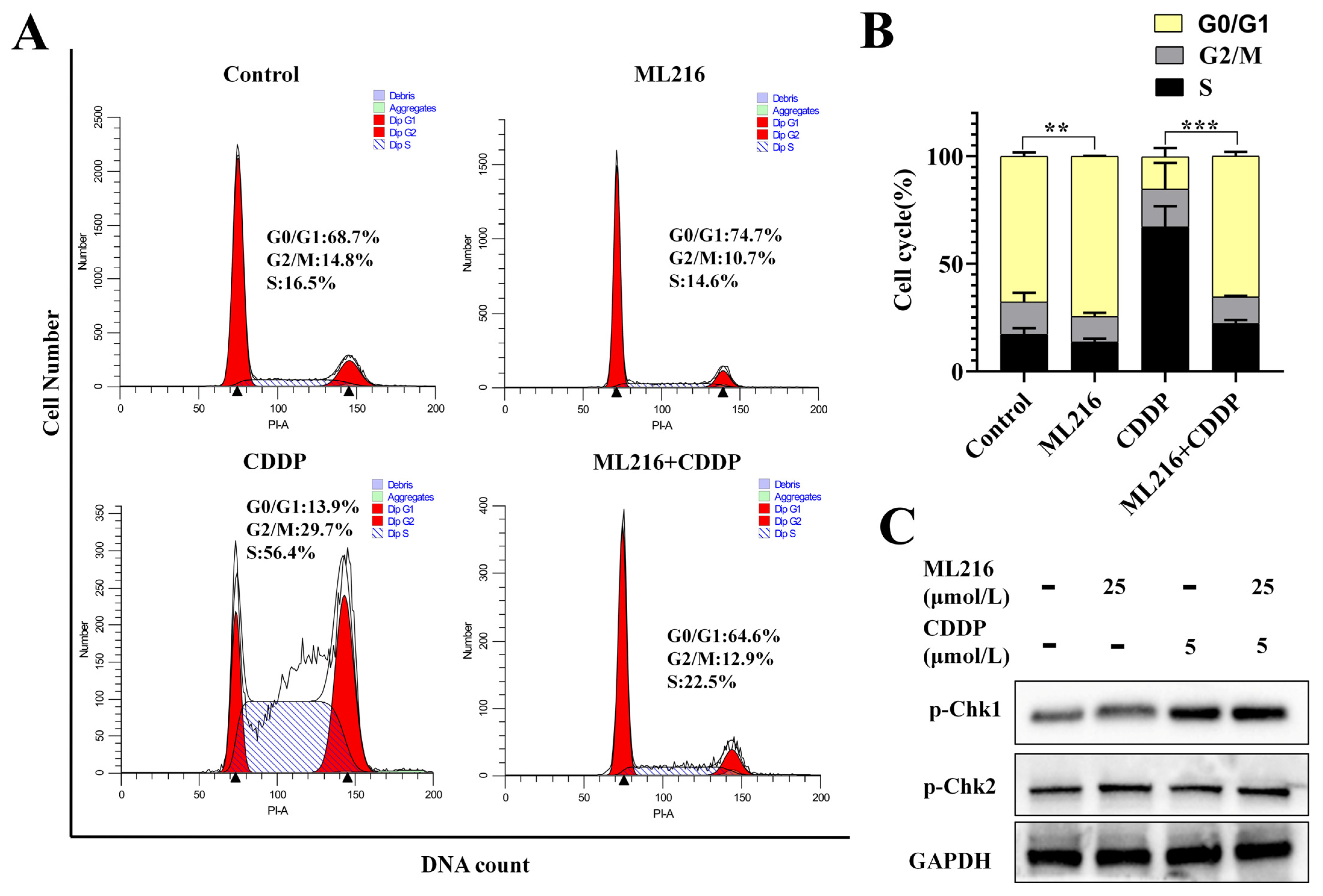
2.5. The Combination of ML216 and CDDP Activates ATM/Chk2 and ATR/Chk1 Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Antibodies and Reagents
4.3. Immunofluorescence (IF) Analysis
4.4. Western Blotting (WB)
4.5. Cell Counting Kit-8 (CCK8) Drug Sensitivity Assessment
4.6. Cell Cycle and Apoptosis Analysis
4.7. Neutral Comet Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Karzai, F.H.; Ning, Y.M.; Adesunloye, B.A.; Huang, X.; Harold, N.; Couvillon, A.; Chun, G.; Cordes, L.; Sissung, T.; et al. Phase II trial of docetaxel, bevacizumab, lenalidomide and prednisone in patients with metastatic castration-resistant prostate cancer. BJU Int. 2016, 118, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Gravis, G.; Boher, J.M.; Chen, Y.H.; Liu, G.; Fizazi, K.; Carducci, M.A.; Oudard, S.; Joly, F.; Jarrard, D.M.; Soulie, M.; et al. Burden of Metastatic Castrate Naive Prostate Cancer Patients, to Identify Men More Likely to Benefit from Early Docetaxel: Further Analyses of CHAARTED and GETUG-AFU15 Studies. Eur. Urol. 2018, 73, 847–855. [Google Scholar] [CrossRef]
- Cheng, H.H.; Pritchard, C.C.; Boyd, T.; Nelson, P.S.; Montgomery, B. Biallelic Inactivation of BRCA2 in Platinum-sensitive Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2016, 69, 992–995. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Spisák, S.; Jia, L.; Cronin, A.M.; Csabai, I.; Ledet, E.; Sartor, A.O.; Rainville, I.; O’Connor, E.P.; Herbert, Z.T.; et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 2017, 123, 3532–3539. [Google Scholar] [CrossRef]
- Schmid, S.; Omlin, A.; Higano, C.; Sweeney, C.; Martinez Chanza, N.; Mehra, N.; Kuppen, M.C.P.; Beltran, H.; Conteduca, V.; Vargas Pivato de Almeida, D.; et al. Activity of Platinum-Based Chemotherapy in Patients With Advanced Prostate Cancer With and Without DNA Repair Gene Aberrations. JAMA Netw. Open. 2020, 3, e2021692. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef]
- Furukawa, N.; Kawaguchic, R.; Kobayashi, H. Use of high-dose cisplatin with aprepitant in an outpatient setting. Eur. J. Cancer Care (Engl.) 2012, 21, 436–441. [Google Scholar] [CrossRef]
- Sakthivel, K.M.; Hariharan, S. Regulatory players of DNA damage repair mechanisms: Role in Cancer Chemoresistance. Biomed. Pharmacother. 2017, 93, 1238–1245. [Google Scholar] [CrossRef]
- Nagel, Z.D.; Kitange, G.J.; Gupta, S.K.; Joughin, B.A.; Chaim, I.A.; Mazzucato, P.; Lauffenburger, D.A.; Sarkaria, J.N.; Samson, L.D. DNA Repair Capacity in Multiple Pathways Predicts Chemoresistance in Glioblastoma Multiforme. Cancer Res. 2017, 77, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.E.; Chung, W.H. Synthetic lethal interaction between oxidative stress response and DNA damage repair in the budding yeast and its application to targeted anticancer therapy. J. Microbiol. 2019, 57, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Conde, M.; Frew, I.J. Therapeutic significance of ARID1A mutation in bladder cancer. Neoplasia 2022, 31, 100814. [Google Scholar] [CrossRef]
- Takeuchi, M.; Tanikawa, M.; Nagasaka, K.; Oda, K.; Kawata, Y.; Oki, S.; Agapiti, C.; Sone, K.; Miyagawa, Y.; Hiraike, H.; et al. Anti-Tumor Effect of Inhibition of DNA Damage Response Proteins, ATM and ATR, in Endometrial Cancer Cells. Cancers 2019, 11, 1913. [Google Scholar] [CrossRef]
- Mazina, O.M.; Rossi, M.J.; Deakyne, J.S.; Huang, F.; Mazin, A.V. Polarity and bypass of DNA heterology during branch migration of Holliday junctions by human RAD54, BLM, and RECQ1 proteins. J. Biol. Chem. 2012, 287, 11820–11832. [Google Scholar] [CrossRef]
- Lansdorp, P.; van Wietmarschen, N. Helicases FANCJ, RTEL1 and BLM Act on Guanine Quadruplex DNA in Vivo. Genes 2019, 10, 870. [Google Scholar] [CrossRef]
- Xue, C.; Daley, J.M.; Xue, X.; Steinfeld, J.; Kwon, Y.; Sung, P.; Greene, E.C. Single-molecule visualization of human BLM helicase as it acts upon double- and single-stranded DNA substrates. Nucleic Acids Res. 2019, 47, 11225–11237. [Google Scholar] [CrossRef]
- Das, T.; Pal, S.; Ganguly, A. Human RecQ helicases in transcription-associated stress management: Bridging the gap between DNA and RNA metabolism. Biol. Chem. 2021, 402, 617–636. [Google Scholar] [CrossRef]
- LaRocque, J.R.; Stark, J.M.; Oh, J.; Bojilova, E.; Yusa, K.; Horie, K.; Takeda, J.; Jasin, M. Interhomolog recombination and loss of heterozygosity in wild-type and Bloom syndrome helicase (BLM)-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11971–11976. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, S. On BLM helicase in recombination-mediated telomere maintenance. Mol. Biol. Rep. 2013, 40, 3049–3064. [Google Scholar] [CrossRef] [PubMed]
- de Renty, C.; Pond, K.W.; Yagle, M.K.; Ellis, N.A. BLM Sumoylation Is Required for Replication Stability and Normal Fork Velocity during DNA Replication. Front. Mol. Biosci. 2022, 9, 875102. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.H.; Dexheimer, T.S.; Rosenthal, A.S.; Chu, W.K.; Singh, D.K.; Mosedale, G.; Bachrati, C.Z.; Schultz, L.; Sakurai, M.; Savitsky, P.; et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem. Biol. 2013, 20, 55–62. [Google Scholar] [CrossRef]
- Wang, C.X.; Zhang, Z.L.; Yin, Q.K.; Tu, J.L.; Wang, J.E.; Xu, Y.H.; Rao, Y.; Ou, T.M.; Huang, S.L.; Li, D.; et al. Design, Synthesis, and Evaluation of New Quinazolinone Derivatives that Inhibit Bloom Syndrome Protein (BLM) Helicase, Trigger DNA Damage at the Telomere Region, and Synergize with PARP Inhibitors. J. Med. Chem. 2020, 63, 9752–9772. [Google Scholar] [CrossRef]
- Yin, Q.K.; Wang, C.X.; Wang, Y.Q.; Guo, Q.L.; Zhang, Z.L.; Ou, T.M.; Huang, S.L.; Li, D.; Wang, H.G.; Tan, J.H.; et al. Discovery of Isaindigotone Derivatives as Novel Bloom’s Syndrome Protein (BLM) Helicase Inhibitors That Disrupt the BLM/DNA Interactions and Regulate the Homologous Recombination Repair. J. Med. Chem. 2019, 62, 3147–3162. [Google Scholar] [CrossRef]
- Zhang, W.M.; Yang, S.; Liu, J.H.; Bao, L.C.; Lu, H.; Li, H.; Pan, W.D.; Jiao, Y.C.; He, Z.X.; Liu, J. Screening antiproliferative drug for breast cancer from bisbenzylisoquinoline alkaloid tetrandrine and fangchinoline derivatives by targeting BLM helicase. BMC Cancer 2019, 19, 1009. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Pragti; Bidyut, K.K.; Suman, M. Target based chemotherapeutic advancement of ruthenium complexes. Coord. Chem. Rev. 2021, 448, 214169. [Google Scholar] [CrossRef]
- Pragti; Kundu, B.K.; Upadhyay, S.N.; Sinha, N.; Ganguly, R.; Grabchev, I.; Pakhira, S.; Mukhopadhyay, S. Pyrene-based fluorescent Ru(II)-arene complexes for significant biological applications: Catalytic potential, DNA/protein binding, two photon cell imaging and in vitro cytotoxicity. Dalton Trans. 2022, 51, 3937–3953. [Google Scholar] [CrossRef]
- Kundu, B.K.; Pragti; Carlton Ranjith, W.A.; Shankar, U.; Kannan, R.R.; Mobin, S.M.; Bandyopadhyay, A.; Mukhopadhyay, S. Cancer-Targeted Chitosan-Biotin-Conjugated Mesoporous Silica Nanoparticles as Carriers of Zinc Complexes to Achieve Enhanced Chemotherapy In Vitro and In Vivo. ACS Appl. Bio Mater. 2022, 5, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Ray, U.; Raghavan, S.C. Inhibitors of DNA double-strand break repair at the crossroads of cancer therapy and genome editing. Biochem. Pharmacol. 2020, 182, 114195. [Google Scholar] [CrossRef] [PubMed]
- Cicconi, A.; Rai, R.; Xiong, X.; Broton, C.; Al-Hiyasat, A.; Hu, C.; Dong, S.; Sun, W.; Garbarino, J.; Bindra, R.S.; et al. Microcephalin 1/BRIT1-TRF2 interaction promotes telomere replication and repair, linking telomere dysfunction to primary microcephaly. Nat. Commun. 2020, 11, 5861. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Wang, Y.; Zhang, C.; Hong, Z.; Han, Z. The synthetic lethality of targeting cell cycle checkpoints and PARPs in cancer treatment. J. Hematol. Oncol. 2022, 15, 147. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Parseghian, C.M.; Parikh, N.U.; Wu, J.Y.; Jiang, Z.Q.; Henderson, L.; Tian, F.; Pastor, B.; Ychou, M.; Raghav, K.; Dasari, A.; et al. Dual Inhibition of EGFR and c-Src by Cetuximab and Dasatinib Combined with FOLFOX Chemotherapy in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 4146–4154. [Google Scholar] [CrossRef]
- Mohammed, S.; Shamseddine, A.A.; Newcomb, B.; Chavez, R.S.; Panzner, T.D.; Lee, A.H.; Canals, D.; Okeoma, C.M.; Clarke, C.J.; Hannun, Y.A. Sublethal doxorubicin promotes migration and invasion of breast cancer cells: Role of Src Family non-receptor tyrosine kinases. Breast Cancer Res. 2021, 23, 76. [Google Scholar] [CrossRef]
- Rosenbaum, J.C.; Bonilla, B.; Hengel, S.R.; Mertz, T.M.; Herken, B.W.; Kazemier, H.G.; Pressimone, C.A.; Ratterman, T.C.; MacNary, E.; De Magis, A.; et al. The Rad51 paralogs facilitate a novel DNA strand specific damage tolerance pathway. Nat. Commun. 2019, 10, 3515. [Google Scholar] [CrossRef]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell. 2017, 65, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.Y.; Xu, H.Q.; Zhao, J.F.; Ruan, Y.; Chen, B. Discovery of a Novel Bloom’s Syndrome Protein (BLM) Inhibitor Suppressing Growth and Metastasis of Prostate Cancer. Int. J. Mol. 2022, 23, 14798. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ML216 (μmol/L) | CDDP (μmol/L) | PC3 | LNCap | 22RV1 | WPMY-1 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Fa | CI | Fa | CI | Fa | CI | Fa | CI | ||
| 200 | 20 | 0.72 | 1.00 | 0.68 | 1.30 | 0.72 | 0.86 | 0.69 | 1.04 |
| 100 | 10 | 0.68 | 0.67 | 0.57 | 1.27 | 0.65 | 0.77 | 0.55 | 1.39 |
| 50 | 5 | 0.55 | 0.81 | 0.49 | 1.00 | 0.45 | 1.69 | 0.44 | 1.51 |
| 25 | 2.5 | 0.51 | 0.53 | 0.41 | 0.79 | 0.46 | 0.79 | 0.41 | 0.95 |
| 10 | 1 | 0.41 | 0.42 | 0.32 | 0.55 | 0.35 | 0.74 | 0.29 | 1.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.-Y.; Zhao, J.-F.; Ruan, Y.; Zhang, W.-M.; Zhang, L.-Q.; Cai, Z.-D.; Xu, H.-Q. ML216-Induced BLM Helicase Inhibition Sensitizes PCa Cells to the DNA-Crosslinking Agent Cisplatin. Molecules 2022, 27, 8790. https://doi.org/10.3390/molecules27248790
Ma X-Y, Zhao J-F, Ruan Y, Zhang W-M, Zhang L-Q, Cai Z-D, Xu H-Q. ML216-Induced BLM Helicase Inhibition Sensitizes PCa Cells to the DNA-Crosslinking Agent Cisplatin. Molecules. 2022; 27(24):8790. https://doi.org/10.3390/molecules27248790
Chicago/Turabian StyleMa, Xiao-Yan, Jia-Fu Zhao, Yong Ruan, Wang-Ming Zhang, Lun-Qing Zhang, Zheng-Dong Cai, and Hou-Qiang Xu. 2022. "ML216-Induced BLM Helicase Inhibition Sensitizes PCa Cells to the DNA-Crosslinking Agent Cisplatin" Molecules 27, no. 24: 8790. https://doi.org/10.3390/molecules27248790
APA StyleMa, X.-Y., Zhao, J.-F., Ruan, Y., Zhang, W.-M., Zhang, L.-Q., Cai, Z.-D., & Xu, H.-Q. (2022). ML216-Induced BLM Helicase Inhibition Sensitizes PCa Cells to the DNA-Crosslinking Agent Cisplatin. Molecules, 27(24), 8790. https://doi.org/10.3390/molecules27248790