
Role of STAT3 and NRF2 in Tumors: Potential Targets for Antitumor Therapy

 ,
, 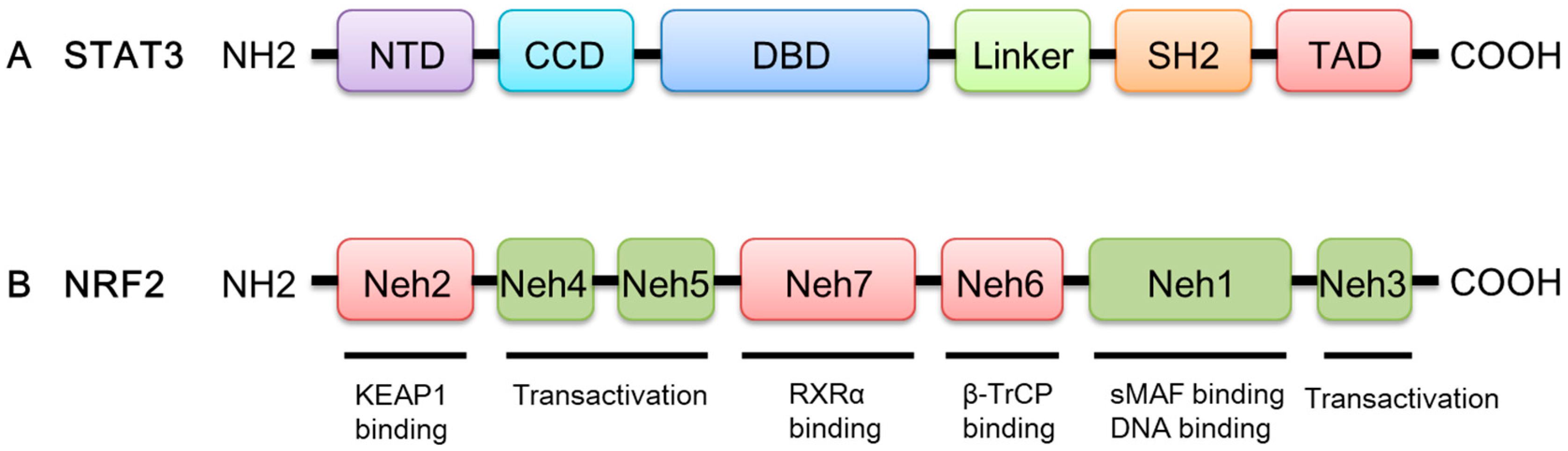{kind=link}
{kind=link}
Abstract
1. Introduction
2. Role of STAT3 Signaling
2.1. STAT3 Signal Transduction Cascade
2.2. STAT3 in Tumor Cells
2.2.1. Functions of STAT3 in Cell Proliferation and Survival in Tumors
2.2.2. Contribution of STAT3 to Tumor Angiogenesis
2.2.3. STAT3 in Immune System Evasion
2.2.4. Function of STAT3 in Cancer Stem Cells
2.3. Dual Roles of STAT3 in Cancer
3. Intricacies of NRF2 Regulation in the Tumor Microenvironment
3.1. NRF2 Signaling Pathway
3.1.1. Canonical Activation of NRF2
3.1.2. Non-Canonical Activation of NRF2
3.2. Dual Roles of NRF2 in Tumor
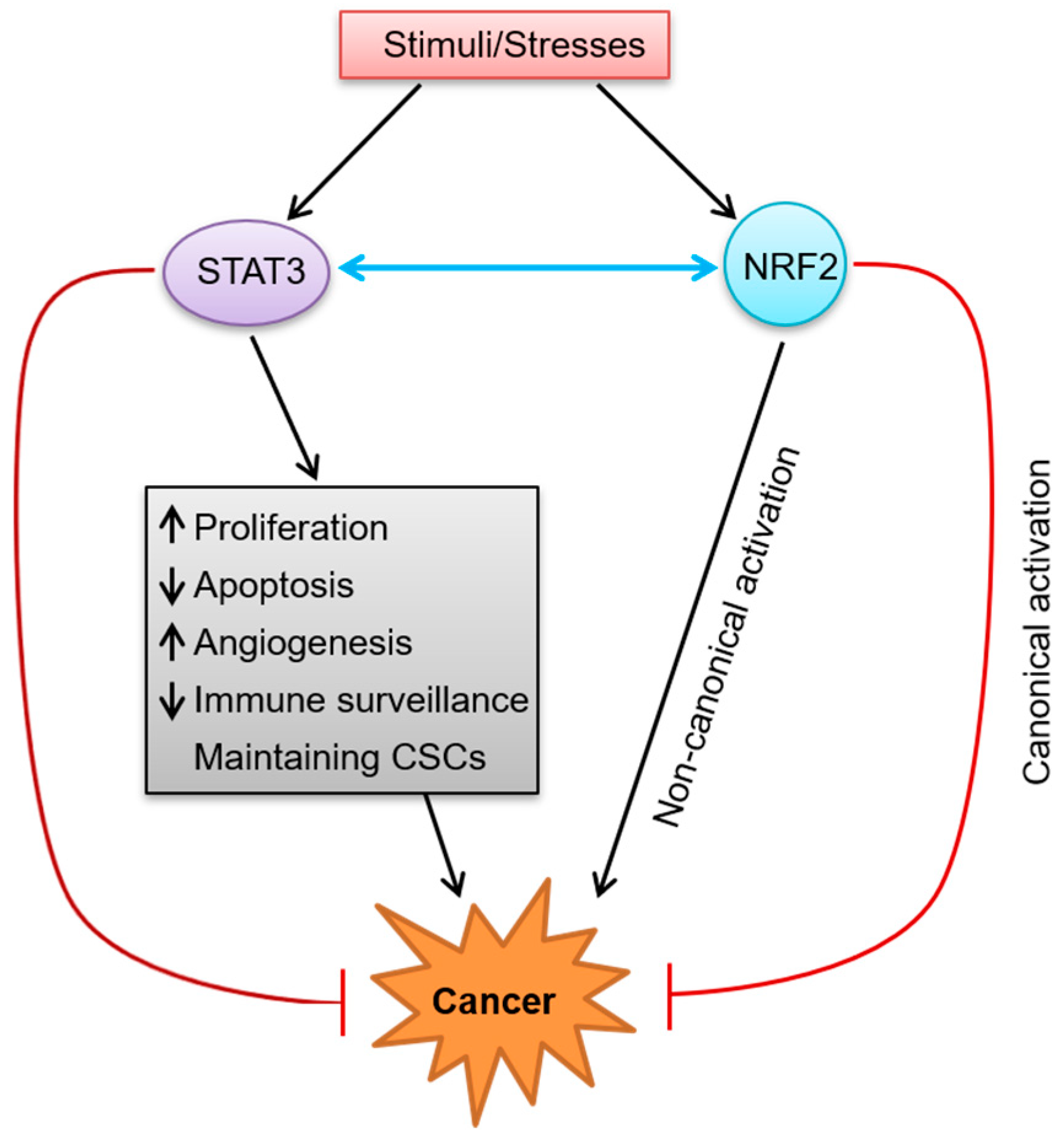
4. Crosstalk between the STAT3 and NRF2 Signaling Pathways in the Tumor Microenvironment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, C.P.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Forbes, L.R.; Milner, J.; Haddad, E. Signal transducer and activator of transcription 3: A year in review. Curr. Opin. Hematol. 2016, 23, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef]
- Sonnenblick, A.; Agbor-Tarh, D.; de Azambuja, E.; Hultsch, S.; Izquierdo, M.; Liu, M.; Pruneri, G.; Harbeck, N.; Piccart, M.; Moreno-Aspita, A.; et al. STAT3 activation in HER2-positive breast cancers: Analysis of data from a large prospective trial. Int. J. Cancer 2021, 148, 1529–1535. [Google Scholar] [CrossRef]
- Yuan, K.; Ye, J.; Liu, Z.; Ren, Y.; He, W.; Xu, J.; He, Y.; Yuan, Y. Complement C3 overexpression activates JAK2/STAT3 pathway and correlates with gastric cancer progression. J. Exp. Clin. Cancer Res. 2020, 39, 9. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, G.; Fan, C.; Xu, J.; Jiang, S.; Yan, X.; Di, S.; Ma, Z.; Hu, W.; Yang, Y. The emerging role of signal transducer and activator of transcription 3 in cerebral ischemic and hemorrhagic stroke. Prog. Neurobiol. 2016, 137, 1–16. [Google Scholar] [CrossRef]
- Xia, Y.; Ling, X.; Hu, G.; Zhu, Q.; Zhang, J.; Li, Q.; Zhao, B.; Wang, Y.; Deng, Z. Small extracellular vesicles secreted by human iPSC-derived MSC enhance angiogenesis through inhibiting STAT3-dependent autophagy in ischemic stroke. Stem. Cell Res. Ther. 2020, 11, 313. [Google Scholar] [CrossRef]
- Cao, X.; Li, B.; Han, X.; Zhang, X.; Dang, M.; Wang, H.; Du, F.; Zeng, X.; Guo, C. Soluble receptor for advanced glycation end-products promotes angiogenesis through activation of STAT3 in myocardial ischemia/reperfusion injury. Apoptosis 2020, 25, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, W.; Lee, L.; Hong, M.; Lee, M.; Chou, G.; Yu, L.; Sui, Y.; Chou, B. Down-regulated microRNA-195-5p and up-regulated CXCR4 attenuates the heart function injury of heart failure mice via inactivating JAK/STAT pathway. Int. Immunopharmacol. 2020, 82, 106225. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Lv, J.; Han, M.; Yang, Z.; Li, T.; Jiang, S.; Yang, Y. STAT3: The art of multi-tasking of metabolic and immune functions in obesity. Prog. Lipid. Res. 2018, 70, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Choi, J.Y.; Li, W.L.; Kouri, R.E.; Yu, J.; Kao, F.T.; Ruano, G. Assignment of the acute phase response factor (APRF) gene to 17q21 by microdissection clone sequencing and fluorescence in situ hybridization of a P1 clone. Genomics 1996, 37, 264–265. [Google Scholar] [CrossRef]
- Hu, T.; Yeh, J.E.; Pinello, L.; Jacob, J.; Chakravarthy, S.; Yuan, G.C.; Chopra, R.; Frank, D.A. Impact of the N-Terminal Domain of STAT3 in STAT3-Dependent Transcriptional Activity. Mol. Cell. Biol. 2015, 35, 3284–3300. [Google Scholar] [CrossRef]
- Chai, E.Z.; Shanmugam, M.K.; Arfuso, F.; Dharmarajan, A.; Wang, C.; Kumar, A.P.; Samy, R.P.; Lim, L.H.; Wang, L.; Goh, B.C.; et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol. Ther. 2016, 162, 86–97. [Google Scholar] [CrossRef]
- Zouein, F.A.; Altara, R.; Chen, Q.; Lesnefsky, E.J.; Kurdi, M.; Booz, G.W. Pivotal Importance of STAT3 in Protecting the Heart from Acute and Chronic Stress: New Advancement and Unresolved Issues. Front. Cardiovasc. Med. 2015, 2, 36. [Google Scholar] [CrossRef]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell. Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Reisman, S.A.; Csanaky, I.L.; Aleksunes, L.M.; Klaassen, C.D. Altered disposition of acetaminophen in Nrf2-null and Keap1-knockdown mice. Toxicol. Sci. 2009, 109, 31–40. [Google Scholar] [CrossRef]
- Iizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A.; et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Sivinski, J.; Zhang, D.D.; Chapman, E. Targeting NRF2 to treat cancer. Semin. Cancer Biol. 2021, 76, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Tajmohammadi, I.; Mohammadian, J.; Sabzichi, M.; Mahmuodi, S.; Ramezani, M.; Aghajani, M.; Ramezani, F. Identification of Nrf2/STAT3 axis in induction of apoptosis through sub-G 1 cell cycle arrest mechanism in HT-29 colon cancer cells. J. Cell. Biochem. 2019, 120, 14035–14043. [Google Scholar] [CrossRef]
- Wu, Y.S.; Chung, I.; Wong, W.F.; Masamune, A.; Sim, M.S.; Looi, C.Y. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 296–306. [Google Scholar] [CrossRef]
- Dai, X.; Ahn, K.S.; Kim, C.; Siveen, K.S.; Ong, T.H.; Shanmugam, M.K.; Li, F.; Shi, J.; Kumar, A.P.; Wang, L.Z.; et al. Ascochlorin, an isoprenoid antibiotic inhibits growth and invasion of hepatocellular carcinoma by targeting STAT3 signaling cascade through the induction of PIAS3. Mol. Oncol. 2015, 9, 818–833. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Sethi, G.; Ahn, K.S. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 2015, 6, 6386–6405. [Google Scholar] [CrossRef]
- Mali, S.B. Review of STAT3 (Signal Transducers and Activators of Transcription) in head and neck cancer. Oral Oncol. 2015, 51, 565–569. [Google Scholar] [CrossRef]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef]
- Sgrignani, J.; Garofalo, M.; Matkovic, M.; Merulla, J.; Catapano, C.V.; Cavalli, A. Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development. Int. J. Mol. Sci. 2018, 19, 1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, B.; Yao, Y.Y.; Zhong, L.X.; Chen, X.Y.; Kong, Q.Y.; Wu, M.L.; Li, C.; Li, H.; Liu, J. PIAS3, SHP2 and SOCS3 Expression patterns in Cervical Cancers: Relevance with activation and resveratrol-caused inactivation of STAT3 signaling. Gynecol. Oncol. 2015, 139, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Ko, J.H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.W.; Lee, J.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef]
- Baek, S.H.; Lee, J.H.; Ko, J.H.; Lee, H.; Nam, D.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Lee, J.; Kim, S.H.; et al. Ginkgetin Blocks Constitutive STAT3 Activation and Induces Apoptosis through Induction of SHP-1 and PTEN Tyrosine Phosphatases. Phytother. Res. 2016, 30, 567–576. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Sun, S.; Steinberg, B.M. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J. Gen. Virol. 2002, 83, 1651–1658. [Google Scholar] [CrossRef]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia; myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.H.; Ou-yang, J.; Xing, Y.; Li, D.Y.; Dong, X.Y.; Liu, R.E.; Xu, R.X. Paeoniflorin inhibits human glioma cells via STAT3 degradation by the ubiquitin-proteasome pathway. Drug Des. Dev. Ther. 2015, 9, 5611–5622. [Google Scholar] [PubMed]
- Hu, F.; Li, G.; Huang, C.; Hou, Z.; Yang, X.; Luo, X.; Feng, Y.; Wang, G.; Hu, J.; Cao, Z. The autophagy-independent role of BECN1 in colorectal cancer metastasis through regulating STAT3 signaling pathway activation. Cell Death Dis. 2020, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Kadye, R.; Stoffels, M.; Fanucci, S.; Mbanxa, S.; Prinsloo, E. A STAT3 of Addiction: Adipose Tissue, Adipocytokine Signalling and STAT3 as Mediators of Metabolic Remodelling in the Tumour Microenvironment. Cells 2020, 9, 1043. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Tang, Q.; Zhang, C.; Wu, J.; Gu, C.; Wu, Z.; Li, X. IL-26 promotes the proliferation and survival of human gastric cancer cells by regulating the balance of STAT1 and STAT3 activation. PLoS ONE 2013, 8, e63588. [Google Scholar] [CrossRef]
- Kanai, M.; Konda, Y.; Nakajima, T.; Izumi, Y.; Kanda, N.; Nanakin, A.; Kubohara, Y.; Chiba, T. Differentiation-inducing factor-1 (DIF-1) inhibits STAT3 activity involved in gastric cancer cell proliferation via MEK-ERK-dependent pathway. Oncogene 2003, 22, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, H.; Guan, L.; Lai, C.; Yu, W.; Lai, M. LL1, a novel and highly selective STAT3 inhibitor, displays anti-colorectal cancer activities in vitro and in vivo. Br. J. Pharmacol. 2020, 177, 298–313. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef]
- Grunstein, J.; Roberts, W.G.; Mathieu-Costello, O.; Hanahan, D.; Johnson, R.S. Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Res. 1999, 59, 1592–1598. [Google Scholar]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.K.; Park, H.J.; Kim, N.H.; Park, S.J.; Park, I.Y.; Kim, I.S. Hypoxia-inducible factor-1alpha enhances haptoglobin gene expression by improving binding of STAT3 to the promoter. J. Biol. Chem. 2011, 286, 8857–8865. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Yu, M.O.; Park, K.J.; Chi, S.G.; Park, D.H.; Chung, Y.G. Activated STAT3 regulates hypoxia-induced angiogenesis and cell migration in human glioblastoma. Neurosurgery 2010, 67, 1386–1395, discussion 1395. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Matsukawa, A.; Kudo, S.; Maeda, T.; Numata, K.; Watanabe, H.; Takeda, K.; Akira, S.; Ito, T. Stat3 in resident macrophages as a repressor protein of inflammatory response. J. Immunol. 2005, 175, 3354–3359. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Saudemont, A.; Jouy, N.; Hetuin, D.; Quesnel, B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood 2005, 105, 2428–2435. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.H.; Ivashkiv, L.B. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 2006, 281, 14111–14118. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef]
- Korkaya, H.; Liu, S.; Wicha, M.S. Regulation of cancer stem cells by cytokine networks: Attacking cancer’s inflammatory roots. Clin. Cancer Res. 2011, 17, 6125–6129. [Google Scholar] [CrossRef]
- Rosen, J.M.; Jordan, C.T. The increasing complexity of the cancer stem cell paradigm. Science 2009, 324, 1670–1673. [Google Scholar] [CrossRef]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003, 113, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I.; Colby, D.; Robertson, M.; Nichols, J.; Lee, S.; Tweedie, S.; Smith, A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 2003, 113, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.P.; Kukekov, V.G.; Reith, J.D.; Tchigrinova, O.; Suslov, O.N.; Scott, E.W.; Ghivizzani, S.C.; Ignatova, T.N.; Steindler, D.A. Stem-like cells in bone sarcomas: Implications for tumorigenesis. Neoplasia 2005, 7, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I. The molecular basis of pluripotency in mouse embryonic stem cells. Cloning Stem Cells 2004, 6, 386–391. [Google Scholar] [CrossRef]
- Vazquez-Santillan, K.; Melendez-Zajgla, J.; Jimenez-Hernandez, L.; Martínez-Ruiz, G.; Maldonado, V. NF-kappaB signaling in cancer stem cells: A promising therapeutic target? Cell. Oncol. 2015, 38, 327–339. [Google Scholar] [CrossRef]
- Wang, S.S.; Jiang, J.; Liang, X.H.; Tang, Y.L. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther. 2015, 8, 2973–2980. [Google Scholar]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef]
- Won, C.; Kim, B.H.; Yi, E.H.; Choi, K.J.; Kim, E.K.; Jeong, J.M.; Lee, J.H.; Jang, J.J.; Yoon, J.H.; Jeong, W.I.; et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015, 62, 1160–1173. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Zhao, C.; Li, H.; Lin, H.J.; Yang, S.; Lin, J.; Liang, G. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol. Sci. 2016, 37, 47–61. [Google Scholar] [CrossRef]
- Villalva, C.; Martin-Lannerée, S.; Cortes, U.; Dkhissi, F.; Wager, M.; Le Corf, A.; Tourani, J.M.; Dusanter-Fourt, I.; Turhan, A.G.; Karayan-Tapon, L. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: A potential for targeted therapy? Int. J. Cancer 2011, 128, 826–838. [Google Scholar] [CrossRef] [PubMed]
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. BBI608 inhibits cancer stemness and reverses cisplatin resistance in NSCLC. Cancer Lett. 2018, 428, 117–126. [Google Scholar] [CrossRef] [PubMed]
- de la Iglesia, N.; Puram, S.V.; Bonni, A. STAT3 regulation of glioblastoma pathogenesis. Curr. Mol. Med. 2009, 9, 580–590. [Google Scholar] [CrossRef] [PubMed]
- de la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, J.C.; Lee, S.E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Grabner, B.; Schramek, D.; Mueller, K.M.; Moll, H.P.; Svinka, J.; Hoffmann, T.; Bauer, E.; Blaas, L.; Hruschka, N.; Zboray, K.; et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat. Commun. 2015, 6, 6285. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef]
- Eggler, A.L.; Luo, Y.; van Breemen, R.B.; Mesecar, A.D. Identification of the highly reactive cysteine 151 in the chemopreventive agent-sensor Keap1 protein is method-dependent. Chem. Res. Toxicol. 2007, 20, 1878–1884. [Google Scholar] [CrossRef]
- Luo, Y.; Eggler, A.L.; Liu, D.; Liu, G.; Mesecar, A.D.; van Breemen, R.B. Sites of alkylation of human Keap1 by natural chemoprevention agents. J. Am. Soc. Mass Spectrom. 2007, 18, 2226–2232. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, S.; Chan, J.Y.; Zhang, D.D. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell. Biol. 2007, 27, 6334–6349. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Su, Z.Y.; Shu, L.; Khor, T.O.; Lee, J.H.; Fuentes, F.; Kong, A.N. A perspective on dietary phytochemicals and cancer chemoprevention: Oxidative stress, nrf2, and epigenomics. Top. Curr. Chem. 2013, 329, 133–162. [Google Scholar]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Slocum, S.L.; Kensler, T.W. Nrf2: Control of sensitivity to carcinogens. Arch. Toxicol. 2011, 85, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Zheng, Y.; Tao, S.; Wang, H.; Whitman, S.A.; White, E.; Zhang, D.D. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol. 2013, 33, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Riley, B.E.; Kaiser, S.E.; Kopito, R.R. Autophagy inhibition engages Nrf2-p62 Ub-associated signaling. Autophagy 2011, 7, 338–340. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Chen, W.; Li, Y.; Villeneuve, N.F.; Zhang, D.D. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: Enhanced Keap1-Cul3 interaction. Toxicol. Appl. Pharmacol. 2008, 230, 383–389. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Sun, R.; Wang, H.; Yang, B.; Wang, F.; Xu, H.; Chen, S.; Zhao, R.; Pi, J.; Xu, Y. Enhanced p62-NRF2 Feedback Loop due to Impaired Autophagic Flux Contributes to Arsenic-Induced Malignant Transformation of Human Keratinocytes. Oxid. Med. Cell. Longev. 2019, 2019, 1038932. [Google Scholar] [CrossRef] [PubMed]
- Schmidlin, C.J.; Shakya, A.; Dodson, M.; Chapman, E.; Zhang, D.D. The intricacies of NRF2 regulation in cancer. Semin. Cancer Biol. 2021, 76, 110–119. [Google Scholar] [CrossRef]
- Iida, K.; Itoh, K.; Kumagai, Y.; Oyasu, R.; Hattori, K.; Kawai, K.; Shimazui, T.; Akaza, H.; Yamamoto, M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004, 64, 6424–6431. [Google Scholar] [CrossRef]
- Ramos-Gomez, M.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis 2003, 24, 461–467. [Google Scholar] [CrossRef]
- Rachakonda, G.; Sekhar, K.R.; Jowhar, D.; Samson, P.C.; Wikswo, J.P.; Beauchamp, R.D.; Datta, P.K.; Freeman, M.L. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene 2010, 29, 3703–3714. [Google Scholar] [CrossRef]
- Satoh, H.; Moriguchi, T.; Taguchi, K.; Takai, J.; Maher, J.M.; Suzuki, T.; Winnard, P.T.; Jr Raman, V.; Ebina, M.; Nukiwa, T.; et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 2010, 31, 1833–1843. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, H.; Fan, L.; Wu, X.; Xin, A.; Ren, H.; Wang, X.J. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic. Biol. Med. 2011, 50, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Matsumoto, T.; Itoi, A.; Kawana, A.; Nishiyama, T.; Ogura, K.; Hiratsuka, A. Enhanced sensitivity of A549 cells to the cytotoxic action of anticancer drugs via suppression of Nrf2 by procyanidins from Cinnamomi Cortex extract. Biochem. Biophys. Res. Commun. 2011, 413, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liang, J.; Li, X.; Liu, L.; Yao, J.; Chen, X.; Chen, R. Renieramycin T Inhibits Melanoma B16F10 Cell Metastasis and Invasion via Regulating Nrf2 and STAT3 Signaling Pathways. Molecules 2022, 27, 5337. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol. Int. 2019, 43, 1245–1256. [Google Scholar] [CrossRef]
- Liu, H.; Ren, G.; Wang, T.; Chen, Y.; Gong, C.; Bai, Y.; Wang, B.; Qi, H.; Shen, J.; Zhu, L.; et al. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial-mesenchymal transition. Carcinogenesis 2015, 36, 459–468. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef]
- Kim, S.J.; Saeidi, S.; Cho, N.C.; Kim, S.H.; Lee, H.B.; Han, W.; Noh, D.Y.; Surh, Y.J. Interaction of Nrf2 with dimeric STAT3 induces IL-23 expression: Implications for breast cancer progression. Cancer Lett. 2021, 500, 147–160. [Google Scholar] [CrossRef]
- Piskin, G.; Sylva-Steenland, R.M.; Bos, J.D.; Teunissen, M.B. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: Enhanced expression in psoriatic skin. J. Immunol. 2006, 176, 1908–1915. [Google Scholar] [CrossRef]
- Sheng, S.; Zhang, J.; Ai, J.; Hao, X.; Luan, R. Aberrant expression of IL-23/IL-23R in patients with breast cancer and its clinical significance. Mol. Med. Rep. 2018, 17, 4639–4644. [Google Scholar] [CrossRef]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar] [PubMed]
- Gao, S.; Zhan, L.; Yang, Z.; Shi, R.; Li, H.; Xia, Z.; Yuan, S.; Wu, Q.P.; Wang, T.; Yao, S. Remote Limb Ischaemic Postconditioning Protects Against Myocardial Ischaemia/Reperfusion Injury in Mice: Activation of JAK/STAT3-Mediated Nrf2-Antioxidant Signalling. Cell Physiol. Biochem. 2017, 43, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.O.; Pratheeshkumar, P.; Divya, S.P.; Zhang, Z.; Shi, X. Nuclear factor erythroid 2-related factor 2 enhances carcinogenesis by suppressing apoptosis and promoting autophagy in nickel-transformed cells. J. Biol. Chem. 2017, 292, 8315–8330. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Liu, H.; Wang, M.; Wang, R.; Yi, G.; Zhang, M.; Chen, R. Role of STAT3 and NRF2 in Tumors: Potential Targets for Antitumor Therapy. Molecules 2022, 27, 8768. https://doi.org/10.3390/molecules27248768
Tian Y, Liu H, Wang M, Wang R, Yi G, Zhang M, Chen R. Role of STAT3 and NRF2 in Tumors: Potential Targets for Antitumor Therapy. Molecules. 2022; 27(24):8768. https://doi.org/10.3390/molecules27248768
Chicago/Turabian StyleTian, Yanjun, Haiqing Liu, Mengwei Wang, Ruihao Wang, Guandong Yi, Meng Zhang, and Ruijiao Chen. 2022. "Role of STAT3 and NRF2 in Tumors: Potential Targets for Antitumor Therapy" Molecules 27, no. 24: 8768. https://doi.org/10.3390/molecules27248768
APA StyleTian, Y., Liu, H., Wang, M., Wang, R., Yi, G., Zhang, M., & Chen, R. (2022). Role of STAT3 and NRF2 in Tumors: Potential Targets for Antitumor Therapy. Molecules, 27(24), 8768. https://doi.org/10.3390/molecules27248768